Tailoring of mechanical properties of derivatized natural polyamino acids through esterification and tensile deformation†
Jessica R.
May
a,
Cristina
Gentilini
a,
David E.
Clarke
a,
Yaroslav I.
Odarchenko
b,
Denis V.
Anokhin
c,
Dimitri A.
Ivanov
*bc,
Kirill
Feldman
d,
Paul
Smith
d and
Molly M.
Stevens
*a
aDepartments of Materials and Bioengineering, Imperial College London, London, SW7 2AZ, UK. E-mail: m.stevens@imperial.ac.uk; Tel: +44(0)20 7594 6804
bInstitut de Sciences des Matériaux de Mulhouse (CNRS UMR 7361), Université de Haute Alsace, 68057, France. E-mail: dimitri.ivanov@uha.fr; Tel: +33(0)3 89 60 88 07
cMoscow State University, Faculty of Fundamental Physical and Chemical Engineering, 119991, Moscow, Russia
dDepartment of Materials, ETH Zürich, 8093, Switzerland
First published on 31st October 2013
Abstract
Tensile deformation was applied to the naturally produced poly-γ-glutamic acid, which can be enzymatically degraded and is, therefore, of interest for biomedical use. However, natural polyamino acids have a similar chemical structure to synthetic polyamides (“nylons”), which are known to feature strong inter-molecular hydrogen bonding that prevents large-scale molecular motion in their solid state. Through esterification, this hydrogen bonding was partially shielded, allowing orientation of the polyamino acid macromolecules through tensile deformation. An increase in Young's modulus and tensile strength was achieved of solution-cast films of the chemically modified poly-γ-glutamic acids, consistent with enhanced uniaxial polymer chain orientation. The latter was confirmed by both wide-angle X-ray scattering and polarized Raman spectroscopy. The films thus produced were found to be non-cytotoxic. These mechanically tailorable, biocompatible polymers may be excellent candidates for use in musculoskeletal tissue engineering applications that have different loading requirements within the body.
Introduction
Natural polymers with mechanical properties that can be tailored hold great potential for regenerative medicine applications as loading requirements of particular tissues vary greatly within the human body.1,2 For example, within a single vertebra, the stiffness – as expressed in terms of the Young's modulus – of the ligamentum flavum3 and anterior longitudinal ligament4 differ by an order of magnitude. Manipulation of such mechanical properties by means of tensile deformation of solid polymers under appropriately selected conditions of temperature and strain rate has been extensively utilized to produce highly oriented fibers and films of synthetic polymers that feature an extremely wide range of stiffness and tensile strength. For instance, in the case of ultra-high molecular weight polyethylene (UHMWPE), employing tensile deformation its Young's modulus has been varied from about 0.5 GPa to well over 200 GPa, a value approaching its theoretical stiffness,5–7 and allowing adaptation of the latter materials for numerous applications ranging from bullet-proof vests to high-performance surgical sutures. Tensile deformation, thus, could present an efficient opportunity to produce biomaterials for tunable regenerative medicine applications.Here, we sought to investigate the application of tensile deformation to naturally produced polyamino acids, more specifically poly-γ-glutamic acid, which can be enzymatically degraded and represents an excellent candidate for biomedical use. It should be borne in mind, though, that natural polyamino acids have a chemical nature similar to that of synthetic polyamides (“nylons”), which have a notoriously low ability to be oriented by tensile deformation due to strong inter-molecular hydrogen bonding that severely hampers rearrangement of the constituent macromolecules in their solid state.8 That considered, polyamino acids can be chemically modified in such a way as to at least partially shield those hydrogen bonds, which would allow for increased macromolecular mobility and permit the polymer to be oriented by deformation in its solid state. In this article, we report that, indeed, mechanical properties of the naturally produced polyamino acid, poly-γ-glutamic acid, can be controlled through esterification and subsequent tensile deformation.
