DOI:
10.1039/C3RA44793F
(Paper)
RSC Adv., 2014,
4, 3355-3363
Salphen metal complexes as tunable G-quadruplex binders and optical probes†
Received
30th August 2013
, Accepted 2nd December 2013
First published on 3rd December 2013
Abstract
A series of seven new metal complexes (metal = NiII, CuII, PtII and VO2+) with substituted salphen ligands have been prepared and their duplex and G-quadruplex DNA affinities determined. The selectivity of the complexes towards a given DNA topology is dictated by several factors including geometry, overall charge and substitution pattern of the complex. We also show that the two platinum(II)–salphen complexes developed as part of this series are emissive. Confocal microscopy studies were carried out with these two complexes using four different cell lines (CHO, HeLa, U2OS and HepG2). These studies showed that the cell permeability and localization are different for the two probes and highly dependent on the cell line used.
Introduction
Guanine-rich sequences of DNA can fold into quadruply-stranded structures known as G-quadruplexes. Recently, these non-canonical DNA structures have received significant attention due to the increasing number of biological roles they have been associated with.1–5 In addition, they have been proposed to be alternative and attractive targets for the development of anticancer drugs.6–8 Therefore, there is continuous interest in developing small molecules (both organic9,10 and metal–organic11) that can interact selectively with these structures and in doing so interfere with biological processes such as gene regulation6,12 and telomere maintenance.13
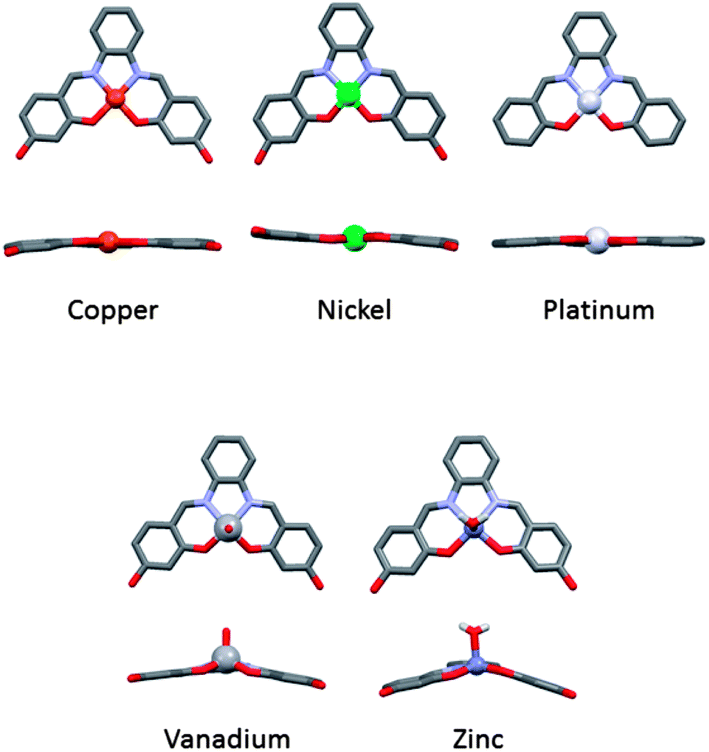
We have previously reported that metal–salphen complexes can be very good G-quadruplex DNA binders.14,15 Their affinity for a given topology of DNA is highly dependent on the overall geometry of the complex as well as the nature of the salphens' substituents. The geometry of these complexes is largely defined by the metal center as highlighted in Fig. 1 which shows the molecular structure of several metal–salphen complexes for which the X-ray crystal structures have been previously determined. In the case of nickel(II),16 copper(II)17 and platinum(II),18 the coordination geometry around the metal cation is square planar. On the other hand, with vanadium(IV),19 the resulting complexes have a square-based pyramidal geometry with an oxo ligand in the axial position. It is worthwhile noting that the phenyl groups of the ligand in this complex are nearly coplanar yielding a practically flat surface (with the potential of displaying π–π stacking interactions). In contrast, the zinc(II) salphen complexes20 have a considerably distorted square-based pyramidal geometry in which the phenyl rings of the ligand are clearly not coplanar.
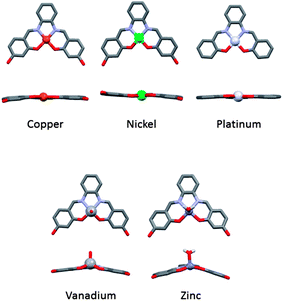 |
| | Fig. 1 X-ray structures of a series of analogous metal salphens.16–20 | |
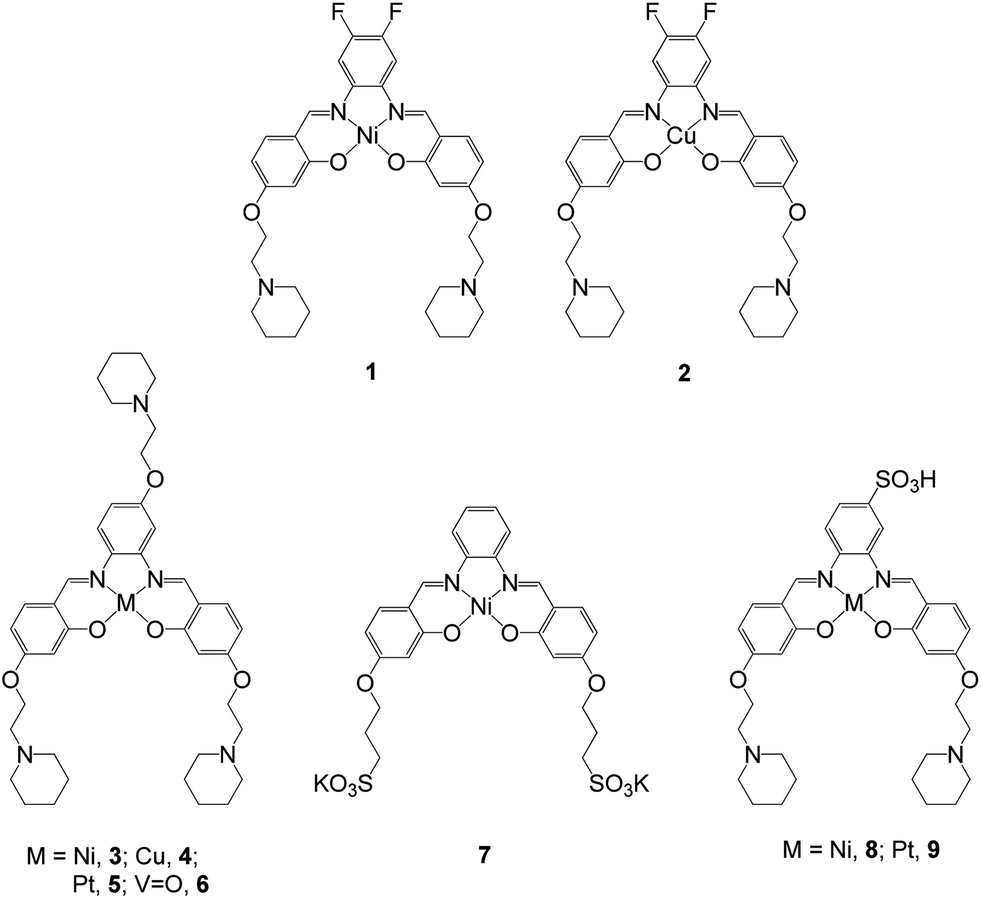
Two square planar metal–salphen complexes (1 and 2 – Fig. 2) have been previously co-crystalized with quadruplex DNA and their X-ray crystal structures determined.21 These studies showed that 1 and 2 bind to the guanine quartet via end-stacking as had been initially predicted.14 The crystal structures also highlighted that the substituents on the metal–salphen core play an important role in defining the exact binding geometry between the metal complexes and G-quadruplex DNA. This in turn, is bound to have a bearing on the affinity and selectivity of the molecules towards a given topology of DNA. Thus, we decided to expand our investigations to study more systematically the effect of different substituents as well as the metal center on the quadruplex DNA binding properties of metal salphen complexes. Herein we report a series of seven new metal complexes with substituted salphen ligands and their duplex and G-quadruplex DNA affinities. We also show that the platinum(II) complexes developed as part of this series are emissive (as previously reported for other platinum(II) salphens22) and cell permeable, which have allowed us to carry out confocal microscopy studies.
