Seed-mediated synthesis of shape-controlled CeO2 nanocrystals
Ruigang Wang* and
Randi Dangerfield
Department of Chemistry, Youngstown State University, One University Plaza, Youngstown OH, USA. E-mail: rwang01@ysu.edu; Fax: +1-330-941-1579; Tel: +1-330-941-2763
First published on 2nd December 2013
Abstract
We report the effect of varying the starting materials and experimental conditions on the shape control of CeO2 nanocrystals using hydrothermal methods, including using different cerium precursors (Ce3+: Ce(NO3)3 and Ce4+: Ce(NH4)2(NO3)6), stirring treatment, and oxidation of cerium(III) hydroxide. It was demonstrated that the formation of Ce(OH)3 nuclei is a key step to growth of different morphological CeO2 nanocrystals.
Introduction
The study of metal nanoparticles on oxide supports is of importance in heterogeneous catalysis because the size and nature of the interaction of a metal nanoparticle with an oxide support are critical in determining catalytic activity and selectivity.1–5 Reduction and oxidation (redox) at elevated temperatures are essential steps for the catalytic reactions. Unlike irreducible inert oxide support like SiO2 or Al2O3, it is well-known that reducible metal oxides (such as CeO2 and TiO2) supported metals, exhibit increased rates for reactions involving redox steps.6 A possible explanation for the increased reaction rates may involve highly mobile surface oxygen species, and/or partially reduced metal oxide sites in reducible oxides at high temperature. Due to the superior oxygen transport capacity, ceria (CeO2) with other oxides constitute an important class of catalysts and/or catalyst supports that can exchange oxygen rapidly under variable reducing or oxidizing conditions, mainly accommodated by a reversible valence change (2Ce(IV)O2 ↔ Ce2(III)O3 + 1/2O2) of the cerium ions with formation or elimination of oxygen vacancies.7–14 This property, characterized as the oxygen storage capacity (OSC) or redox functionality, has made it a material of considerable interest in applications such as vehicle three-way exhaust clean-up,7–13 in water-gas shift reactions,4,14 in gas sensors,15,16 and in fuel cell electrodes.17–20In order to enhance the low temperature activity of CeO2 support, hydrothermal synthesis of shape/size-controlled CeO2 nanocrystals has drawn much attention due to the controlled and mild reaction conditions (T < 250 °C).21–28 The ability to precipitate and crystallize nanoparticles directly from solution allows the control of rate and uniformity of crystal nucleation and growth, via systematic variation of processing variables, such as different starting materials, temperature, dwell time, pH, and concentrations of reactants and additives. The results are a much better control of crystal morphology, a narrow crystallite size distribution, and substantially reduced aggregation levels that cannot be achieved with most of the high temperature solid state reaction based processes.29,30
However, some inconsistent or conflicting results have been reported for the synthesis of CeO2 and cerium-based mixed oxide nanocrystals using the same or similar starting chemicals under hydrothermal reaction conditions. For example, Lin et al.23 synthesized CeO2 nanocrystals with irregular shape at 180 °C for 24 h, while Mai et al.21 reported the shape-controlled synthesis of CeO2 nanorods, nanocubes and nanopolyhedra using a similar hydrothermal reaction condition between 100 °C and 180 °C. Using different cerium precursors, Liu et al.24 reported that CeO2 nanorods were prepared by Ce(NO3)3, while spherical-like nanoparticles were obtained from Ce(NH4)2(NO3)6 and Ce(SO4)2. It seems that the significant differences in their preparation protocols were the selection of different cerium precursors, the treatment of the starting mixture suspension, and the hydrothermal temperature.
It is well documented that Ce(NO3)3 reacts with base solutions (i.e., NaOH or NH4OH) to form Ce(OH)3 initially.21–23 Ce(OH)3 is not stable in air even at room temperature, and tends to decompose and oxidize to CeO2. Recently, we observed a continuous color change of the mixture of Ce(NO3)3 and NaOH solutions during the stirring when the flask was open to air. As the formed Ce(OH)3 serves as a crystal seed during hydrothermal reactions, we hypothesize that the origin of the inconsistent results reported regarding the CeO2 shapes may be related to the decomposition and oxidation of Ce(OH)3. In other words, the formation and/or oxidation of crystal seeds may play a critical role in controlling the shape during subsequent hydrothermal reactions.