Materials and methods
Chemicals
All starting polymer samples (γ-D,L-PGA Na+ salt, produced by Bacillus subtilis) were purchased from Natto Biosciences (Shanghai, China). All reagents and solvents were acquired from Aldrich (Gillingham, UK) and used as received, unless otherwise specified.Polymer synthesis
γ-D,L-PGA-Na+ salt was acidified using HCl in distilled water to pH 1.5 at room temperature RT and subsequently lyophilized to yield γ-PGA-H. Esterification of γ-PGA-H was performed as previously described,9,10 using ethyl, propyl, or benzyl bromide to yield γ-PGA-Et, γ-PGA-Pr, and γ-PGA-Bn, respectively.Chemical and thermal characterization
Chemical structures of the polymers were analyzed using 1H-NMR and 13C-NMR spectroscopy. NMR spectra were recorded on a Bruker Biospin GmbH AV500 (operating at 500 MHz for 1H; 125.76 MHz for 13C) using DMSO-d6 as deuterated solvent. Successful conjugation of γ-PGA-H into γ-PGA ethyl, proyl and benzyl esters was confirmed by integration of the –NH peak from the 1H-NMR spectra (cf. Fig. S1†). ATR-FTIR spectra were obtained with a Spectrum 100 FTIR Spectrophotometer (Perkin Elmer, Seer Green, UK), from 500 to 4000 cm−1 using the Universal diamond attenuated total reflectance (ATR) top-plate and reported elsewhere.11 Size-exclusion chromatography (SEC) was performed with esterified γ-PGA using a PL-GPC 220 integrated gel permeation chromatograph/size exclusion chromatograph (SEC) fitted with a differential refractometer, a PL-220R viscometer, and a PL-PD2040 light scattering detector (all Varian, Inc.; Palo Alto, CA, USA). Solutions of esterified γ-PGA (1.5 mg mL−1 in dimethylformamide (DMF) containing 1 g L−1 LiBr) were prepared and injected through two 10 μm PLgel MIXED-B columns connected in series using DMF (1 mL min−1 flow rate, 45 °C) with monodisperse poly(methyl methacrylate) (PMMA) solutions as reference standards. SEC of γ-PGA-H form was performed using sodium polyacrylate standards, in phosphate buffer (pH 7) at Smithers Rapra (UK). Dynamic thermogravimetric analysis (TGA) was performed on 5–7 mg γ-PGA polymer samples at a scanning rate of 2 °C min−1 under N2 at 50 mL min−1 from 25–300 °C with a TGA/SDTA851e instrument (Mettler-Toledo, Greifensee, Switzerland), calibrated with indium and zinc. Glass transition (Tg) and melting (Tm) temperatures were determined with a Mettler-Toledo DSC822e differential scanning calorimeter (Mettler-Toledo, Greifensee, Switzerland), calibrated with indium and zinc. Samples of 5–7 mg sealed in aluminum crucibles were heated under nitrogen at a rate of 10 °C min−1 from −25 °C until 10–20 °C above Tm. Melting temperatures reported refer to the maximum in the endothermal peaks. The second heating on the same sample was treated as melt crystallized, and glass transition temperatures were measured as the midpoint change in slope of this trace.Film casting
Esterified γ-PGA samples were dissolved at RT in HFIP (2% w/w), subsequently poured into fluorinated glass petri dishes, covered and the solvent was allowed to evaporate at RT for 48 hours, followed by 24 hours under vacuum at 60 °C to remove any remaining solvent. γ-PGA-H was dissolved in diH2O (2% w/w), allowed to evaporate at RT for 96 hours, and then placed under vacuum to prevent rehydration. Sample concentrations were calculated to produce films approximately 100 μm thick.RT tensile testing
Initial mechanical testing of as-cast, dried films was performed at RT with an Instron Tensile Tester Model 5864 (Norwood, USA) with hydraulic clamps (5 bar) at a cross-head speed of 2 mm min−1. Dumbbell shaped strips of 1.2 mm width and 4.5 mm gauge length were cut from the dried films.Tensile deformation of esterified γ-PGA polymers
Tensile deformation of the dried, esterified γ-PGA films was performed above their respective Tg, using an Instron Tensile Tester (Model 5864) fitted with an environmental chamber (Model EC43; Norwood, USA) at a cross-head speed of 2 mm min−1. Dumbbell shaped strips were cut as for RT testing; ink marks were printed at 1 mm intervals prior to drawing to determine draw ratios. γ-PGA-Et and γ-PGA-Pr samples were drawn at 125 °C while γ-PGA-Bn samples were drawn at 115 °C, as maximum draw ratios (λmax) were achieved at these respective temperatures.Mechanical testing of oriented polymer films
Ink mark displacement was employed to determine local draw ratios, samples were then cut and glued using Al-Fix glue (Novatio; Olen, Belgium) between card frames (separation distance = 10 mm), and allowed to dry for 2 hours under 200 g weights to ensure a strong bond. Sample widths were measured using an optical microscope, while thicknesses were determined using a micrometer. Mechanical testing of the different samples was performed at RT using hydraulic clamps as above, at a cross-head speed of 5 mm min−1.Optical microscopy
All optical microscopy was carried out with a Leica DMRX polarising microscope equipped with a Leica DFC 480 Camera (Wetzlar, Germany).Wide-angle X-ray scattering (WAXS)
WAXS experiments were performed on the BM26 beamline of the ESRF (Grenoble, France) using a FReLoN detector positioned at approximately 6 cm from the sample. This sample-to-detector distance allowed recording of the signals in the s-range (s = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinΘ/λ, where Θ is the Bragg angle) from 0.03 to 0.52 Å−1 using the wavelength of 1.04 Å. Additional measurements were carried out on a custom-built SAXS/WAXS machine (Molecular Metrology) coupled to a Rigaku MicroMax-007HF rotating anode generator. The 2D WAXS data were collected in vacuum using Fuji image plates with the pixel size of 100 × 100 μm2. The modulus of the scattering vector was calibrated using diffraction orders of silver behenate in both setups. The measurements were performed in transmission mode on the oriented samples (films with a thickness in the range 0.1–0.3 mm). The structure of cast polymer films was addressed by WAXS using a PANalytical MPD X-ray diffractometer (Almelo, Netherlands) and reported elsewhere.11 Measurements were taken using a Cu-Kα source with a secondary graphite monochromator (step size = 0.03°, 50 s count time per step).
sinΘ/λ, where Θ is the Bragg angle) from 0.03 to 0.52 Å−1 using the wavelength of 1.04 Å. Additional measurements were carried out on a custom-built SAXS/WAXS machine (Molecular Metrology) coupled to a Rigaku MicroMax-007HF rotating anode generator. The 2D WAXS data were collected in vacuum using Fuji image plates with the pixel size of 100 × 100 μm2. The modulus of the scattering vector was calibrated using diffraction orders of silver behenate in both setups. The measurements were performed in transmission mode on the oriented samples (films with a thickness in the range 0.1–0.3 mm). The structure of cast polymer films was addressed by WAXS using a PANalytical MPD X-ray diffractometer (Almelo, Netherlands) and reported elsewhere.11 Measurements were taken using a Cu-Kα source with a secondary graphite monochromator (step size = 0.03°, 50 s count time per step).