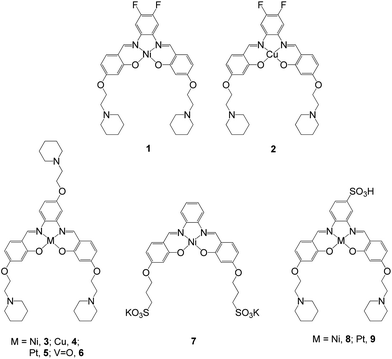 |
| | Fig. 2 Structures of metal–salphen complexes under study. Complexes 1 and 2 have been previously reported and shown by X-ray crystallography to bind to quadruplex DNA via end-stacking.21 Complexes 3–9 are new compounds that are the focus of the current study. | |
Results and discussion
Synthesis of metal–salphen complexes
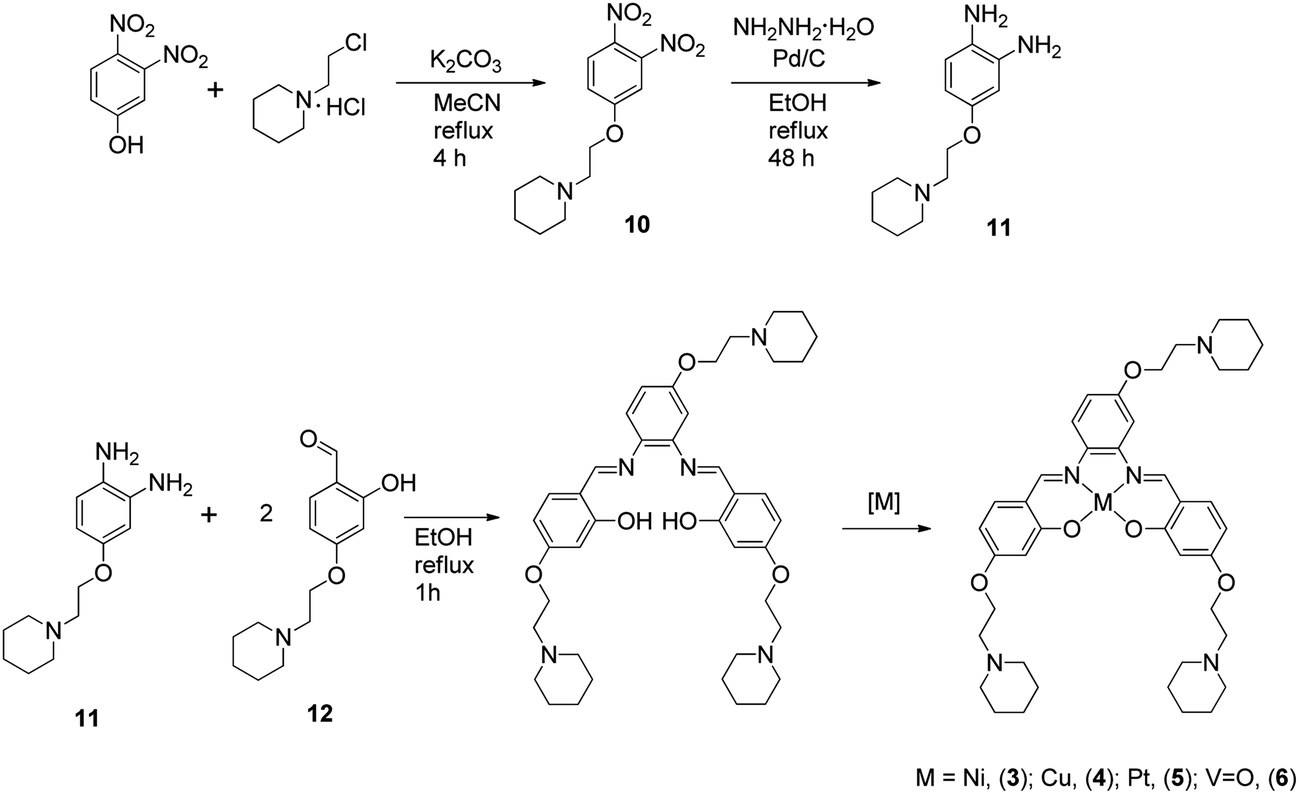
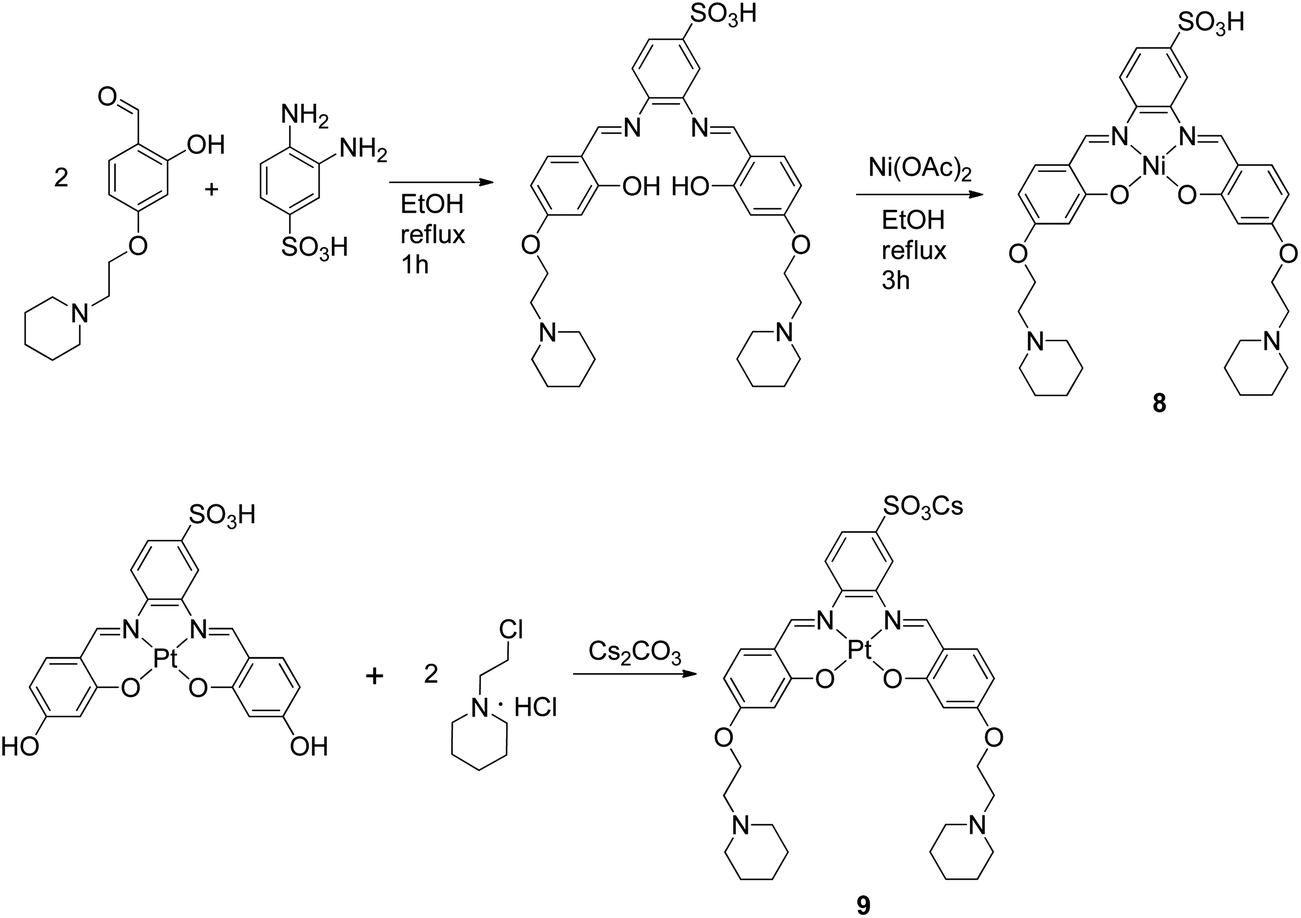
The metal–salphen complexes shown in Fig. 2 were synthesized as outlined in Schemes 1–3. In a first series of complexes (3–6) three ethyl-piperidine substituents were added to nickel(II), copper(II), platinum(II) and vanadyl–salphen cores. This type of substituent is well documented to confer enhanced quadruplex DNA binding properties to planar molecules. We have previously synthesized several metal–salphen complexes containing two ethyl-piperidine substituents.15 The interest in adding a third substituent to the ligand was to maximize the possible contacts of the resulting complexes with quadruplex DNA's grooves and loops. In addition, we rationalized that having a substituent on each of the three phenyl rings of the salphen, would reduce the possibility of the resulting metal complexes intercalating with duplex DNA and therefore enhance their quadruplex selectivity. The third piperidine substituent was also found to increase considerably the water solubility of the salphen complexes. These four complexes (3–6) were synthesized as shown in Scheme 1 and characterized by spectroscopic and analytical techniques (see Experimental section for details).