In this study, we exploited the effect of different starting cerium precursors, oxidation and stirring of the Ce(OH)3/Ce(OH)4 precursor on controlling the shape of CeO2 nanocrystals. Then, a possible explanation was proposed based on a series of comparison experiments. Finally, we demonstrated the results about tuning the shape/size of CeO2 by varying the hydrothermal experimental parameters.
Experimental section
All samples were synthesized using a hydrothermal method. The cerium precursors were 99.99% purity for cerium(III) nitrate hexahydrate and ammonium cerium(IV) nitrate, from Alfa Aesar. Sodium hydroxide pellets was >98% purity from Sigma-Aldrich. Typically, 0.1 M Ce(NO3)3·6H2O (Alfa Aesa, 99.99%) was loaded to a conical flask, and then 6 M NaOH solution was added. The solution was then stirred. The stirring step is critical for growing different morphological CeO2 nanocrystals (more details are discussed in the text). The resulting suspension solutions were heated in 200 mL stainless steel Teflon-lined autoclaves (∼50% filled) at temperatures between 50 °C and 220 °C with different dwell times (6 h, 48 h, and 96 h). At the end of the reaction, the autoclaves were cooled to room temperature and the precipitates were separated, filtered, and washed repeatedly using 500 mL distilled water to remove co-precipitated salts (for example, NaNO3), and then washed with ethanol to avoid hard agglomeration between the nanoparticles during drying. The resulting powders were then dried at 50 °C for 12 h.Powder X-ray diffraction (XRD) analysis was performed on a Rigaku Miniflex II diffractometer using Cu Kα radiation with a wavelength of 1.5408 Å. The samples were loaded on an aluminum plate and the data were collected over a 2θ range of 10°–90° with a step size of 0.5° min−1. The recorded patterns were analyzed using JADE software to determine peak positions, lattice constant, and average crystalline sizes by use of Scherrer's formula.
A JEOL 2100 transmission electron microscope (TEM) was used for structural and chemical analysis of the nanocrystals at atomic level, operated at 200 kV and equipped with an EDAX detector for chemical composition analysis and annular dark-field detector. High resolution transmission electron microscopy (HRTEM) was used to determine the exposed crystal facets in individual CeO2 nanocrystals. TEM samples were prepared by using dilute suspensions of the nanopowder samples. These solutions were obtained by ultrasonicating particles in ethanol for 10 min then dropping the suspension of the sample powders onto an ultrathin carbon film/holey carbon, 400 mesh copper grid (from Ted Pella) and letting it air dry for several hours.
Results and discussion
Color change of suspension during stirring
In a typical synthesis procedure to prepare CeO2 before hydrothermal reaction, 8 mL of 6 M NaOH solution was added dropwise into 88 mL of 0.1 M Ce(NO3)3 solution with vigorous stirring, in which the stirring was used to homogenize the two reactants and facilitate the reaction. It was observed that there was a continuous color change in the mixture of Ce(NO3)3 and NaOH suspension with stirring, as shown in Fig. 1. When NaOH was added to a vigorously stirred Ce(NO3)3 solution in a conical flask, the color of the solution changed from colorless (Ce(NO3)3), to dark gray (step I, up to 6 min), and then to pale yellow (step II). | ||
| Fig. 1 The color change of the precipitate suspension (6 M NaOH + 0.1 M Ce(NO3)3) with different stirring time: (a) before addition of NaOH; (b) 0 min; (c) 3 min; (d) 6 min; (e) 9 min; (f) 12 min; (g) 15 min; (h) 20 min. | ||
Initially, a lilac precipitate formed, and with vigorous stirring the lilac precipitate changed to a dark gray suspension in about 6 min (step I: Fig. 1a–d). Continuing to stir the suspension, the dark gray suspension turned lighter and eventually came to a yellow color in 20 min (step II: Fig. 1e–h). No further color change was observed by continuing the stirring up to 2 hours. During the stirring, the continuous color changes indicate the occurrence of some chemical reactions. It should be pointed out that the conical flask with suspension shown in Fig. 1 was left open to air during the stirring.