Cell cytotoxicity ISO 10993:5 test
All reagents for cell culture were used as purchased from Invitrogen (Paisley, UK) unless stated otherwise. Salts were purchased from Sigma-Aldrich (Gillingham, UK), the Triton X-100 from BDH laboratories (Poole, UK).Following the ISO 10993:5 protocol, modified γ-PGA films were sterilized for 2 hours on each side under UV light and prepared with a material surface area to culture medium ratio of 6 cm2 mL−1 according to thin-film requirements outlined in ISO10993:12. Similarly, controls were calculated with an extraction ratio of 3 cm2 mL−1 [negative, non-toxic PVC (Med7539 noDop) tubing; positive, organo-tin stabilized PVC sheet, t > 0.5 mm; both kindly supplied by Raumedic (Munchberg, Germany)], sterilized using 70% ethanol for 40 minutes and allowed to air dry.
The mouse osteoblast cell line MC3T3-E1 (European Collection of Cell Cultures; Salisbury, U.K.) was routinely cultured under standard conditions (37 °C, 5% CO2, 100% humidity) in Alpha Minimum Essential Medium (α-MEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 2 mM L-glutamine (all Invitrogen; Paisley, U.K.).
Sterile polymer film and control samples were soaked in α-MEM for 7 days at 37 °C, 5% CO2 to make the conditioned media and test for the release of any cytotoxic agents from the films. MC3T3-E1 cells were seeded into 96 well plates at a density of 20000 cells cm−2 in standard culture medium (as above). After 24 hours, the medium was exchanged with conditioned culture medium (supplemented with 10% v/v FBS and 1% v/v L-glutamine) and diluted by factors of 2, 4, 8, or 16 with standard culture medium for 24 hours, upon which the cellular metabolic activity was assessed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide).
For the assay, 20 μL of a sterile-filtered 5 mg mL−1 solution of MTT in Dulbecco's phosphate buffered saline (DPBS, pH 7.4) was added to the culture media and incubated for 2 hours. Upon completion of incubation, media was removed, cells washed with PBS, and 200 μL DMSO was pipetted in wells to dissolve the formazan product. Finally, 150 μL of formazan/DMSO was transferred to a fresh 96 well plate and read on a colorimetric plate reader at 592 nm (background subtraction at 620 nm). All results are normalized to negative control conditioned, media-treated MC3T3-E1 cell metabolic activity.
Results and discussion
Chemical modification
The highly hydrophilic poly-γ-glutamic acid (γ-PGA-H) was esterified using ethyl, propyl, or benzyl bromide as previously described,9,10 yielding, resp. γ-PGA-Et, γ-PGA-Pr, and γ-PGA-Bn (Fig. 1). The weight-average molecular weight (Mw), glass transition-(Tg), melting-(Tm) and onset-of-decomposition (Td, taken at 5% weight loss) temperatures of the resulting materials are presented in Table 1. | ||
| Fig. 1 Chemical structures of polymers explored. | ||
| Polymer | M w | T g | T m | T d |
|---|---|---|---|---|
| a GPC performed under different conditions than for esterified γ-PGAs. b Degradation detected by TGA prior to melting. c Double endotherm, second peak at 240 °C. | ||||
| γ-PGA-Ha | 4.89 × 105 | 124 | 240b | 165 |
| γ-PGA-Et | 2.75 × 105 | 66 | 260c | 238 |
| γ-PGA-Pr | 1.97 × 105 | 40 | 228 | 230 |
| γ-PGA-Bn | 3.22 × 105 | 40 | 226 | 231 |
Not unexpectedly, esterification was accompanied by a loss of the molecular weights of the materials; although respectable and useful values remained. The thermal properties of the modified polyamino acids were beneficially affected by the esterification procedure. Their glass transition temperature was reduced, generally leading to less brittle materials. The values of Tm remained desirably high, and their thermal stability actually was significantly enhanced.