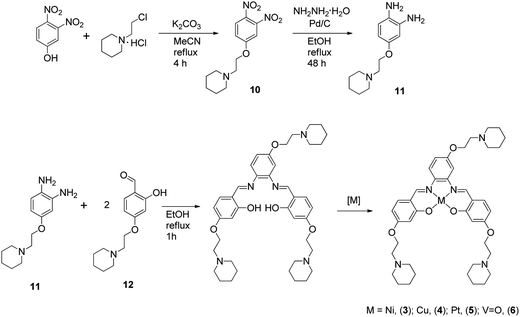 |
| | Scheme 1 Synthetic route for the preparation of complexes 3–6. | |
 |
| | Scheme 2 Synthetic route for the preparation of complex 7. | |
 |
| | Scheme 3 Synthetic route for the preparation of complexes 8 and 9. | |
A second approach to increase the selectivity of salphen complexes for quadruplex vs. duplex DNA was investigated. This was based on the complexes' functionalization with groups that are negatively charged at physiological pH. The rationale was that this would in principle reduce non-specific electrostatic interactions with DNA (which is negatively charged) and direct the compound to the topology with which other non-covalent interactions such as π–π stacking were more favorable. Thus, complexes 7–9 with either one or two sulphonate groups were prepared as shown in Schemes 2 and 3. In complex 7 two propyl-sulphonate groups (instead of ethyl piperidine) were attached yielding a complex with an overall negative charge. In complexes 8 and 9 a sulphonic group was introduced in the ligand's backbone while maintaining two piperidine substituents. These three complexes were characterized by 1H and 13C NMR spectroscopies which showed the expected number of signals with the right chemical shift and integration for each of the compounds (see Experimental details). The formulations and purity of the compounds were further confirmed by mass spectrometry and elemental analyses.
Interaction of metal complexes with DNA
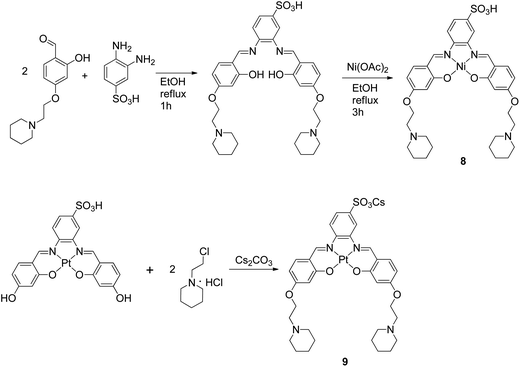
In order to establish the affinity of the new metal–salphen complexes towards HTelo quadruplex DNA, Fluorescence Resonance Energy Transfer (FRET) melting assays were carried out using the doubly-labeled oligonucleotide 5′-FAM-d(GGGTTAGGGTTAGGGTTAGGG)-TAMRA-3′ (where FAM = fluorescein and TAMRA = tetramethylrhodamine).23 The ΔTm values (see Table 1) show that the tri-ethylpiperidine square planar complexes 3–5 are excellent HTelo quadruplex binders; these are followed by the square planar sulphonate-substituted complexes 8 and 9 (which also show considerable quadruplex stabilization). The square pyramidal complex 6 and the negatively charged complex 7 show only a modest ΔTm, as expected considering their geometry and charge.
Table 1 ΔTm (°C) determined by FRET of F21T-HTelo quadruplex DNA upon addition of 1 μM complex (3–9). Values are average of three independent measurements
| Comp. |
ΔTm (°C) |
| 3 |
26.8 ± 1.5 |
| 4 |
22.9 ± 1.0 |
| 5 |
25.4 ± 1.0 |
| 6 |
6.4 ± 0.9 |
| 7 |
5.2 ± 1.0 |
| 8 |
17.4 ± 0.2 |
| 9 |
15.9 ± 0.2 |
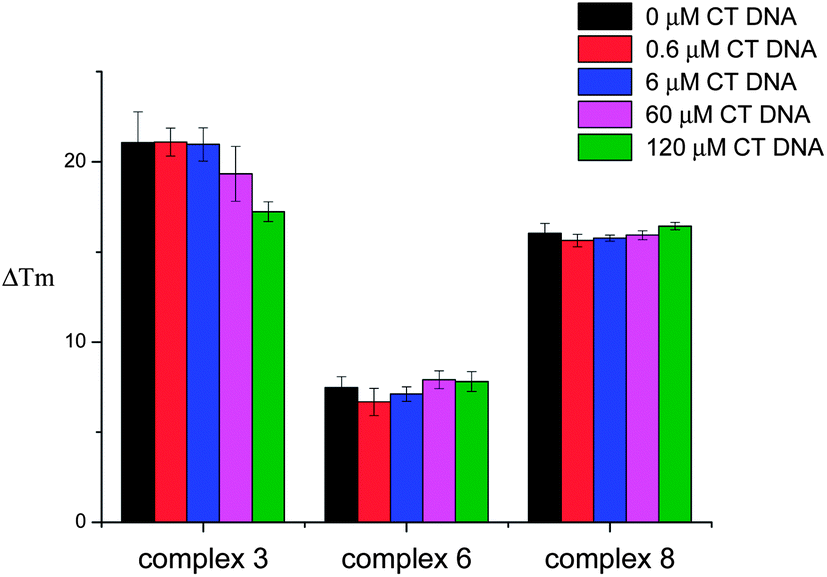
FRET competition assays were carried out for complexes 3, 6 and 8 (as representatives of the series) to investigate their selectivity for HTelo quadruplex vs. duplex DNA. Complex 3 was selected as an example to probe the square planar tri-ethylpiperidine complexes (3–5). Complex 6 (also a tri-ethylpiperidine metal complex) has a square-based pyramidal structure and therefore it was of interest to investigate whether having only one planar ‘face’ would improve its selectivity for quadruplex vs. duplex DNA. The third compound investigated by FRET competition assays was complex 8 which features a negatively-charged sulfonate group and hence might display a different selectivity profile than e.g. complex 3 – the overall charge of complex 8 at physiological pH is 1+ while that of complex 3 is 3+. In the FRET competition assays the ΔTm of HTelo quadruplex DNA (0.4 μM) was measured for a fixed concentration of metal complex (1 μM) in the presence of an increasing amounts of ct-DNA (from 0 to 120 μM). These studies (see Fig. 3) show that while the tri-ethylpiperidine complex 3 has the highest affinity for quadruplex HTelo DNA, its selectivity is not as good as that of the other two compounds. The square-based pyramidal complex 6 shows a relatively low quadruplex affinity but it is highly selective since the ΔTm of HTelo quadruplex DNA in the presence of this compound does not change even if a large excess of ct-DNA is added. As per complex 8, it shows high affinity and selectivity for HTelo quadruplex DNA. These results highlight the importance of the compounds' geometry and substitution pattern in defining their selectivity and affinity for quadruplex DNA.
 |
| | Fig. 3 Plot of ΔTm of F21T-HTelo upon addition of 1 μM concentration of complex 3, 6 or 8 and increasing concentration of CT DNA (0–120 μM). Results shown are average of three independent measurements. | |
Emission studies
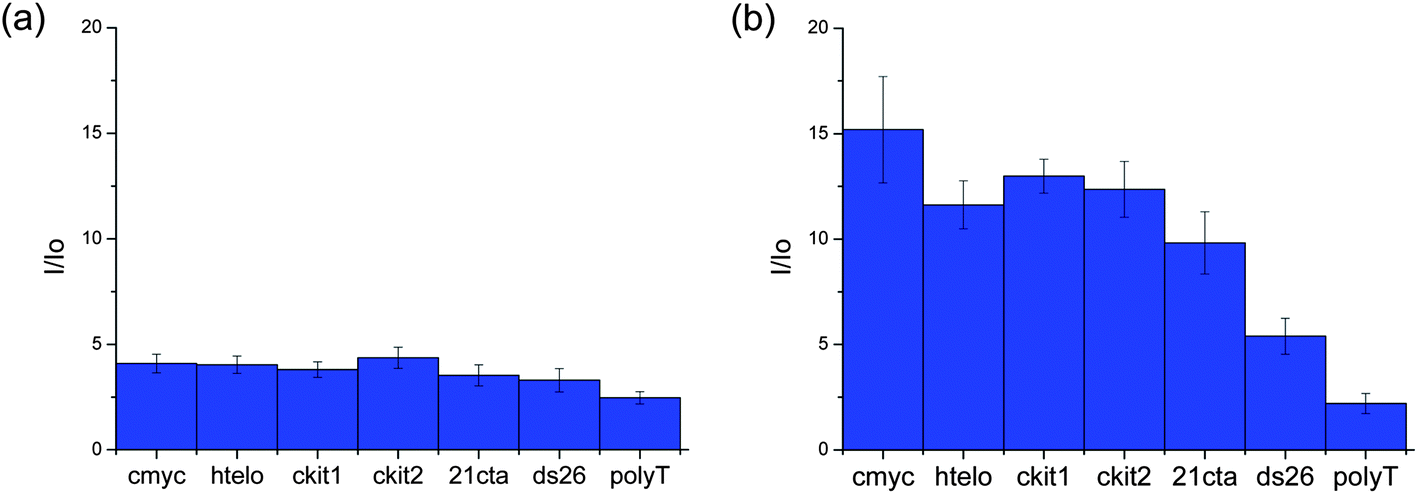
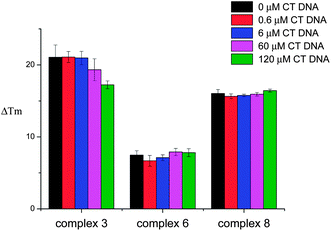
Several metal complexes have been previously reported to change their optical properties upon binding to quadruplex nucleic acids.22,24–29 An interesting feature of the platinum(II) complexes 5 and 9 is that they are luminescent and their emission in aqueous solution is greatly enhanced upon addition of DNA. Therefore, these complexes were further investigated by emission spectroscopy in the presence of different oligonucleotides. We first recorded the emission of the complexes (in aqueous buffered solution) upon addition of 10 equivalents of several quadruplex-forming oligonucleotides (namely G-rich sequences in the promoter regions of proto-oncogenes c-myc, c-kit1 and c-kit2, telomeric DNA (hTelo) and modified telomeric DNA 21CTA – for the specific sequences, see Experimental details) as well as duplex DNA (ds26) and a 25-base singly-stranded oligonucleotide (poly(dT)25). In all cases, an increase in the emission intensity was observed (see Fig. 4). While complex 5 is not selective for any given DNA topology (Fig. 4(a)), complex 9 showed to have some selectivity for quadruplex structures over duplex and singly-stranded DNA (Fig. 4(b)).
