
Effects of pyrolysis parameters on hydrogen formations from biomass: a review
Abstract
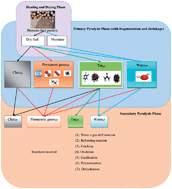
This study investigates the effects of different parameters such as biomass composition, moisture content, particle size, heating rate, temperature, inert gas, reactor system, and catalyst on the production of hydrogen gas (HG) and other gases (OGs) such as CO2, CO, CH4, C2H6, and so on. The reformation of OGs into H2 via the shift reaction significantly increases the total HG formation during biomass pyrolysis. Biomass raw material is capable of producing different proportions of HG at different temperatures because the raw material structure varies from one material to another. It is very puzzling that the formation of HG and total gas (TG) could either increase or decrease with moisture and this result varied between researchers. Smaller particles are more suitable than larger ones in terms of HG and TG formation. Additionally, longer residence times and higher temperatures favor good HG yield while the heating rate is a function of heat flux and particle size of biomass fuel is supposed to increase the pyrolytic gases and properties. Moreover, the heating rate would play a smaller role on the process when the system is introduced with inert gas and if the purpose is to maximize the production of TGs, secondary reactions such as thermal cracking, re-polymerization, and re-condensation should be maximized because the inert gas removes the volatiles from the pyrolysis environment. Therefore, the ultimate product of biomass conversion is dependent on the reactor design and type of feedstock in the presence of appropriate catalysts.
 Please wait while we load your content...
Please wait while we load your content...