Mechanical properties
Mechanical properties of the native γ-PGA-H and esterified γ-PGA films were analyzed in standard tensile tests performed at room temperature (RT). Typical stress–strain curves recorded for these samples are presented in Fig. 2. Mechanical characteristics derived from them, i.e. Young's modulus (E), tensile strength (σ), and strain at break (ε), are listed in Table 2. As expected, due to hydrogen bonding between the polymer's carboxylic groups, films of the native γ-PGA-H were found to be relatively brittle when tested at RT (cf.Fig. 2).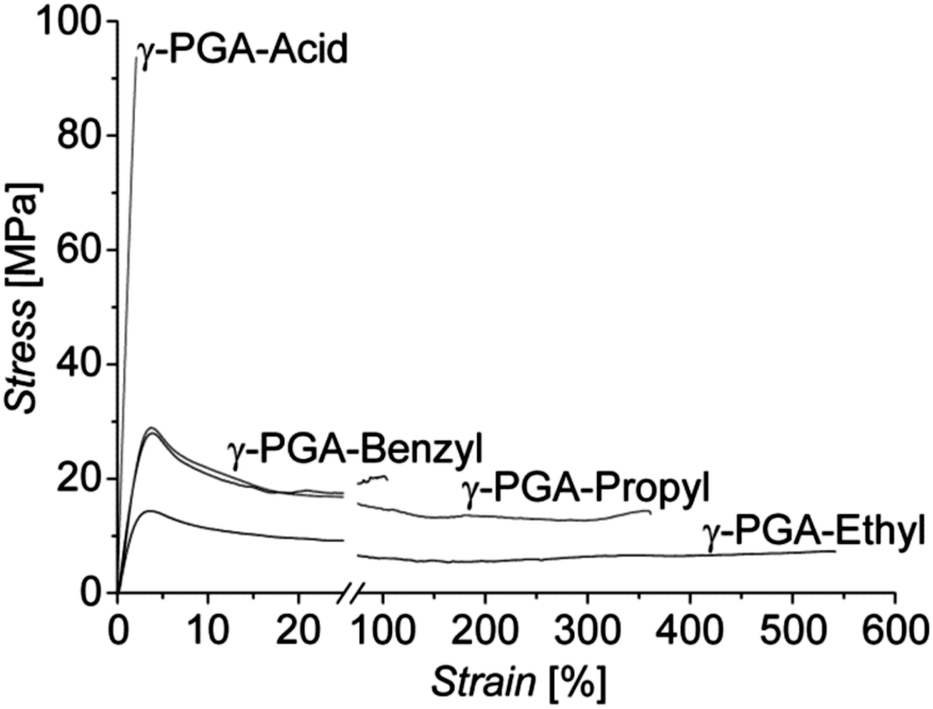 | ||
| Fig. 2 Representative nominal stress–strain curves of native, brittle γ-PGA-H and the esterified γ-PGA polymers recorded at RT, displaying high strains at break. | ||
| Polymer | E [GPa] | σ [MPa] | ε [%] |
|---|---|---|---|
| γ-PGA-H | 5.0 | 94 | 2 |
| γ-PGA-Et | 0.9 | 15 | 542 |
| γ-PGA-Pr | 1.4 | 29 | 360 |
| γ-PGA-Bn | 1.2 | 28 | 106 |
Esterification of γ-PGA-H introducing alkyl side chains, therewith inhibiting the formation of H-bonds between the macromolecules, resulted in highly enhanced ductility of films fabricated from them, with a dramatic increase in the strain at break of up to approximately 50, 150, and 250 times that of γ-PGA-H observed for γ-PGA-Bn, γ-PGA-Pr, and γ-PGA-Et, respectively (Fig. 2). The increased strain at break was, however, expectedly accompanied by a decrease in values of the stiffness E, and of both the yield and tensile strengths (Table 2).
Tensile deformation
In subsequent experiments, stress–strain curves of the various γ-PGA films were recorded at different temperatures in the range from RT to the melting temperature (Tm) in order to establish the maximum draw ratio of the different polymers λmax, which is defined as:| λmax = (ε/100) + 1 | (1) |
Films of γ-PGA-H could not be drawn to high draw ratios (λ ≤ 4) at RT and elevated temperatures due to hydrogen bonding between the macromolecular chains. By contrast, films of all of the esterified γ-PGAs could readily be drawn to λ ≥ 10 at their respective optimum temperature (125 °C for both γ-PGA-Et and γ-PGA-Pr, and 115 °C for γ-PGA-Bn), thereby significantly surpassing the “natural” draw ratio of γ-PGA-H and synthetic polyamides (λ = 4–6).8,12 Above these optimum temperatures, λmax rapidly decreased due to excessive macromolecular mobility, as is commonly observed for weakly interacting polymers such as polyethylene.13,14
Subsequently, films of the esterified γ-PGAs were drawn at their respective optimum deformation temperatures to various draw ratios and then subjected to tests at RT to determine their mechanical properties (Fig. 3). Common to all esterified polymer samples, both Young's modulus and tensile strength rapidly increased with increasing draw ratio, while the strain at break decreased, as is commonly observed for oriented polymers. To produce a comparable set of results between the polymers with different side chains, films were drawn to λ ∼ 10. Nominal stress–strain curves recorded at RT for these drawn films are presented in Fig. 3. Maximum values of the mechanical characteristics determined for these samples are presented in Table 3.