 |
| | Fig. 4 Bar chart depicting the enhancement in the emission of complex 5 (a) and 9 (b) (in Tris–KCl buffer) upon addition of 10 equivalents of several DNA quadruplex (hTelo, c-myc, c-kit1, c-kit2 and 21CTA) duplex (ds26) and ssDNA (polyT) respectively. | |
To assess more accurately the selectivity of complex 9 towards DNA, we determined the binding constants of this complex for HTelo and c-myc quadruplex DNA as well as ds26 duplex DNA. This was done by titrating complex 9 with increasing amounts of the corresponding oligonucleotide (see ESI† for titration plots) to give the following values: K = 0.99 ± 0.07 × 105 (c-myc); 1.15 ± 0.23 × 105 (HTelo); 2.98 ± 0.28 × 103 (ds26).
Confocal microscopy
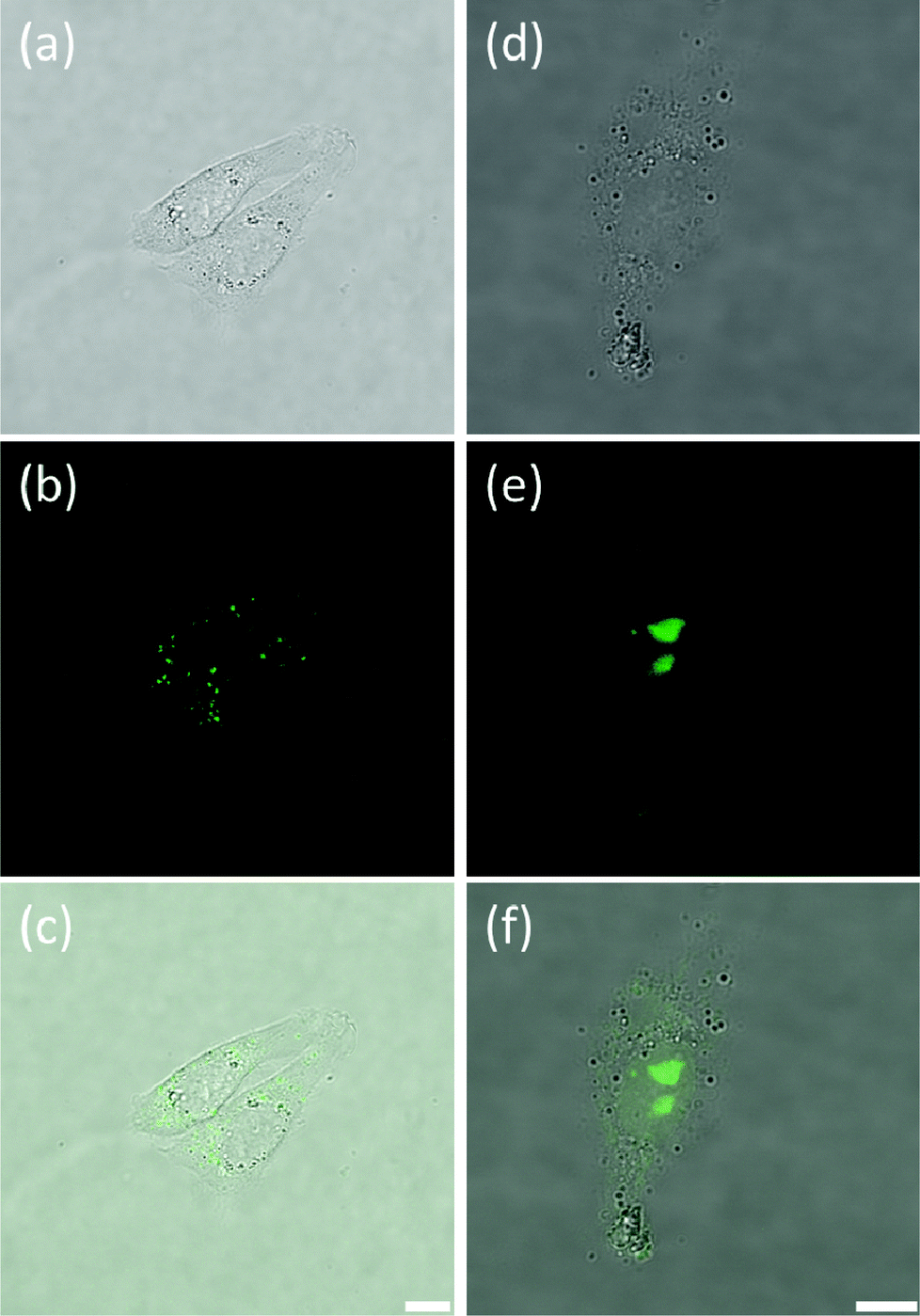
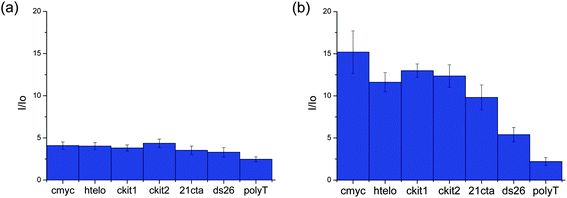
Once the spectroscopic properties of complexes 5 and 9 were established, we were interested in determining via confocal microscopy whether they are cell permeable and, if so, what their cellular localization is. For this, complexes 5 and 9 (at concentrations between 0.5 and 16 μM) were incubated with the following cell lines: Chinese Hamster Ovary (CHO), human osteosarcoma (U2OS), human cervical cancer (HeLa) and human hepatocellular carcinoma (HepG2) for up to 24 hours and confocal microscopy images recorded. Interestingly, some differences in uptake and localization were observed depending on the cell line used. Neither of the two platinum(II) complexes were taken up by U2OS cells under the experimental conditions used. For the other three cell lines, we observed that complex 5 was readily taken up while complex 9 was only partially cell permeable after long incubation times. We have also observed variations in the localization of the complexes in the different cell lines. With HeLa cells, complex 5 was shown to localize mainly in the nucleus, more specifically in the nucleoli (see Fig. 5(a and b)). Co-staining experiments using the nuclear dye DRAQ5 were carried out which confirmed the cellular localisation of the probe (Fig. 5(c and d)).
 |
| | Fig. 5 Fluorescence microscopy of live HeLa cells stained with complex 5 (10 μM, 30 min incubation). (a) shows the transmitted light image, (b) is the fluorescence intensity image, (c) shows DRAQ5 nuclear staining (5 μM) and (d) represents the merge of the three channels. Clear fluorescence is seen in the nucleus of all cells indicating that the dye localises to this region and becomes fluorescent in the presence of DNA. Scale bar 20 microns. | |
Complex 5 was also taken up by CHO cells where strong fluorescence was observed from the nuclei (Fig. S4†). The nuclear fluorescence in CHO cells appeared homogeneous rather than specifically localized in nucleoli as was the case with HeLa cells. Interestingly, staining of HepG2 cells with 5 shows that, while the compound is also taken up readily by the cells, its localization is different (see Fig. S5†). The compound accumulates in small pockets outside the nucleus and co-staining experiments with DRAQ5 indicate that the two dyes co-localize in these pockets.