Origin of the color change
When using trivalent cerium precursor (Ce(NO3)3), the expected Ce(OH)3 precipitate was unstable in air at even room temperature, and the final product showed a yellow color originated from lilac after 20 min stirring. This indicates the occurrence of oxidation of the sample as only CeO2 or Ce(OH)4 with tetravalent Ce4+ possess yellow color.31,32 In order to understand the origin of the color change, two comparison experiments were performed.In comparison experiment 1, two conical flasks both with 88 mL 0.1 M Ce(NO3)3 and a stir bar were put on two magnetic hot plates, and then the solutions were stirred as 8 mL 6 M NaOH solution was added to each flask. For the first flask, after adding NaOH, the flask was sealed with Parafilm, and the second flask was left unsealed (exposed to air) after adding NaOH. In comparison experiment 2, everything was set up the same as in experiment 1 except using 0.1 M Ce(NH4)2(NO3)6 instead of 0.1 M Ce(NO3)3 as the starting cerium precursor. Fig. 2 shows the results for these two comparison experiments. In experiment 1 using Ce(NO3)3, the unsealed sample showed the same color change as seen in Fig. 1, from lilac to dark gray and then to yellow after 20 min stirring. However, the sealed sample did not show the same color change, and the final suspension still presented a dark gray color. The only difference between these two suspensions in the flasks, shown in Fig. 2, was the accessibility of air (oxygen) to the suspension solution, so the color change to yellow for the unsealed sample clearly indicates an occurrence of oxidation during the stirring. A possible explanation for the color change when using trivalent cerium precursor Ce(NO3)3 such as in Fig. 2i, could be as follows, involving a precipitation step (I) of the cerium precursor, followed by a dehydration/oxidation step (II):
| Precipitation I: Ce3+ + 3OH− → Ce(OH)3 (s) |
| Oxidation II: 4Ce3+ + 12OH− + O2 → 4CeO2 (s) + 6H2O |
 | ||
| Fig. 2 The color change of the precipitate suspension for the sealed and unsealed samples with continuous stirring for a mixture of 6 M NaOH solution with different cerium precursors ((i) Ce(NO3)3 and (ii) Ce(NH4)2(NO3)6). The left flask was sealed and the right one was not sealed after NaOH addition during the 20 min stirring. | ||
The actual occurrence of these two reactions depends on the availability of the dissolved oxygen in the solution and oxygen at the air–liquid interface. Ce(OH)3 nuclei form as soon as Ce3+ ions are mixed with NaOH solution. Ce(OH)3 is not stable in air, and thus when vigorously stirring the solution in air, Ce(OH)3 can react with oxygen and then become oxidized to CeO2. The role of stirring may relate to the kinetics of oxygen–Ce(OH)3 gas–solid interaction, thus favoring the formation of CeO2 by introduction of stirring. For the unsealed sample, both steps I and II can proceed completely resulting in the formation of yellow CeO2, while for the sealed sample, the availability of oxygen was limited, so only a relatively small quantity of Ce(OH)3 can be transformed to CeO2, leading to no color change after about 6 min during stirring.