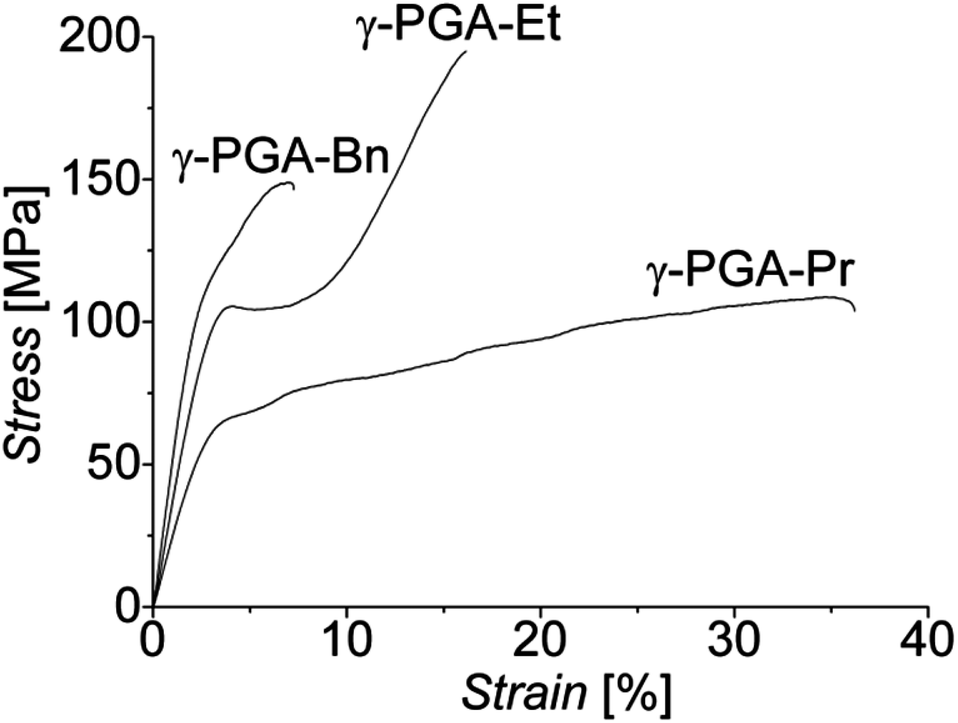 | ||
| Fig. 3 Nominal stress–strain curves recorded at RT for esterified γ-PGA films drawn to λ ∼ 10. | ||
| Polymer | E [GPa] | σ [MPa] | ε [%] |
|---|---|---|---|
| γ-PGA-Et | 3.9 | 196 | 16 |
| γ-PGA-Pr | 2.5 | 109 | 36 |
| γ-PGA-Bn | 5.4 | 149 | 7 |
By tensile deformation to λ ∼ 10, the RT modulus of γ-PGA-Et films increased by approximately a factor of 4.5 and the tensile strength by over a factor of 13. Benzyl esterified γ-PGA offered the highest modulus, with a 4.3 fold increase to 5.4 GPa following tensile deformation to the same draw ratio. The tensile strength achieved with γ-PGA-Bn was found to be 149 MPa, a significant increase of more than 5-fold, but still inferior to that of γ-PGA-Et. Drawing γ-PGA-Pr resulted in close to doubling the value of its modulus and a tripling of the tensile strength, but with a 10-fold decrease in strain at break.
In accord with results obtained for tensile drawn common oriented polymers, such as polyesters, nylons, polyolefins, etc., the above results clearly indicate that all esterified γ-PGA films significantly benefitted in a similar fashion from this mechanical treatment.
A plot of the tensile strength vs. the Young's modulus of all present samples is presented in Fig. 4. This classical graph demonstrates that, importantly, the properties achieved with these materials approach values similar to those of soft human tissues (cf.Fig. 4); of relevance is that their strain at break are at or above the physiologically relevant 7%.15–17
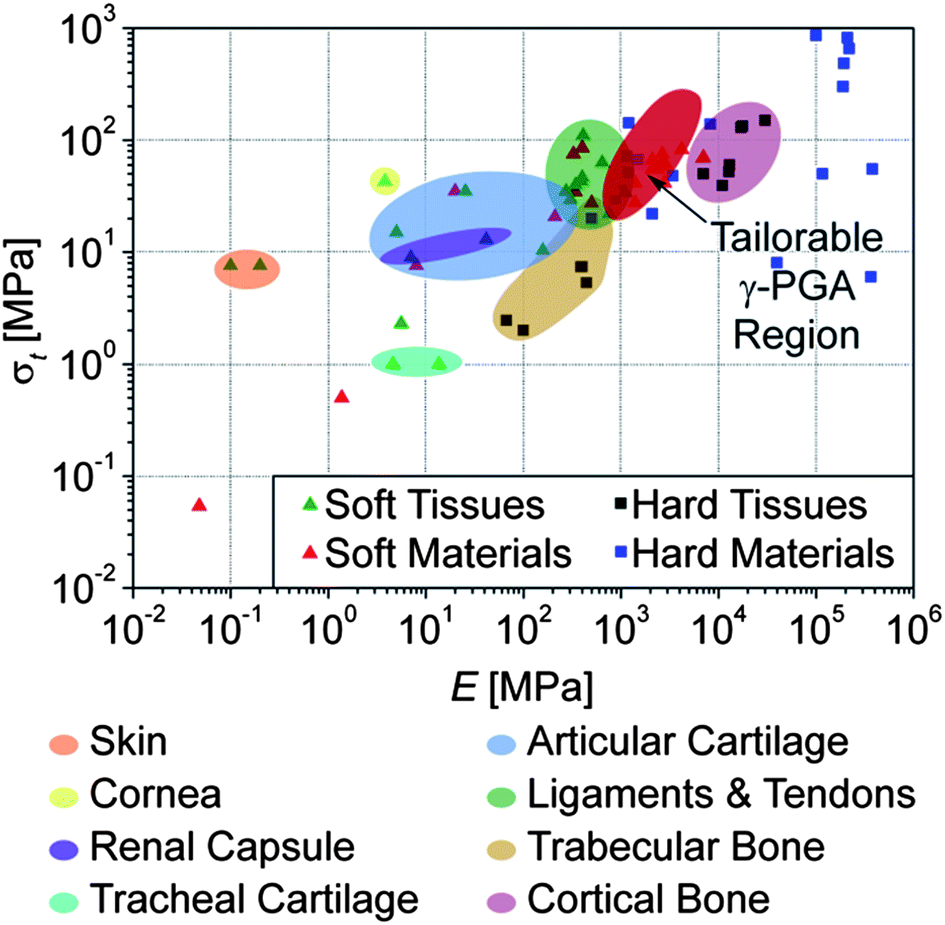 | ||
| Fig. 4 Tensile strength vs. stiffness of all modified γ-PGA polymer samples, isotropic and tensile drawn to λ ≤ 10, compared with soft and hard (mineralized) human tissues, as well as materials commonly used as replacements (References available in ESI, Table S1†). | ||
An interesting phenomenon was discovered upon testing drawn esterified γ-PGA polymer samples at RT. For films drawn to ratios approximately λ > 5, a double yield point was observed in the stress–strain curves for all three polymer species (Fig. 3). This double yield point could be indicative of an α-helix conformation of the macromolecules.18 This feature and the increased macromolecular orientation were further investigated using wide-angle X-ray diffraction (WAXD).
