
An operationally simple, palladium catalysed dehydrogenative cross-coupling reaction of pyridine N-oxides and thiazoles “on water”†
Abstract
Herein we describe the first reported example of dehydrogenative cross-coupling of heteroaromatic moieties achieved “on water”. This reaction is catalytic in palladium, site selective and operationally simple.
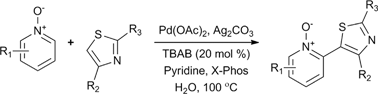
 Please wait while we load your content...
Please wait while we load your content...