The comparison experiment 2 further clarified the effect of oxidation of Ce(OH)3 during stirring treatment, by using tetravalent cerium precursor 0.1 M Ce(NH4)2(NO3)6 as the starting material. As Ce ion possesses +4 valence state in Ce(NH4)2(NO3)6, the addition of 6 M NaOH to the Ce(NH4)2(NO3)6 solution results in a precipitation reaction to form Ce(OH)4 (yellow color). When the cerium source was changed from Ce(NO3)3 to Ce(NH4)2(NO3)6, a soluble source of Ce(IV), no such color change was observed as shown in Fig. 2ii, except in the early stage of reaction (0 to ∼3 minutes of stirring). This early color change could be due to the formation of Ce(OH)4 from Ce(NH4)2(NO3)6. After this initial period, the color of the precipitate suspension remained unchanged, even after dehydration of Ce(OH)4 to CeO2 because no change of oxidation state was involved in the process. A possible mechanism for the observation shown in Fig. 2ii may involve the following precipitation and dehydration steps:
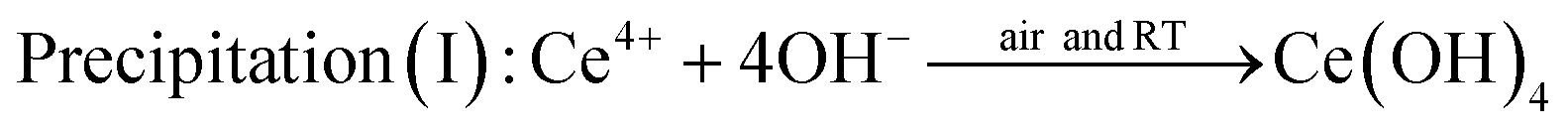
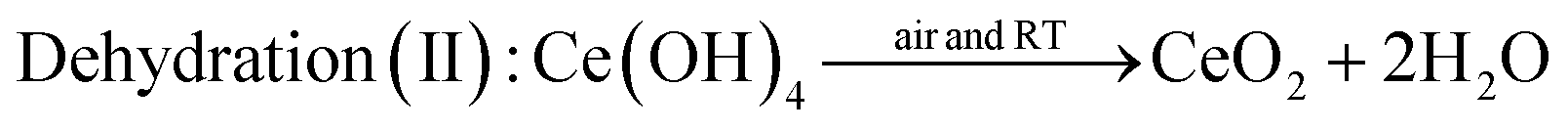
Fig. 3 shows the XRD and TEM results of the nanopowders prepared by hydrothermal reaction at 150 °C for 48 hours using the four suspension solutions in Fig. 2i-l and ii-l. The XRD patterns revealed that while the XRD peaks of all four as-synthesized samples are in agreement with a face-centered cubic CeO2 structure (JCPDS-34-0394), the sealed and unsealed samples using Ce(NH4)2(NO3)6 precursor showed a smaller crystalline size according to the Scherrer equation (Table 1), compared to the samples prepared using Ce(NO3)3, as evidenced by significantly broader XRD reflections and from the TEM images. The sealed and unsealed samples using Ce(NH4)2(NO3)6 precursor show a similar irregular shape of individual nanoparticles and 2.8 nm in size (Table 1).
 | ||
| Fig. 3 Powder X-ray diffraction patterns (i) and TEM images (ii) of sealed and unsealed samples after a hydrothermal reaction at 150 °C for 48 h. (a and b) The sealed and unsealed samples using Ce(NH4)2(NO3)6, respectively. (c and d) The sealed and unsealed samples using Ce(NO3)3, respectively. | ||
| Cerium precursor | Sealed | Unsealed |
|---|---|---|
| Ce3+: Ce(NO3)3 | 7.3 nm | 3.4 nm |
| Ce4+: Ce(NH4)2(NO3)6 | 2.8 nm | 2.8 nm |
Fig. 4 compares the color difference of the suspension solutions between the sealed and unsealed samples using Ce(NO3)3 before and after the hydrothermal reactions at 150 °C for 48 h. After the hydrothermal reactions, both of the samples show white color, but high resolution TEM (HRTEM) images show that the nanoparticles for the unsealed sample have an irregular shape with a size of 3–4 nm, while most of the nanoparticles for the sealed sample display nanocubes/nanocuboids with a size of 6–10 nm. The sealed sample using Ce(NO3)3 presents the best crystallinity and largest particle size (Fig. 3 and 4(2-A)). The diffractogram pattern taken from HRTEM images from individual cerium oxide nanocubes/nanocuboids reveals the single crystal nature of the cubes with {100} facets.