Structure of the drawn films
Typical wide-angle X-ray diffraction patterns of drawn (λ ∼ 10) esterified γ-PGA samples are presented in Fig. 5a–d. All patterns clearly indicate a high degree of uniaxial orientation in the direction of deformation. It is noteworthy that the diffraction patterns of γ-PGA-Et and γ-PGA-Pr are very similar. For example, for γ-PGA-Pr a weak meridional peak (i.e. in the fiber direction) appears on the second layer line, which indicates a 2/1 helical conformation of the backbone with the helix subunit parameter of 4.66 Å, or the c-parameter (fiber repeat) of 9.23 Å (Fig. 5a). To ensure that the chain conformation is indeed that of the 2/1 helix, the meridional direction of the pattern was explored by tilting the oriented film in the incidence plane by about 20°. It was found that the only higher order meridional peak visible for the tilted sample (004) is less intense than the mentioned 002 peak (cf.Fig. 5b), which shows indeed that the conformation is most likely a 2/1 helix. The value of the a-parameter equals 20.07 Å, which is greater than the length of the fully extended lateral ester groups (14.5 Å) corresponding to spread in the opposite directions. This indicates that the unit cell accommodates more than one chain.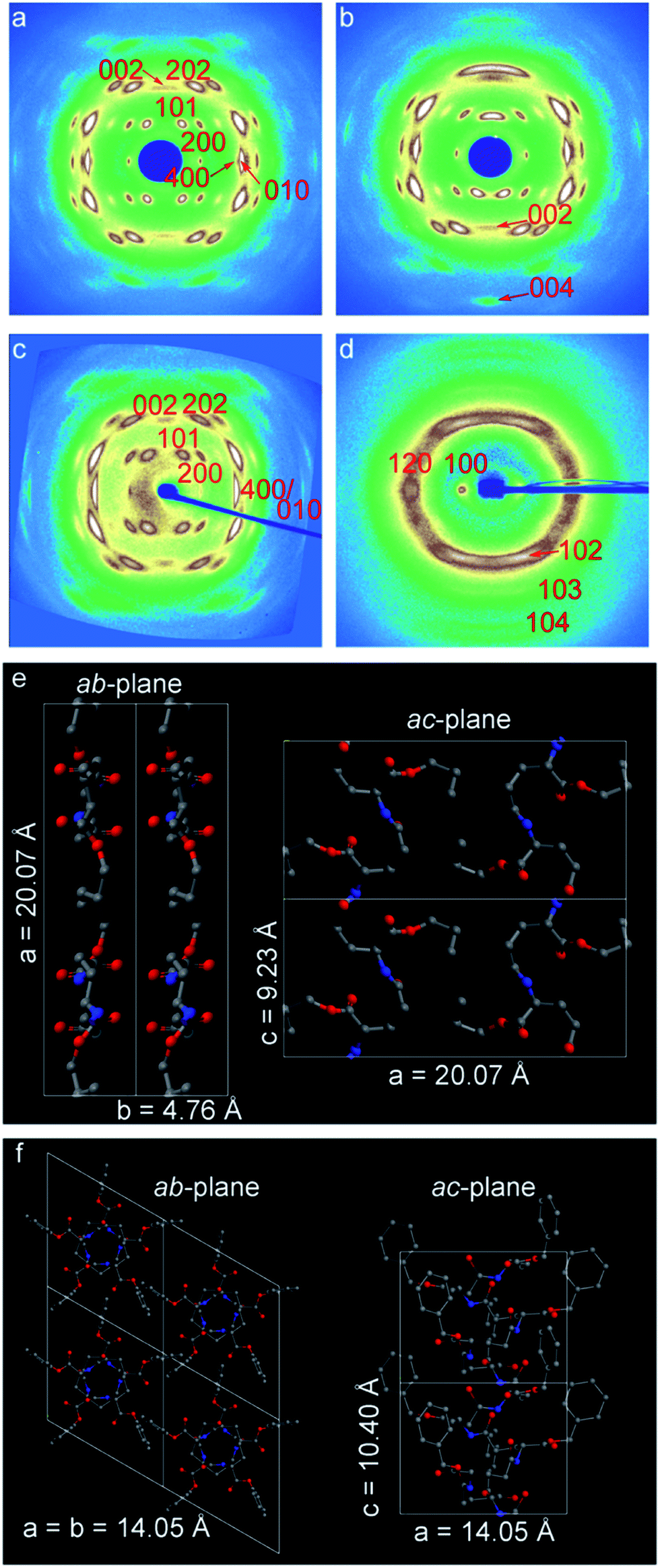 | ||
| Fig. 5 2D-WAXD patterns recorded for highly oriented samples of (a) γ-PGA-Pr, (b) the same film as in (a) tilted by 20°, (c) γ-PGA-Et and (d) γ-PGA-Bn. The patterns are given as a function of the norm of the reciprocal space vector s, which ranges between −0.48 to 0.48 Å−1. Schematic representations of the 2/1 helix for (e) γ-PGA-Pr and 5/2 helix for (f) γ-PGA-Bn. | ||
The proposed unit cells for γ-PGA-Et and γ-PGA-Pr polymers are both orthorhombic; the unit cell parameters are a = 18.40 Å, b = 4.55 Å, c = 9.13 Å and a = 20.07 Å, b = 4.76 Å, c = 9.23 Å, respectively. The unit cells contain two chains and 4 monomers in total, which is in agreement with density measurements. The fact that it is mainly the a parameter that is affected by the lateral chain length, means that lateral chains are positioned within the hydrogen-bonded sheets and determine the inter-chain distance. A table summarizing both the observed and calculated d-spacings is presented in the ESI (Table S2†).