As indicated above, in contrast to 5, complex 9 showed to have only limited cell permeability with CHO, HeLa and HepG2. The low levels of 9 that are taken up by these three cell lines show some nuclear localization (see Fig. 6, S6 and S7† for CHO, HeLa and HepG2 cells respectively). As can be seen in Fig. 6, two distinct staining regimes were observed. Fig. 6(a–c) respectively show transmitted light, fluorescence and merged images for the first – and most common – staining regime where dim fluorescence is observed in discrete regions of the cytoplasm and no fluorescence is seen in the nucleus. This implies that the compound is taken up by the cells but cannot penetrate the nuclear membrane. Hence, it does not encounter any oligonucleotides and, as such, does not become fluorescent. The second type of staining observed is documented in Fig. 6(d–f). In this case the dye has reached the nucleus and fluorescence is observed in the nucleoli, suggesting some selective location in these small regions of the nucleus. The fluorescence is considerably more intense than in the first staining regime (Fig. 6(a–c)) and we note that the transmission images appear dimmer in Fig. 6(d–f) solely because the laser power was reduced in this case due to the increased fluorescence intensity. This second type of staining was less common than the first type and it is worth noting that the cell shown in Fig. 6(d–f) appears less viable than the cells presented in Fig. 6(a–c). Thus, it may be the case that complex 9 can only penetrate the nuclear membrane and reach the nucleus when the cells are beginning to undergo apoptosis (likely due to the cytotoxicity of the compound itself) and the permeability of the nuclear membrane is starting to increase. Despite this, it is still interesting to note that complex 9 locates specifically to the nucleoli and not simply to the entire nucleus equally. Indeed, this was also the case with complex 5 in HeLa but not in CHO cell line.
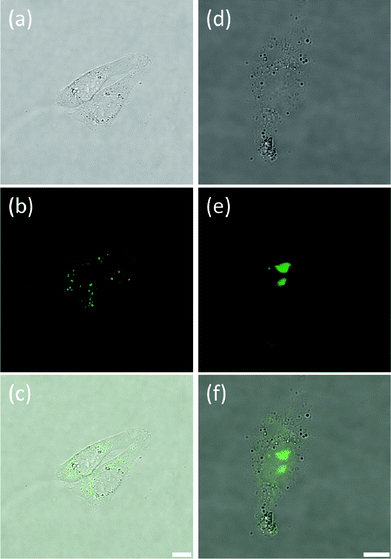 |
| | Fig. 6 Confocal microscopy images of live CHO cells stained with complex 9, showing transmitted light images (a and d), fluorescence intensity images (b and e) and merged fluorescence and transmission images (c and f). (a–c) show two healthy cells with dim fluorescence mainly emanating from discrete regions within the cytoplasm. Most cells observed exhibited staining similar to this. (d–f) show images of a single cell with nuclear staining. In this case the dye has reached the nucleus. Scale bar for (a–c) 10 microns and for (d–f) 7.5 microns. | |
Conclusions
Seven new salphen-based metal complexes have been prepared, characterized and their DNA affinities studied. The affinity of these complexes towards HTelo quadruplex DNA is determined by the geometry around the metal center with the square planar complexes (Ni, Cu and Pt) having the higher affinities. Interestingly, the vanadyl complex with a square-based pyramidal structure, while it has a lower affinity towards quadruplex than the analogue square planar complexes, it displays higher selectivity for quadruplex over duplex DNA – as we reported previously for another vanadyl complex.15 This is likely to be a consequence of the axial ligand which would prevent easy intercalation into duplex DNA but still allow the complex to interact by end-stacking (using its one planar face) with a guanine tetrad. Our results also show that the nature and number of substituents on the salphen, play an important role in defining the affinity and selectivity of the resulting complexes. A particularly interesting observation is the increased selectivity of complexes 8 and 9 (with a negatively charged sulfonate group in the ligand backbone) for quadruplex over duplex DNA as compared to the complexes with no sulfonate and either two or three ethyl-piperidino substituents. An interesting feature of the platinum(II) complexes is that they are highly emissive under the right conditions. This was successfully used to investigate their DNA binding profile and study their cellular uptake and localization by confocal microscopy. These studies showed that both 5 and 9 are taken up by cells (CHO, HeLa and HepG2) and localize in the nucleus (in CHO and HeLa cell lines); this is more prominent in the case of complex 5 – with three ethyl-piperidino groups. It is likely that the negative charge on the sulfonate group of 9 reduces its ability to cross biological membranes and therefore is not taken up by cells as readily as complex 5. Further studies are currently underway to modify this platinum(II) complex (which is selective for quadruplexes) to increase its affinity as well as cell permeability.
Experimental details
General
The oligonucleotides 21CTA (5′-GGGCTAGGGCTAGGGCTAGGG-3′), c-kit1 (5′-GGGAGGGCGCTGGGAGGAGGG-3′), c-kit2 (5′-GGGCGGGCGCGAGGGAGGGG-3′) and calf thymus DNA were purchased from Sigma Aldrich. H-telo DNA, (5′-AGGGTTAGGGTTAGGGTTAGGG-3′), c-myc (5′-GGGGAGGGTGGGGAGGGTGGG-3′), ds26 (5′-CAATCGGATCGAATTCGATCCGATTG-3′) and polyT (5′-T25-3′) were purchased from Eurogentec. The labelled F21T oligo (5′-FAM GGGTTAGGGTTAGGGTTAGGG-TAMRA-3′) was purchased from IBA. Test compounds were dissolved in a mixture of DMSO (95% by volume) and 1 mM HCl aqueous solution (5% by volume) to give 10 mM stock solutions. All solutions were stored at −20 °C and defrosted and diluted immediately before use. Prior to use, the compounds were diluted to 1 mM using DMSO. This was then further diluted using suitable buffer to the appropriate concentrations.
Emission titrations
The corresponding DNA (i.e. c-myc, HTelo and ds26) was dissolved in 50 mM Tris–HCl (pH 7.4) buffer containing 100 mM KCl and annealed at 95 °C for 5 min before cooling to room temperature overnight. The concentration of DNA was checked using the molar extinction coefficients 216![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900, 228500 and 6600 dm3 mol−1 cm−1 for c-myc, HTelo and ds26 respectively. Annealing concentrations were approximately 1 mM for c-myc and HTelo. For emission titrations, complex 9 (10 μM) in the same buffer was titrated with the corresponding DNA until saturation of fluorescence. Strand concentration was used for c-myc and HTelo whilst ds26 was considered in base pair concentration. The emission spectra were recorded between 460 and 750 nm with an excitation wavelength of 440 nm in 1 cm path-length quartz cuvettes. The emission spectra were recorded on a Varian Cary Eclipse Spectrometer. Spectra were smoothed using the Savitzky–Golay algorithm and emission maxima were fitted to 1:1 binding model using the Levenberg–Marquardt algorithm and equations reported previously by Thordarson in Matlab 7.12.0.30
900, 228500 and 6600 dm3 mol−1 cm−1 for c-myc, HTelo and ds26 respectively. Annealing concentrations were approximately 1 mM for c-myc and HTelo. For emission titrations, complex 9 (10 μM) in the same buffer was titrated with the corresponding DNA until saturation of fluorescence. Strand concentration was used for c-myc and HTelo whilst ds26 was considered in base pair concentration. The emission spectra were recorded between 460 and 750 nm with an excitation wavelength of 440 nm in 1 cm path-length quartz cuvettes. The emission spectra were recorded on a Varian Cary Eclipse Spectrometer. Spectra were smoothed using the Savitzky–Golay algorithm and emission maxima were fitted to 1:1 binding model using the Levenberg–Marquardt algorithm and equations reported previously by Thordarson in Matlab 7.12.0.30
Determination of quantum yields
Complex 5 and 9 absorption and emission spectra were measured in 50 mM Tris–HCl (pH 7.4) buffer containing 100 mM KCl. The absorbance at 436 nm was plotted against the integrated emission intensity from 530 to 800 nm and fitted to the equation y = mx + c (R2 > 0.98) for both complexes and the reference [Ru(bpy)3]Cl2 (aerated water ϕ = 0.028).31 Absorbance above the excitation wavelength was kept below 0.1. Quantum yields were determined by the equation ϕx = ϕref(mx/mref). The absorbance and emission spectra were recorded on Perkin-Elmer UV-vis and Horiba Jobin Yvon Fluorolog spectrometers. ϕ (complex 5, λex = 436 nm; λex = 602 nm) = 0.0019 and ϕ (complex 9, λex = 436 nm; λex = 572 nm) = 0.0007.
FRET assay
Labelled DNA F21T-HTelo was dissolved as a 20 μM stock solution in MilliQ water, then annealed as a 400 nM concentration in 60 mM potassium cacodylate buffer (pH 7.4) at 95 °C for 5 min, and allowed to cool slowly to room temperature overnight. Compounds were dissolved from stock solutions (see above) to final concentrations in the buffer. Each well of a 96-well plate (Applied Biosystem) was prepared with 40 μL, with a final 200 nM DNA concentration and increasing concentration of tested compound (0–4 μM). Measurements were performed on a PCR Stratagene Mx3005P (Agilent Technologies) with excitation at 450–495 nm and detection at 515–545 nm. Readings were taken from 25 °C to 95 °C at interval of 0.7 °C maintaining a constant temperature for 30 seconds before each reading. Each measurement was done in triplicate. The normalised fluorescence signal was plotted against the compound concentration and the ΔTm for 1 μM compound concentration were determined.
FRET competition assay
F21T-HTelo (see above) was annealed as a 400 nM concentration in 60 mM potassium cacodylate buffer (pH 7.4) at 95 °C for 5 min, and allowed to cool slowly to room temperature overnight. Compounds were dissolved from stock solutions (see above) to final concentrations in the buffer. Each well of a 96-well plate (MJ Research, Waltham, MA) was prepared with a final 200 nM oligo concentration, 1 μM compound concentration, and the ct-DNA concentration to test (0 to 120 μM). Measurements were performed under the same conditions as the FRET melting assay.
Cell culture
HeLa, HepG2 and U2OS cells were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C with 10% CO2 in the air. For microscopy, cells were seeded at 2 × 104 in 200 μL per well (0.8 cm2) in phenol red free media. For 24 h treatments, after cell adhesion (6 h) the medium was replaced with complexes 5 or 9 diluted at the specified concentration in phenol red-free medium (200 μL). For 30 min treatments, the media was replaced 28 h after seeding with complexes 5 or 9 diluted at the specified concentration in phenol red-free medium (200 μL). Prior to imaging, the cells were washed with PBS and fresh phenol red free media was added. Cells were co-stained by aspirating the medium and replacing with the DRAQ-5 (5 μM, 200 μl, PBS) for 10 min.
CHO cells were grown in serum-free Ham's F-12 Nutrient Mix (Invitrogen) at 37 °C with 10% CO2 in the air. Cells were transferred to the chambers of an 8-well plate (Lab-Tek Chamber Slide System, Nunc, Denmark) in a density of 10000 cells per well. After 6 h incubation for adhesion, the media was replaced with complexes 5 and 9 diluted at the specified concentration in Ham's F-12 media (200 μL) and then, cells were incubated for 24 h.
Cellular imaging
Cells were imaged using a confocal fluorescence microscope (TCS SP5 Confocal, Leica Microsystems GmbH, Germany). A variety of complex concentrations were tested in every well in order to ascertain the optimal conditions for cell imaging (0, 0.5, 4, 8 and 16 μM). After incubation, the cells were washed with PBS and 8-well plate then mounted on the microscope. Using a 63× magnification microscope objective (water immersion, NA = 1.2) and an excitation wavelength of 458 nm, images of the cells were recorded in both transmission and fluorescence modes. For the fluorescence images, the detection band was 525–700 nm, which covered the entire emission range of these metal complexes.
General procedures for characterization of compounds
1H NMR and 13C NMR spectra were recorded on either a Bruker Avance 400 MHz Ultrashield NMR spectrometer or a Bruker Avance 500 MHz NMR spectrometer. Mass spectrometric analysis was performed under either ESI+ or ESI− condition on a LCT Premier mass spectrometer. IR spectra were recorded on a Perkin-Elmer Spectrum 100 FT-IR spectrometer. Compounds 12, 13 and 3,4-diaminophenyl sulfonic acid were prepared following previously reported procedures.14,15,32
Synthesis of 1-(2-(piperidin-1-yl)ethoxy)-3,4-dinitrobenzene (10). 3,4-Dinitrophenol (1.250 g, 6.8 mmol), 1-(2-chloroethyl)piperidine hydrochloride (1.499 g, 8.2 mmol) and K2CO3 (1.576 g, 11.4 mmol) were heated at 100 °C under N2 in acetonitrile for 4 h. After that time, the solvent was removed under reduced pressure. The crude was then purified through column chromatography using mixture of CHCl3:ethanol 9:1 on silica to obtain 10 as an orange oil. Yield: 1.04 g, 51.9%. 1H NMR (400 MHz, CDCl3): δ 1.48–1.51 (br, 2H, piperidine-H), 1.60–1.66 (br, 4H, piperidine-H), 2.52–2.53 (br, 4H, piperidine-H), 2.81 (t, 3JHH = 6 Hz, 2H, –CH2N–), 4.24 (t, 3JHH = 6 Hz, 2H, –CH2O–), 7.16 (dd, 3JHH = 8.8 Hz, 4JHH = 2.4 Hz 1H, ArH), 7.81 (d, 4JHH = 2.4 Hz, 1H, ArH), 8.05 (d, 3JHH = 8.8 Hz, 1H, ArH). 13C NMR (400 MHz, CDCl3): δ 24.0, 25.8, 55.0, 57.3, 68.0, 111.1, 117.2, 127.5, 134.2, 145.4, 163.1. ESI(+)-MS m/z calcd for C13H17N3O5: 295.29 a.m.u.; found 296 a.m.u. corresponding to [M + 1]+.
Synthesis of 3,4-diamino-1-(2-(piperidin-1-yl)ethoxy)benzene (11). Compound 10 (0.160 g, 0.50 mmol) was first dissolved in 40 mL degassed EtOH under nitrogen. Palladium on carbon (0.050 g, 0.01 mmol) was added to this solution. NH2NH2·H2O (0.18 mL, 3.70 mmol) was slowly added to it and the reaction mixture was stirred at reflux for 48 h. The solvent and excess of NH2NH2 were removed under reduced pressure to obtain 11 as green oil. Yield: 0.10 g, 85.0%. 1H NMR (400 MHz, CDCl3): δ 1.43–1.49 (br, 2H, piperidine-H), 1.60–1.65 (br, 4H, piperidine-H), 2.52 (br, 4H, piperidine-H), 2.76 (t, 3J = 6 Hz, 2H, –CH2N–), 4.04 (t, 3JHH = 6 Hz, 2H, –CH2O–), 6.27 (dd, 3JHH = 8.4 Hz, 4JHH = 2.4 Hz, 1H, ArH), 6.35 (d, 4JHH = 2.4 Hz 1H, ArH), 6.64 (d, 4JHH = 8.4 Hz, 1H, ArH). 13C NMR (400 MHz, CDCl3): δ 23.7, 25.2, 54.8, 57.7, 65.5, 103.7, 105.0, 118.2, 127.7, 137.0, 153.3. ESI(+)-MS m/z calcd for C10H10O3: 235.3 a.m.u.; found 236 a.m.u. corresponding to [M + 1]+.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-(2-(piperidin-1-yl)ethoxy)-1,2-phenylenediamine-nickel(II) (3). Compound 12 (0.479 g, 1.9 mmol) and 11 (0.226 g, 1.0 mmol) were dissolved in ethanol and heated at reflux for 1 h. Ni(OAc)2·4H2O (0.239 g, 1.0 mmol) was then added to this solution and the reaction mixture was refluxed for another 3 h. The solvent was removed under reduced pressure yielding a red solid. The solid was then recrystallised from CH2Cl2–pentane to yield 3 as a dark red solid. Yield: 0.74 g, 98.0%. 1H NMR (400 MHz, CDCl3): δ 1.52 (br, 6H, piperidine-H), 1.70 (br, 12H, piperidine-H), 2.67 (br, 12H, piperidine-H), 2.93 (br, 6H, –CH2N–), 4.18 (br, 6H, –CH2O–), 6.28 (d, 3JHH = 9.2 Hz, 1H, ArH), 6.30 (d, 3JHH = 8.4 Hz, 1H, ArH), 6.56 (s, 2H, ArH), 6.74 (d, 3JHH = 8 Hz, 1H, ArH), 7.13 (d, 3JHH = 9.2 Hz 1H, ArH), 7.15 (s, 1H, ArH), 7.21 (d, 3JHH = 8.8, 1H, ArH), 7.56 (d, 3JHH = 9.2 Hz, 1H, ArH), 7.90 (s, 1H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 8.01 (s, 1H, –CHN–). 13C NMR (400 MHz, CDCl3): δ 54.4, 54.6, 57.0, 57.1, 65.1, 65.2, 65.7, 99.54, 103.12, 108.2, 108.8, 113.5, 114.6, 114.8, 114.9, 134.0, 134.4, 136.6, 143.6, 151.2, 152.5, 157.6, 157.8, 164.2, 164.7, 167.3, 168.3. IR (cm−1): 1581 (CN), 1182 (C–O), 2924 (CH2–), 2850 (CH–O), 1431 (CC), 1121 (CC), 1371 (CC). ESI(+)-MS m/z calcd for C41H53N5O5Ni: 754.58. Found: 754.44. Anal. calcd for C41H53N5O5Ni·CH2Cl2: C, 60.09 H, 6.60 N, 8.34. Found: C, 60.50 H, 6.81 N, 8.55%.
N–), 8.01 (s, 1H, –CHN–). 13C NMR (400 MHz, CDCl3): δ 54.4, 54.6, 57.0, 57.1, 65.1, 65.2, 65.7, 99.54, 103.12, 108.2, 108.8, 113.5, 114.6, 114.8, 114.9, 134.0, 134.4, 136.6, 143.6, 151.2, 152.5, 157.6, 157.8, 164.2, 164.7, 167.3, 168.3. IR (cm−1): 1581 (CN), 1182 (C–O), 2924 (CH2–), 2850 (CH–O), 1431 (CC), 1121 (CC), 1371 (CC). ESI(+)-MS m/z calcd for C41H53N5O5Ni: 754.58. Found: 754.44. Anal. calcd for C41H53N5O5Ni·CH2Cl2: C, 60.09 H, 6.60 N, 8.34. Found: C, 60.50 H, 6.81 N, 8.55%.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-(2-(piperidin-1-yl)ethoxy)-1,2-phenylenediamine-copper(II) (4). Compounds 12 (0.439 g, 1.8 mmol) and 11 (0.207 g, 0.9 mmol) were dissolved in ethanol and heated to reflux for 1 h. Cu(OAc)2·H2O (0.176 g, 0.9 mmol) was then added to this solution and the reaction mixture was refluxed for another 3 h. The solvent was removed under reduced pressure to yield a dark green solid. The solid was then recrystallised from CHCl3–hexane to give 4 as dark green solid. 0.379 g, 56%. ESI(+)-(MS) m/z calcd for C41H53N5O5Cu: 759.4; found: 759.4 a.m.u. IR (cm−1): 1581 (CN), 1187 (C–O), 2928 (CH2–), 2854 (CH–O), 1430 (CC), 1121 (CC), 1372 (CC). Anal. calcd for C41H53N5O5Cu·CHCl3: C, 57.40 H, 6.19 N, 7.97. Found: C, 57.22 H, 6.62 N, 7.59%.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-(2-(piperidin-1-yl)ethoxy)-1,2-phenylenediamine-platinum(II) (5). Compounds 12 (0.200 g, 0.8 mmol) and 11 (0.095 g, 0.4 mmol) were dissolved in ethanol and heated at 60 °C for 4 h. Afterward, the solvent was removed under reduced pressure. Acetonitrile (40 mL) was added to this solid and the resulting reaction mixture was stirred for 5 min. Sodium acetate as a solid and K2PtCl4 (0.168 g, 0.4 mmol) in DMSO (2 mL) were added to the solution. The reaction mixture was heated to 40 °C for 24 h. The solvent was removed under reduced pressure to and the resulting solid was recrystallised from CH2Cl2–pentane to give 5 as a dark red solid. Yield: 0.055 g, 17.0%. 1H NMR (400 MHz, DMSO-d6): δ 1.4 (m, 6H, piperidine-H), 1.50–1.54 (m, 12H, piperidine-H), 2.50–2.52 (m, 12H, piperidine-H), 2.73 (br, 6H, –CH2N–), 4.14–4.18 (m, 4H, –CH2O–), 4.22 (t, 3JHH = 5.6 Hz, 2H, –CH2O–), 6.43–6.48 (m, 2H, ArH), 6.56 (d, 4JHH = 2.4 Hz, 1H, ArH), 6.57 (d, 4JHH = 2.4 Hz, 1H, ArH), 7.55 (d, 3JHH = 9.2 Hz, 1H, ArH), 7.67 (d, 3JHH = 8.8 Hz, 1H, ArH), 7.73 (d, 3JHH = 9.2 Hz, 1H, ArH), 7.88 (s, 1H, ArH), 8.23 (d, 3JHH = 9.2 Hz, 1H, ArH), 9.12 (s, 1H, –CHN–), 9.28 (s, 1H, –CHN–). IR (cm−1): 1580 (CN), 1179 (C–O), 2928 (CH2–), 2831 (CH–O), 1421 (CC), 1125 (CC), 1373 (CC). ESI(+)-MS m/z calcd for C41H53N5O5Pt: 890.97; found: 891.0 a.m.u. Anal. calcd for: C41H53N5O5Pt·2CH2Cl2: C, 48.68; H, 5.42; N, 6.60; found: C, 48.91; H, 5.75; N, 6.14%.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-(2-(piperidin-1-yl)ethoxy)-1,2-phenylenediamine-vanadium(II) (6). To a mixture of 12 (0.500 g, 2 mmol) and 11 (0.235 g, 1 mmol) methanol (10 mL) was added and the resulting mixture was refluxed for 1 h. When the solution became yellow a solution of vanadium sulphate (0.163 g, 1 mmol) in water and triethyl amine (0.3 mL) were added to the reaction mixture. The resulting mixture was kept for reflux for another 3 h. After this time, the green precipitate formed was filtered and washed with water to give 6 as a yellow-green solid. Yield: 0.609 g, 80%. IR (cm−1): 1606 (CN); 1575 (Ar); 960 (VO). ESI MS calcd for C41H53N5O6V: 762.34. Found: 763.0 a.m.u. Analysis calculated for C41H53N5O6V: C, 64.55; H, 7.00; N, 9.18; found: C, 64.91; H, 7.03; N, 9.36%.
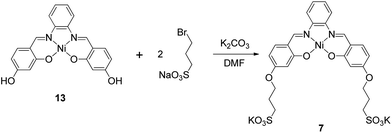
Synthesis of N,N′-bis[4-[[1-(2-propyl)sulfonic]oxy]salicylidene]-1,2-phenylenediamine-nickel(II) (7). A suspension of complex 13 (0.483 g, 1.2 mmol), 3-bromopropanesulfonic acid sodium salt (1.080 g, 4.8 mmol) and K2CO3 (0.828 g, 6.0 mmol) in dry DMF (35 mL) was stirred for 12 h at 90 °C temperature. After this period of time the reaction mixture was filtered to remove all solid material and the DMF from the filtrate was evaporated under reduced pressure. The resulting solid was dissolved in water a purified by reversed phase flash chromatography using a water–methanol mixture (gradient from 1:0 to 0:1) as eluent affording a red solid which was characterised as the di-potassium salt of 7. Yield: 0.34 g, 45%. 1H NMR (400 MHz, DMSO-d6): δ 2.00 (t, 3JHH = 7.3 Hz, 4H, CH2), 2.56 (dd, 3JHH 8.4, 6.4 Hz, 4H, CH2), 4.04 (t, 3JHH = 6.6 Hz, 4H, CH2), 6.26 (dd, 3JHH = 8.9, 4JHH = 2.3 Hz, 2H, ArH), 6.40 (d, 4JHH = 2.3 Hz, 2H, ArH), 7.26 (dd, 3JHH = 6.3, 4JHH = 3.2 Hz, 2H, ArH), 7.48 (d, 3JHH = 8.9 Hz, 2H, ArH), 8.05 (dd, 3JHH 6.1, 4JHH = 3.3 Hz, 2H), 8.67 (s, 2H). 13C NMR (400 MHz, DMSO-d6): δ 26.5, 48.2, 67.3, 102.3, 108.0, 115.2, 116.0, 127.1, 135.6, 142.7, 154.7, 165.21, 167.8. ESI(+)-MS m/z calcd ESI(+)-MS m/z calcd for C26H24K2N2NiO10S2: 724.0; found 763.0 a.m.u. corresponding to [M + K]+. Anal. calcd for C26H24K2N2NiO10S2·2.5H2O: C, 40.53; H, 3.79; N, 3.64. Found: C, 40.35; H, 3.71; N, 3.87%.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-sulfonic-1,2-phenylenediamine-nickel(II) (8). Compound 12 (0.381 g, 1.5 mmol) and 3,4-diaminophenyl sulfonic acid (0.130 g, 0.7 mmol) were dissolved in ethanol and heated at reflux for 1 h. Ni(OAc)2·4H2O (0.378 g, 1.5 mmol) was then added to the solution and the reaction mixture was refluxed for another 3 h. The solvent was removed under reduced pressure to yield a red solid. The solid was then washed with ethanol, CH2Cl2 and diethyl ether to give 8 as an orange solid. Yield: 0.42 g, 85.0%. 1H NMR (400 MHz, DMSO-d6): δ 1.48 (br, 4H, piperidine-H), 1.65 (br, 8H, piperidine-H), 2.52 (br, 12H, piperidine-H), 4.24 (br, 4H, –CH2O–), 6.32–6.39 (m, 4H, ArH), 7.48–7.52 (m, 2H, ArH), 7.68 (d, 3JHH = 8.6 Hz, 1H, ArH), 7.98 (d, 3JHH = 8.7 Hz, 1H, ArH), 8.23 (s, 1H, ArH), 8.68 (s, 1H, –CHN–), 8.74 (s, 1H, –CHN–). 13C NMR (400 MHz, DMSO-d6): δ 22.5, 23.8, 53.5, 101.7, 107.4, 112.6, 114.8, 115.2, 124.2, 135.3, 135.7, 141.7, 142.2, 154.8, 167.2. ESI(−)-MS m/z calcd for C34H39N4NiO7S: 705.19. Found: 705.0 a.m.u. Anal. calcd for C34H40N4NiO7S·1.5H2O: C, 55.60; H, 5.90; N, 7.63. Found C, 55.55; H, 5.81; N, 7.52%.
Synthesis of N,N′-bis[4-[[1-(2-ethyl)piperidine]oxy]salicylidene]-4-sulfonic-1,2-phenylenediamine-platinum(II) (9). 3,4-Diaminophenyl sulfonic acid (0.096 g, 0.5 mmol) and 2,4-dihydroxybenzaldehyde (0.140 g, 1 mmol) were suspended in ethanol–methanol (1:1, 10 mL) and heated to reflux for 3 h. After cooling to room temperature, a yellow solid formed which was recovered by filtration and washed with MeCN (mass of crude product: 0.178 g). A portion of these solid (0.080 g) was then added to a suspension of K2PtCl4 (0.095 g, 0.2 mmol) and sodium acetate (0.075 g, 0.9 mmol) in DMSO–MeCN (1:10, 11 mL) and heated to 76 °C for 24 h. After cooling to room temperature, an orange solid was recovered by filtration and washed with MeCN. The crude product (0.118 g), 1-(3-chloroethyl)piperidine hydrochloride (0.070 g, 0.4 mmol) and Cs2CO3 (0.154 g, 0.5 mmol) were placed under an inert atmosphere before the addition of dry DMF (15 mL). The reaction was stirred for 72 h at 30 °C. The solution was filtered and the filtrate was evaporated to dryness. The resultant solid was washed with dichloromethane, acetone, acetonitrile and hexane to give 9 as a red solid. Yield: 0.031 g, 18% (overall yield calculated with respect to 3,4-diaminophenyl sulfonic acid). IR (neat) 1181 and 1035 (st asymmetric and symmetric, Ar-SO3−), 1598 and 1571 (st, CN), 2792 (br st, N–H+), 3432 (br st, HO–H) cm−1. 1H NMR (400 MHz, DMSO-d6): δ 1.24 (s, 1H, piperidine-H), 1.33–1.44 (m, 4H, piperidine-H), 1.46–1.58 (m, 7H, piperidine-H), 2.36–2.50 (br, 8H, piperidine-H), 2.69 (t, J = 5.7 Hz, 4H, –CH2N–), 4.17 (t, J = 5.7 Hz, 4H, –CH2O–), 6.43 (dd, J = 9.1, 2.2 Hz, 1H, ArH), 6.47 (dd, J = 9.1, 2.2 Hz, 1H, ArH), 6.59 (d, J = 2.2 Hz, 1H, ArH), 6.60 (d, J = 2.2 Hz, 1H, ArH), 7.61 (d, J = 8.8 Hz, 1H, ArH), 7.74 (d, J = 9.1 Hz, 1H, ArH), 7.93 (d, J = 9.1 Hz, 1H, ArH), 8.31 (d, J = 8.8 Hz, 1H, ArH), 8.53 (s, 1H, ArH), 9.31 (s, 1H, –CHN–), 9.38 (s, 1H, –CHN–). 13C NMR (500 MHz, DMSO-d6): δ 24.2, 25.8, 54.6, 57.4, 66.1, 102.8, 102.9, 108.3, 108.6, 11.4, 115.7, 116.9, 117.0, 124.7, 137.0, 137.5, 144.0, 144.5, 147.6, 150.0, 150.2, 165.0, 166.7. ESI(+)-MS m/z calcd for C34H40N4O7PtS: 843.2. Found 844.2 a.m.u corresponding to [M + H]+. Anal. calcd for C34H39CsN4O7PtS·1HCl·2H2O: C, 38.96; H, 4.23; N, 5.34. Found: C, 38.81; H, 3.91; N, 5.21%.
Acknowledgements
The UK's Engineering and Physical Sciences Research Council (EPSRC) is thanked for financial support including Fellowships to M. K. K. (grant number: EP/E038980/1) and to R. V. (grant number: EP/H005285/1). We are also grateful to EC for a Marie Curie Fellowship (O. M.), to the Royal Society–Newton Fellowship (S. G.) and to the Malaysian Government for a PhD studentship (N. H. A. K.). Johnson Matthey is thanked for a lone of platinum.
References
- N. Maizels, Nat. Struct. Mol. Biol., 2006, 13, 1055 CAS.
- N. Maizels and L. T. Gray, PLoS Genet., 2013, 9, e1003468 CAS.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182 CrossRef CAS PubMed.
- A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727 CrossRef CAS PubMed.
- H. J. Lipps and D. Rhodes, Trends Cell Biol., 2009, 19, 414 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261 CrossRef CAS PubMed.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867 RSC.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345 CrossRef CAS PubMed.
- A. Arola and R. Vilar, Curr. Top. Med. Chem., 2008, 8, 1405 CrossRef CAS.
- D. Monchaud and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627 CAS.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020 CrossRef CAS PubMed.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459 CrossRef CAS PubMed.
- S. Neidle, FEBS J., 2010, 277, 1118 CrossRef CAS PubMed.
- J. E. Reed, A. A. Arnal, S. Neidle and R. Vilar, J. Am. Chem. Soc., 2006, 128, 5992 CrossRef CAS PubMed.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910 CrossRef CAS PubMed.
- H.-K. Fun, R. Kia, V. Mirkhani and H. Zargoshi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1181 CAS.
- M. Niu, S. Fan, K. Liu, Z. Cao and D. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m77 CAS.
- C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS PubMed.
- M. J. Xie, S. P. Yan, D. Z. Liao, Z. H. Jian and P. Chen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1530 CAS.
- N. E. Eltayeb, S. G. Teoh, S. Chantrapromma, H.-K. Fun and K. Ibrahim, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2838 CAS.
- N. H. Campbell, N. H. A. Karim, G. N. Parkinson, M. Gunaratnam, V. Petrucci, A. K. Todd, R. Vilar and S. Neidle, J. Med. Chem., 2012, 55, 209 CrossRef CAS PubMed.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem.–Eur. J., 2009, 15, 13008 CrossRef CAS PubMed.
- A. De Cian, L. Guittat, M. Kaiser, B. Sacca, S. Amrane, A. Bourdoncle, P. Alberti, M.-P. Teulade-Fichou, L. Lacroix and J.-L. Mergny, Methods, 2007, 42, 183 CrossRef CAS PubMed.
- J. Alzeer, B. R. Vummidi, P. J. C. Roth and N. W. Luedtke, Angew. Chem., Int. Ed., 2009, 48, 9362 CrossRef CAS PubMed.
- B. R. Vummidi, J. Alzeer and N. W. Luedtke, ChemBioChem, 2013, 14, 540 CrossRef CAS PubMed.
- K. Suntharalingam, A. Leczkowska, M. A. Furrer, Y. Wu, M. K. Kuimova, B. Therrien, A. J. P. White and R. Vilar, Chem.–Eur. J., 2012, 18, 16277 CrossRef CAS PubMed.
- D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131, 1835 CrossRef CAS PubMed.
- A. Granzhan, N. Saettel, F. Hamon, E. Largy, C. Guetta and M.-P. Teulade-Fichou, Collect. Symp. Ser., 2011, 12, 77 CAS.
- T. Wilson, M. P. Williamson and J. A. Thomas, Org. Biomol. Chem., 2010, 8, 2617 CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- K. Nakamaru, Bull. Chem. Soc. Jpn., 1982, 55, 2697 CrossRef CAS.
- F. da Silva Miranda, A. M. Signori, J. Vicente, B. de Souza, J. P. Priebe, B. Szpoganicz, N. S. Goncalves and A. Neves, Tetrahedron, 2008, 64, 5410 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44793f |
| ‡ These authors contributed equally to the paper. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.