 | ||
| Fig. 4 A comparison of color difference and corresponding TEM images between the sealed and unsealed suspension solutions using Ce(NO3)3 before and after hydrothermal reaction. Both of the samples were stirred for 20 min. Sample a/A was sealed during the stirring, while sample b/B was not. | ||
Shape-controlled hydrothermal synthesis of CeO2 nanocrystals
It is commonly assumed that continuous stirring will lead to a more thorough dispersion of newly formed nuclei during the precipitation reaction and result in more compositionally homogeneous products. The observation above demonstrated that the formation (and/or shape/size) of Ce(OH)3 crystal seeds play a critical role in synthesizing shape/size-controlled CeO2 nanocrystals. Based on this strategy, we prepared CeO2 nanocrystals of different morphology at different temperatures and dwell times, in which the Ce(NO3)3/NaOH suspension solutions were not stirred and quickly transferred to autoclaves for the hydrothermal reactions at different temperatures and dwell times. Fig. 5 shows representative TEM images of CeO2 nanocrystals with controlled morphology. Table 2 displays the crystalline size calculated using (111) diffraction peaks in the powder XRD patterns and the BET surface area of CeO2 nanocrystals prepared by different hydrothermal reaction temperature and dwell time. The well-dispersed CeO2 nanocrystals with different shapes and sizes including nanosheets, nanorods, nanocubes, and nanocuboids can be obtained by the control of reaction temperature and dwell time. In general, the CeO2 nanocrystals seemed to prefer the morphology of nanosheets and nanorods at low-temperature (≤130 °C) hydrothermal conditions, while at higher temperature the nanocube-like or irregular shapes were observed. | ||
| Fig. 5 TEM images of CeO2 nanocrystals prepared by a hydrothermal method at different temperatures ((a) 70 °C; (b) 110 °C; (c) 130 °C; (d) 210 °C) and dwell time (1: 6 h; 2: 48 h; 3: 96 h). | ||
| 6 h | 48 h | 96 h | ||||
|---|---|---|---|---|---|---|
| Crystalline size (nm) | BET surface area (m2 g−1) | Crystalline size (nm) | BET surface area (m2 g−1) | Crystalline size (nm) | BET surface area (m2 g−1) | |
| 70 °C | 4.1 | 77 | 4.3 | 76 | 5.0 | 69 |
| 110 °C | 5.3 | 78 | 7.3 | 70 | 14.1 | 68 |
| 130 °C | 5.3 | 71 | 11.4 | 65 | 15.2 | 52 |
| 210 °C | 17.7 | 47 | 17.7 | 46 | 17.0 | 52 |
According to the results above, Fig. 6 shows a schematic diagram, which summarizes the shape-controlled synthesis strategy of CeO2 nanocrystals in this study. The synthesis mechanisms may be described as follows. A precipitate is obtained by adding NaOH to Ce(NO3)3 or Ce(NH4)2(NO3)6. If using Ce(NH4)2(NO3)6 as cerium precursor, the formation of Ce(OH)4 and subsequent decomposition to CeO2 leads to the growth of irregular nanocrystals during hydrothermal reactions. If using Ce(NO3)3 as cerium precursor, the oxidation (to Ce(OH)4 or CeO2) or disturbance of Ce(OH)3 nucleation also results in formation of irregular CeO2 nanocrystals during hydrothermal reactions. Shape-controlled CeO2 nanorods and nanocubes can be only obtained by fast mild stirring or without stirring treatment to avoid oxidation of Ce(OH)3 before sealing the mixture of NaOH and Ce(NO3)3 for hydrothermal reaction. Therefore, the controllable formation of Ce(OH)3 from solution seems to be a key to growing different morphological CeO2 nanocrystals, although the synthetic mechanism of CeO2 nanosheets, nanorods, and nanocubes obtained during the hydrothermal reaction is still not well understood and needs further investigation.
 | ||
| Fig. 6 Schematic diagram showing the shape-controlled synthesis of CeO2 nanocrystals. | ||
Conclusion
In summary, we reported that the nucleation and oxidation of Ce(OH)3 are critical steps which needs careful control in order to synthesize different morphological CeO2 nanocrystals. The combination of stirring and oxidation of suspension could destroy the nucleation and growth of Ce(OH)3, resulting in the formation of irregular CeO2 nanocrystals during the hydrothermal reactions. We demonstrated a general route for shape-controlled synthesis of CeO2 nanocrystals via mediation of the Ce(OH)3 seed before hydrothermal reactions.Acknowledgements
The financial support from American Chemical Society Petroleum Research Fund (#52323), the Department of Transportation (DoT-CTME project), the Department of Energy (EE0004094), YSU University Research Council grant and the use of TEM facilities at the Center of Excellence in Materials Science and Engineering at Youngstown State University are gratefully acknowledged.Notes and references
- I. E. Washs, Catal. Today, 1996, 27(3–4), 437–455 Search PubMed.