The WAXD pattern of γ-PGA-Bn differs significantly from that of γ-PGA-Et and γ-PGA-Pr. The former pattern was indexed to a hexagonal unit cell (a = b = 14.05 Å, c = 10.40 Å), which is in agreement with the previously published data for γ-PGA-Bn.18,19 In this case, the chain conformation is that of 5/2 helix, which corresponds to a strongly non-planar backbone. Schematics of the 2/1 helix of γ-PGA-Pr and 5/2 helix of γ-PGA-Bn are shown in Fig. 5(e and f). The corresponding simulated fiber X-ray patterns show a reasonable agreement with the actual diffractograms (cf. Fig. S4†).
Importantly, the difference in the chain conformation between the γ-PGA-Et, γ-PGA-Pr and γ-PGA-Bn can be conveniently addressed with Raman spectroscopy. The measurements of dichroic ratios of the amide and ester peaks allow identifying the direction of hydrogen bonds with respect to the backbone. Thus, for γ-PGA-Et and γ-PGA-Pr the hydrogen bonds are found to be arranged perpendicular to the orientation direction, i.e. perpendicular to the plane of the sheets (cf. Fig. S5†), whereas for γ-PGA-Bn they are parallel to it.19 The latter observation is more compatible with predominant formation of the intra-molecular hydrogen bonds.
Gratifyingly, the mechanical properties, in particular the Young's modulus, recorded for the different, oriented γ-PGA films appear consistent with above X-ray analysis. While it is readily envisioned that the stiffness of γ-PGA-Pr is below that of γ-PGA-Et due to the increased cross-sectional area of the former macromolecule, at first sight, one may be puzzled by the fact that the Bn-derivative with its largest transverse chain dimension, featured the higher Young's modulus. That observation is now readily understood given the conclusion that in γ-PGA-Et and -Pr, hydrogen bonds are perpendicular to the chain direction while those in γ-PGA-Bn are parallel to it.
Cytocompatibility
Esterification of natural polymers is an accepted approach for biomedical materials. For example, hyaluronic acid has been esterified to produce the clinical grade HYAFF® polymers, which are modified with ethyl or benzyl esters in order to reduce their sensitivity to water.20,21We have reported elsewhere the controlled chemical functionality of these materials when formed into 3-D electrospun fibre scaffolds as versatile, enzymatically-degraded scaffolds for tissue engineering applications.11 However, in order to assess whether the esterified γ-PGA polymers would release any cytotoxic dissolution products that would have any adverse effect on cytocompatibility, we conducted the ISO 10993:5 test on the modified polymers.22 This standard test assesses whether the materials release any toxic dissolution products that cause a detectable reduction in cellular metabolic activity level, as assessed using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.
Mouse pre-osteoblastic cells (MC3T3-E1) displayed a spread, normal morphology when cultured in conditioned medium soaked with either the negative control (medically approved PVC) or esterified γ-PGA films, while those exposed to positive control (cytotoxic organo-tin stabilized PVC) conditioned medium appeared abnormally round. Metabolic activities of cells exposed to all experimental esterified γ-PGA film soaked media were similar to that of the negative control, while MTT activity of positive control treated cells was significantly lower than all other tested groups (p < 0.001; Fig. 6). The fact that all esterified γ-PGA polymers pass the ISO 10993:5 test is highly encouraging for their potential use in tissue engineering scaffolds.
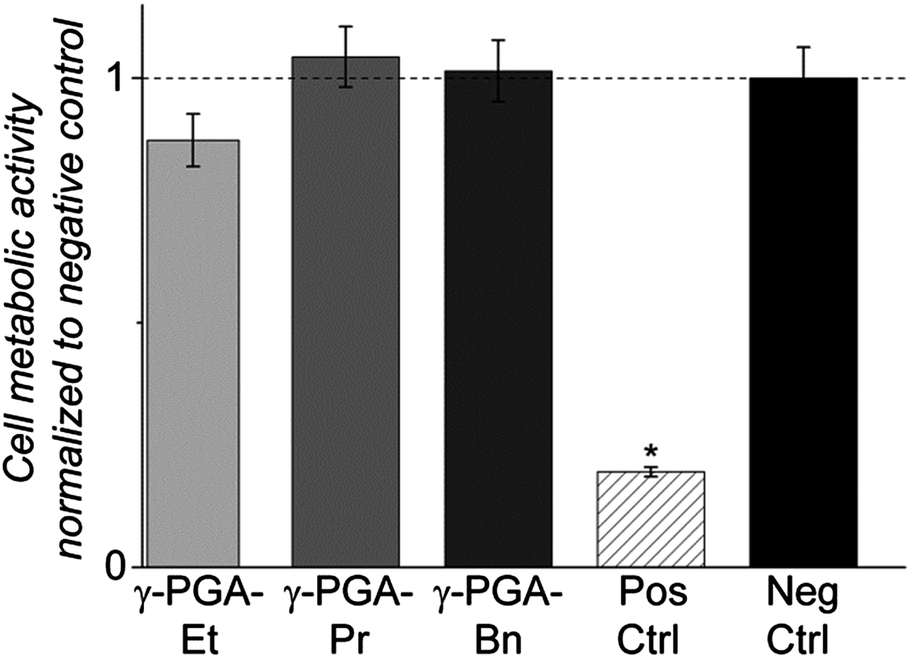 | ||
| Fig. 6 ISO 10993:5 pre-clinical test of esterified γ-PGA films, normalized to negative control. All polymers passed and metabolic activities were not significantly different to the negative control, but were significantly different from the positive control (cytotoxic PVC) (* p < 0.001). | ||
Conclusions
In conclusion, through chemical modification of the naturally produced polymer poly-γ-glutamic acid, we have developed a set of materials and a wide range of mechanical properties that can readily be tailored through common tensile deformation procedures and with in vitro biocompatibility. These polymers may be excellent candidates for use in musculoskeletal tissue engineering applications that have different loading requirements within the body. We anticipate that increasing the molecular weight of these polymers will further increase the range over which they may be customized is the subject of ongoing study.Acknowledgements
The authors acknowledge the Rosetrees Trust for funding this work and Geordies' at ETH Zürich for inspiration and stimulating discussions. The authors are also grateful to Wim Bras and Giuseppe Portale from the DUBBLE beamline (ESRF, France) for fruitful discussions and excellent technical support. The authors thank the financial support provided by the Russian Ministry of Science and Education (grants 11.G34.31.0055 and 14.518.11.7013) and the European project Interreg IV Rhin-Solar (no C25).References
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- C. R. Ethier and C. A. Simmons, Introductory biomechanics: from cells to organisms, Cambridge University Press, New York, 2007 Search PubMed.
- P. Neumann, T. S. Keller, L. Ekström, L. Perry, T. H. Hansson and D. M. Spengler, Journal of Biomechanics, 1992, 25, 1185–1194 CrossRef CAS.
- P. A. Irvine and P. Smith, Macromolecules, 1986, 19, 240–242 CrossRef CAS.
- I. M. Ward, Mechanical properties of solid polymers, Wiley, Chichester, Sussex, New York, 1983 Search PubMed.
- T. Kanamoto, A. Tsuruta, K. Tanaka, M. Takeda and R. S. Porter, Polym. J., 1983, 15, 327–329 CrossRef CAS.
- J. Smook, G. J. H. Vos and H. L. Doppert, J. Appl. Polym. Sci., 1990, 41, 105–116 CrossRef CAS.
- H. Kubota, Y. Nambu and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2877–2878 CrossRef CAS.
- H. Kubota, Y. Nambu and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 85–88 CrossRef CAS.
- C. Gentilini, Y. Dong, J. R. May, S. Goldoni, D. E. Clarke, B.-H. Lee, E. T. Pashuck and M. M. Stevens, Adv. Healthcare Mater., 2012, 1, 308–315 CrossRef CAS PubMed.
- Y. Termonia, Macromolecules, 1996, 29, 4891–4894 CrossRef CAS.
- G. Capaccio, T. A. Crompton and I. M. Ward, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 301–309 CrossRef CAS.
- P. Smith and P. J. Lemstra, J. Mater. Sci., 1980, 15, 505–514 CrossRef CAS.
- K.-N. An, Functional Tissue Engineering, Springer-Verlag, New York, 2003 Search PubMed.
- B. D. Beynnon, B. C. Fleming, R. J. Johnson, C. E. Nichols, P. A. Renstrom and M. H. Pope, The American Journal of Sports Medicine, 1995, 23, 24–34 CrossRef CAS PubMed.
- M. L. Hull, G. S. Berns, H. Varma and H. A. Patterson, Journal of Biomechanics, 1996, 29, 199–206 CrossRef CAS.
- T. Iwata, M. Fujita, Y. Aoyagi, Y. Doi and T. Fujisawa, Biomacromolecules, 2005, 6, 1803–1809 CrossRef CAS PubMed.
- J. Melis, D. Zanuy, C. Alemán, M. García-Alvarez and S. Muñoz-Guerra, Macromolecules, 2002, 35, 8774–8780 CrossRef CAS.
- D. Campoccia, P. Doherty, M. Radice, P. Brun, G. Abatangelo and D. F. Williams, Biomaterials, 1998, 19, 2101–2127 CrossRef CAS.
- V. Vindigni, R. Cortivo, L. Iacobellis, G. Abatangelo and B. Zavan, Int. J. Mol. Sci., 2009, 10, 2972–2985 CrossRef CAS PubMed.
- H. J. Johnson, S. J. Northup, P. A. Seagraves, P. J. Garvin and R. F. Wallin, J. Biomed. Mater. Res., 1983, 17, 571–586 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra; images of representative polarized optical microscopy of highly oriented polymer films; representative first heating differential scanning calorimetry thermograms; chemical properties of polymers used in this work; detailed resources used to compile data for Fig. 4; observed and calculated X-ray diffraction d-spacings corresponding to γ-PGA-Et, γ-PGA-Pr and γ-PGA-Bn; equations used for calculation of simulated fiber X-ray patterns; and polarized Raman spectra corresponding to oriented films. See DOI: 10.1039/c3ra44865g |
| This journal is © The Royal Society of Chemistry 2014 |