- A. K. Datye, A. D. Logan and N. J. Long, J. Catal., 1988, 109(1), 76–88 CrossRef CAS.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- R. J. Gorte, AIChE J., 2010, 56, 1126–1135 CAS.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS PubMed.
- M. Cargnello, V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341(6147), 771–773 CrossRef CAS PubMed.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 389(4), 439–520 Search PubMed.
- J. Kaspar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50(2), 285–298 CrossRef CAS.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50(2), 353–367 CrossRef CAS.
- S. Carrettin, P. Concepcion, A. Corma, J. M. L. Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 319, 2538–2540 CrossRef PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309(5735), 752–755 CrossRef CAS PubMed.
- E. Aneggi, M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli, J. Alloys Compd., 2006, 408, 1096–1102 CrossRef PubMed.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309(5735), 713–714 CrossRef CAS PubMed.
- Q. Fu and H. Saltsburg, Active nonmetallic Au and Pt species on ceria-based water–gas shift catalysts, Science, 2003, 301(5635), 935–938 CrossRef CAS PubMed.
- E. L. Brosha, R. Mukundan, D. R. Brown, F. H. Garzon and J. H. Visser, Solid State Ionics, 2002, 148(1–2), 61–69 CrossRef CAS.
- I. Kosacki, T. Suzuki, H. U. Anderson and P. Colomban, Solid State Ionics, 2002, 149(1–2), 99–105 CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129(1–4), 95–110 CrossRef CAS.
- Z. P. Shao and S. M. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed.
- H. L. Tuller, Solid State Ionics, 2000, 131(1–2), 143–157 CrossRef CAS.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ionics, 2000, 129(1–4), 63–94 CrossRef CAS.
- H. X. Mai, L. D. Sun, Y. W. Zhang, R. Si, W. Feng, H. P. Zhang, H. C. Liu and C. H. Yan, J. Phys. Chem. B, 2005, 109(51), 24380–24385 CrossRef CAS PubMed.
- K. Zhou, X. Wang, X. Sun, Q. Peng and Y. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS PubMed.
- M. Lin, Z. Y. Fu, H. R. Tan, J. P. Y. Tan, S. C. Ng and E. Teo, Cryst. Growth Des., 2012, 12(6), 3296–3303 CAS.
- L. Liu, Y. Cao, W. Sun, Z. Yao, B. Liu, F. Gao and L. Dong, Catal. Today, 2011, 175, 48–54 CrossRef CAS PubMed.
- M. M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18(16), 3808–3812 CrossRef CAS.
- M. Hirano and E. Kato, J. Am. Ceram. Soc., 1996, 82(3), 786–788 CrossRef.
- L. Yan, R. B. Yu and X. R. Xing, Cryst. Growth Des., 2008, 8(5), 1474–1477 CAS.
- K. Kaneko, K. Inoke, B. Freitag, A. B. Hungria, P. A. Midgley, T. W. Hansen, J. Zhang, S. Ohara and T. Adschiri, Nano Lett., 2007, 7(2), 421–425 CrossRef CAS PubMed.
- F. Li, X. H. Yu, H. J. Pan, M. L. Wang and X. Q. Xin, olid State Sci., 2000, 2, 767–772 CAS.
- X. H. Yu, F. Li, X. R. Ye, X. Q. Xin and Z. L. Xue, J. Am. Ceram. Soc., 2000, 83(4), 964–966 CrossRef CAS.
- Q. Yuan, H. H. Duan, L. L. Li, L. D. Sun, Y. W. Zhang and C. H. Yan, J. Colloid Interface Sci., 2009, 335, 151–167 CrossRef CAS PubMed.
- K. S. Lin and S. Chowdhury, Int. J. Mol. Sci., 2010, 11, 3226–3251 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |