 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocrystallisation of the 2C–2′C dimer of a triphenylimidazolyl radical†
Robert M.
Edkins‡
,
Michael R.
Probert§
,
Craig M.
Robertson¶
,
Judith A. K.
Howard
and
Andrew
Beeby
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: andrew.beeby@durham.ac.uk
First published on 13th December 2013
Abstract
The structure of an unusual dimerisation mode of a triphenylimidazolyl radical has been elucidated, revealing a relatively long C–C bond. The diffraction-quality single crystals were produced under continuous irradiation by photocrystallisation in an adaption of the method that should be widely applicable to other photochromic systems.
Introduction
Neutral triphenylimidazolyl radicals (TPIRs) have attracted significant attention1–7 since they were first reported by Hayashi and Maeda in 1960 (ref. 8) due to their remarkable photo-, piezo- and thermochromism. These various forms of chromism have a common origin: reversible formation of two coloured TPIRs from a colourless dimer.9 The dimer is a hexaarylbiimidazole (HABI) that has six potential isomeric modes (Scheme 1).10 Early studies attempted to establish the HABI speciation in solution and in the solid state and to correlate the identified modes to particular types of chromism, with some conflicting results.11–14 More recent assessments suggest that the most common HABIs are the 2C–1′N mode, which is both photo- and thermochromic, and the 4C–4′C mode, for which thermo- or piezochromic materials have been described.1,15 Concern for their long-term stability has recently been raised through the isolation of deleterious peroxide side-products, but they are in general reasonably robust.16,17 Modes other than these six have been observed occasionally in sterically hindered systems.18 | ||
| Scheme 1 Potential dimerisation modes of two TPIRs bound through combinations of the atoms of the imidazole rings to form HABIs. | ||
Single-crystal X-ray diffraction (SC-XRD) studies have been indispensable in furthering the understanding of the dimerisation of TPIRs, which otherwise has relied principally on the less definitive method of IR spectroscopy.19 Several 2C–1′N dimers have been characterised by SC-XRD,20 and it has been concluded that this is usually the most stable form. Irradiation of a crystal of the unsubstituted 2C–1′N HABI led to partial conversion, through a “molecular swapping” process, to a mixture of the radical and its 2C–2′C isomer (QIWXID).21 However, the low conversion (14%) for this process made structural analysis of the dimer challenging. Very recently, a non-chromic 2C–2′C dimer was structurally characterised and a similar structure was inferred for a negative photochromic derivative.22 In these examples, the two TPIRs are held in close proximity by a tether and cannot form the usual 2C–1′N dimer. Low temperature NMR of an in situ irradiated sample has also been used recently to observe the 2C–2′C dimer.23 However, the 2C–2′C dimer mode has not been isolated in a pure form for a non-tethered TPIR and it is thus of interest to identify conditions under which it is accessible.24 In the work presented below, we describe the use of a photocrystallisation method to produce single crystals of a 2C–2′C HABI, which has allowed us to characterise this normally unstable mode by SC-XRD.
Results and discussions
We were interested in preparing a TPIR derivative with extended conjugation and, hence, a red-shifted absorption band. To this end, we designed an acetylenic TPIR derivative, ETPI, with a rigid, π-conjugated 4-tert-butylphenylethynylphenyl substituent at the 2-position of the imidazole ring (Scheme 2). The synthesis of ETPI comprised four steps. The intermediate 4′-(4-tert-butylphenylethynyl)benzaldehyde (EBA) was synthesised by conventional Sonogashira cross-coupling and deprotection methodology in 63% yield (two steps). This compound was then condensed with benzil and ammonium acetate in a microwave-heated reaction25 to give the imidazole ETPI-H in 64% yield; this compound was characterised fully, including the SC-XRD structure of its ethanol disolvate (ESI, Fig. S1†).|| Finally, ETPI-H was oxidised by K3[Fe(CN)6] in a biphasic mixture of CH2Cl2 and aqueous KOH, forming a dark green solution initially, from which a light green solid was isolated (see ESI† for full details). | ||
| Scheme 2 Synthesis of extended TPIR derivative ETPI and the conditions used to obtain single crystals of its isomeric dimers ETPI-CN and ETPI-CC. | ||
The tBu group was selected for two purposes: to increase solubility and to provide a readily interpretable set of NMR signals, characteristic of the HABI symmetry in solution. Signals corresponding to two inequivalent tBu groups were observed by 1H and 13C{1H} NMR, and two inequivalent acetylene groups were identified by 13C{1H} NMR, indicating an asymmetric dimerisation mode (Fig. 1). SC-XRD of pale green crystals grown in the dark from acetone/hexanes (anti-solvent), confirmed this compound to be the common 2C–1′N HABI (ETPI-CN) (Fig. 2).||26 This compound was found to be both photo- and thermochromic in CH2Cl2 solution, but exhibited no obvious piezochromism in the solid state, consistent with other 2C–1′N derivatives. Fig. 3 shows the change in the UV-visible absorption spectra following irradiation at 365 nm. The coloured species generated has two additional bands in the visible region: a structured, but very broad, band between 500 and 800 nm (λmax = 721 nm) with an energy spacing of ca. 2000 cm−1 between the two vibronic features, and a higher energy peak at λmax = 445 nm. An isosbestic point, indicative of only two species present in equilibrium in solution, was observed at ca. 330 nm. Compared to the unsubstituted TPIR,27 there is a bathochromic shift of ca. 170 nm (4300 cm−1) in the low-energy band, demonstrating the colour tuning of these photochromic materials (from violet to green in this case). This spectral tuning of ETPI relative to the parent TPIR is intermediate to that of a previously reported vinylbithiophene derivative, which absorbs at λmax = 862 nm (low-energy band).28
 | ||
| Fig. 1 The inequivalent (a) acetylene and (b) tBu signals in the 101 MHz 13C{1H} NMR (C6D6) spectrum of the initial ETPI dimer, consistent with the asymmetric 2C–1′N HABI dimer ETPI-CN. | ||
 | ||
| Fig. 2 SC-XRD molecular structure of ETPI-CN obtained from a crystal grown in the absence of light from acetone/hexanes. Hydrogen atoms are omitted for clarity; ADPs are drawn at 50% probability.26 | ||
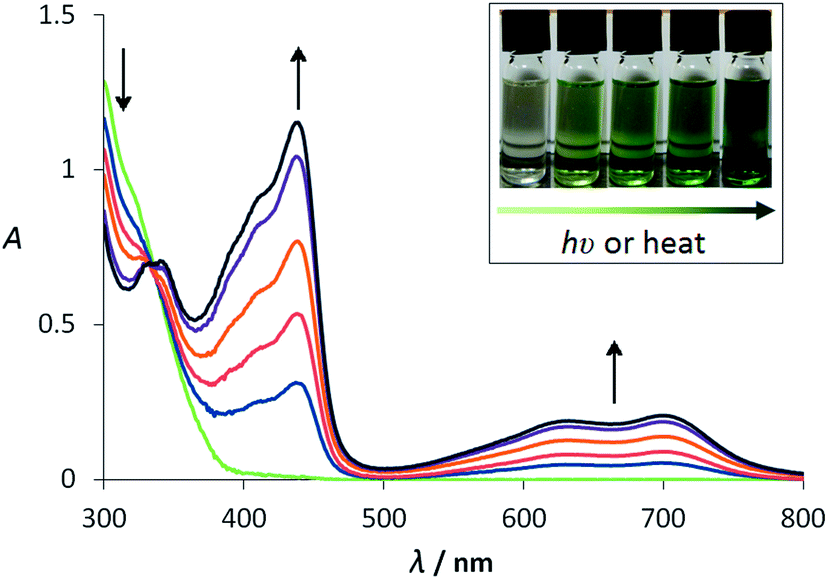 | ||
| Fig. 3 UV-visible absorption spectra recorded during the conversion of ETPI-CN (ca. 5 × 10−6 M) to ETPI in CH2Cl2 by UV (365 nm) irradiation at room temperature. Interval of ca. 30 s. Inset: photograph of the visible colour change from colourless to dark green. | ||
Irradiation (365 nm) of an ethyl acetate solution of ETPI-CN at room temperature generated a dark green deposit at the point of irradiation on the wall of the vial. Abe and co-workers reported previously the structure of the quinoidal form of a bisimidazole diradical from a crystal obtained during low temperature (200 K) irradiation at 360 nm of a toluene solution of the parent dimer.29,30 Similarly, during the study of phenalenyl neutral radicals, Haddon and co-workers obtained a novel dimerisation mode when a sample crystallised under ambient visible light conditions.31 These reports of unusual materials being formed under irradiation, encouraged us to elucidate the structure of the solid. After considerable experimentation, single crystals suitable for diffraction were produced from a ca. 10 mM CHCl3 solution of ETPI-CN by 365 nm irradiation focused on the vial wall.32 The structure of the crystallised material was determined by SC-XRD to be the 2C–2′C HABI (ETPI-CC) (Fig. 4),|| previously only observed as a small proportion of an irradiated crystal or in tethered systems (vide supra).21,22 Importantly, the SC-XRD structure and NMR spectra of ETPI-CN from the bulk sample confirm that the 2C–2′C dimer is not the favoured mode for this derivative under standard conditions.
 | ||
| Fig. 4 SC-XRD molecular structure of ETPI-CC obtained from a crystal grown under irradiation at 365 nm in CHCl3 solution. Hydrogen atoms are omitted for clarity; ADPs are drawn at 50% probability. The molecule lies on a crystallographic inversion centre; thus, the primed atoms are generated by the symmetry operation (1 − x, 1 − y, 1 − z). | ||
The 2C–2′C bond length of ETPI-CC is 1.601(4) Å, which is relatively long compared to the typical C(sp3)–C(sp3) bond length of 1.54 Å.33 It is also longer than the 2C–2′C bond of the recently reported biphenyl bridged compound (1.562(2) Å),22 which is attributable to structural constraint in the tethered system. Long bonds are of interest for furthering our understanding of chemical bonding,34,35 and while this C–C bond is not as long as some previously reported systems designed to induce long bonds through ring strain (1.72 Å)36,37 or steric bulk (1.63–1.65 Å),38 it is unusual in that it is a system where neither of these driving forces are present.31,39,40 Dispersion interactions have been used to stabilise a bulky alkane with a bond length of 1.71 Å, but this does not seem an obvious stabilising factor for ETPI-CC.41,42 A radical cation dimer with a bond length of 1.637(5) Å has also been reported very recently,43 which highlights the continued interest in this area. The long bond is suggestive of weak association between the constituent parts of the dimers. Consistent with this, dissolution of ETPI-CC in CH2Cl2 immediately produced a dark green solution. The UV-visible absorption spectrum of this solution was indistinguishable from an irradiated solution of ETPI-CN, suggesting that ETPI-CC dissociates rapidly to give ETPI. After leaving the solution of ETPI-CC in the dark for ca. two hours, the colour disappeared and the absorption spectrum matched that of freshly dissolved ETPI-CN. This is very similar behaviour to that described for the 4C–4′C mode, which is said to be only stable in the solid state, dissociating to the radical when dissolved. It is noted that the structure of a 4C–4′C mode has never been confirmed by SC-XRD, with IR and NMR23 spectra presented as verification. We did not observe any evidence for the existence of this, or any other, additional dimerisation mode of ETPI.
ETPI-CC and ETPI-CN have strong Raman acetylenic stretches centred at 2217 and 2214 cm−1, respectively, and ring modes at 1602 and 1605 cm−1, respectively. Both have bands at 1115 cm−1. Characteristically, ETPI-CC has a weak band at 910 cm−1, absent for ETPI-CN, while ETPI-CN has additional aromatic stretches at 1472 and 1538 cm−1 due to its lower symmetry. Raman spectra of CH2Cl2 solutions of ETPI-CN before and after irradiation are indistinguishable, both from each other and from a solution of ETPI-CC immediately following dissolution. The instability of ETPI-CC in solution and the absence of a bulk quantity inhibited further spectroscopic investigation.
We have performed DFT geometry optimisations of ETPI (unrestricted), ETPI-CN and ETPI-CC using the B3LYP and M06-2X44 functionals and 6-31+G(d) basis set (tBu substituted for Me groups on the dimers). The connecting C–C bond length of ETPI-CC is calculated to be 1.618 Å (B3LYP)/1.633 Å (M06-2X), reproducing moderately well the experimentally observed long bond of this isomer. The calculated elongation of 0.02–0.03 Å is likely a result of the difference between the structure in the solid state and the gas phase, or is otherwise due to a minor deficiency in the method chosen. The Wiberg bond index45 of this C–C bond is 0.81 (M06-2X)/0.85 (B3LYP), indicating a relatively weak bond (the other three bonds to these carbon atoms have Wiberg bond indices of 0.97–0.98). According to previous calculations on a related phenanthroline fused-TPIR system,15 the order of thermodynamic stability of the dimers is 1N–1′N > 2C–1′N > 2C–2′C > all other potential modes, and we calculate, in agreement with this, that ETPI-CN is lower in energy than ETPI-CC by 27 kJ mol−1 (B3LYP)/35 kJ mol−1 (M06-2X). Dimerisation through the 2-position of the imidazolyl ring disrupts conjugation with the phenylethynyl moiety; therefore, the lower energy of ETPI-CN compared to ETPI-CC is rationalised as loss of conjugation in one half versus both halves of the respective dimers.
Due to the radical nature of ETPI (as shown for several other derivatives by EPR measurements16,28), its spin-density distribution must be considered in order to understand its dimerisation behaviour. In the DFT-calculated spin-density distribution (Fig. 5) it can be seen that there is a significant contribution at the 2-position of the imidazolyl ring through which both ETPI-CN and ETPI-CC dimerise. The radical is delocalised onto the 4-phenylethynyl moiety in the 2-position, but the terminal phenyl ring shows only a minor contribution. The symmetry related 4- and 5-positions also carry significant spin density, but dimerisation through these positions was not observed, tentatively ascribed to steric hindrance. The low spin density on the nitrogen atoms has been suggested to kinetically inhibit the formation of the 1N–1′N mode.28 It would, however, also be expected to contribute to a lower rate of formation of ETPI-CN than of ETPI-CC. Under the non-equilibrium conditions of constant irradiation used in the crystallisation of ETPI-CC, a high local concentration of ETPI is produced, which potentially could form either of these dimer modes; however, kinetic dimerisation leads to a greater proportion of ETPI-CC, and it is thus this mode that crystallises, localised at the site of irradiation. This method of producing crystalline material also relies on the relative solubility difference between the photogenerated and initial species in the particular solvent.
 | ||
| Fig. 5 UB3LYP/6-31+G(d) DFT calculated spin density distribution of ETPI. Black and white shading show positive and negative spin density, respectively. Isovalue: ±5 × 10−4 e a0−3. | ||
Crystallisation following a photochemical reaction has precedence, both in the gas phase46 and in solution, having been used in the crystallisation of the photochemical product of benzophenone (benzopinacol)47 and of proteins.48,49 Photocrystallisation has been also been used to form charge transfer salts following photoreduction of TCNQ in the presence of a sacrificial electron donor.50,51 However, these prior reports resulted in non-reversible formation of a photoproduct rather than perturbing an equilibrium as we demonstrate here for the formation of ETPI-CC. Non-photochemical irradiation has also been used to induce nucleation of both inorganic52–55 and organic materials,56–60 although the suggested origin of this effect differs greatly.61 We believe the adaption of the photocrystallisation method outlined here should be a viable route to single crystals for many systems that switch between different structural forms following irradiation.
An unrestricted TD-DFT calculation (UB3LYP/6-31+G(d)) was used to assign the broad, low-energy band observed in the absorption spectrum of ETPI as the D1 ← D0 transition. This transition is described by a β-highest occupied spin orbital to a β-lowest unoccupied spin orbital (β-LUSO ← β-HOSO) excitation (calculated 720 nm, observed 721 nm), while the higher energy band (calculated 462 nm, observed 445 nm) is assigned as chiefly an α-LUSO ← α-HOSO transition (see ESI† for orbital diagrams, a summary of the other low-energy transitions and calculated UV-visible absorption spectra). The band at ca. 720 nm is classified as predominantly charge transfer (CT) in character; in contrast, the more intense band at ca. 445 nm is identified as a π–π* transition. The difference in the transition type can be seen most clearly in the electron-density-difference maps (EDDMs) (Fig. 6). TD-DFT calculations of both dimers predict, as expected, no low-energy absorption bands: instead they have higher energy CT and π–π* transitions (see ESI†). Therefore, the immediate colouration upon dissolution of ETPI-CC can be concluded to arise from fast dissociation to ETPI in solution before reformation of the ETPI-CN dimer upon standing in the dark. The dark green colouration of the ETPI-CC crystals can be attributed to photochromism on their surface, which occurs even under minimal exposure to ambient light. The slower rate of colour formation following dissolution and irradiation of ETPI-CN confirms its greater kinetic stability relative to ETPI-CC.
 | ||
| Fig. 6 EDDMs of (a) the λ = 720 nm (f = 0.405) and (b) λ = 463 nm (f = 0.639) transitions of ETPI. UB3LYP/6-31+G(d) TD-DFT calculation. Green and red shading show increase and decrease in electron density, respectively. Hydrogen atoms are omitted for clarity. Isovalue: ±4 × 10−4 e a0−3. | ||
In conclusion, we have identified that photocrystallisation can be used to produce the thermodynamically less stable 2C–2′C HABI isomer. This method has facilitated the production of diffraction-quality single crystals of this unusual dimer mode for a colour-tuned TPIR derivative, allowing the determination of its SC-XRD structure and revealing its relatively long C–C bond (1.60 Å). This technique should be widely applicable to other photochromic systems in which a less stable (and less soluble) form of a molecule is produced upon irradiation. We have also identified that non-tethered 2C–2′C dimers are only stable in the solid state, dissociating rapidly to the radical upon dissolution before reforming the thermodynamically more stable 2C–1′N form in the dark.
Acknowledgements
R.M.E. thanks Durham University for the award of a Durham Doctoral Fellowship. We thank the EPSRC for funding (Grant: EP/C536436/1 (M.R.P. and C.M.R.)). We thank Diamond Light Source for synchrotron beam time allocated to the regional team of Newcastle and Durham Universities.Notes and references
- R. Dessauer, Photochemistry, History and Commercial Applications of Hexaarylbiimidazoles: All About HABIs, Elsevier, Amsterdam, 1st edn, 2006 Search PubMed.
- Y. Kishimoto and J. Abe, J. Am. Chem. Soc., 2009, 131, 4227–4229 CrossRef PubMed.
- H. Miyasaka, Y. Satoh, Y. Ishibashi, S. Ito, Y. Nagasawa, S. Taniguchi, H. Chosrowjan, N. Mataga, D. Kato, A. Kikuchi and J. Abe, J. Am. Chem. Soc., 2009, 131, 7256–7263 CrossRef PubMed.
- Y. Harada, S. Hatano, A. Kimoto and J. Abe, J. Phys. Chem. Lett., 2010, 1, 1112–1115 CrossRef.
- K. Mutoh and J. Abe, Chem. Commun., 2011, 47, 8868–8870 RSC.
- K. Mutoh and J. Abe, J. Phys. Chem. A, 2011, 115, 4650–4656 CrossRef PubMed.
- H. Yamashita and J. Abe, J. Phys. Chem. A, 2011, 115, 13332–13337 CrossRef PubMed.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1960, 33, 565–566 CrossRef.
- K. Maeda and T. Hayashi, Bull. Chem. Soc. Jpn., 1970, 43, 429–438 CrossRef.
- E. H. White and M. J. C. Harding, J. Am. Chem. Soc., 1964, 86, 5686–5687 CrossRef.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1962, 35, 2057–2058 CrossRef.
- D. M. White and J. Sonnenberg, J. Org. Chem., 1964, 29, 1926–1930 CrossRef.
- H. Tanino, T. Kondo, K. Okada and T. Goto, Bull. Chem. Soc. Jpn., 1972, 45, 1474–1480 CrossRef.
- T. Goto, H. Tanino and T. Kondo, Chem. Lett., 1980, 431–434 CrossRef.
- E. Kiepek, Y. Zhou, S. Hoz, E. Rozental, P. M. Kazmaier and E. Buncel, Can. J. Chem., 2005, 83, 1448–1459 CrossRef.
- S. Hatano and J. Abe, Phys. Chem. Chem. Phys., 2012, 14, 5855–5860 RSC.
- R. M. Edkins, M. R. Probert, K. Fucke, J. A. K. Howard and A. Beeby, Phys. Chem. Chem. Phys., 2013, 15, 7848–7853 RSC.
- S. Hatano, K. Sakai and J. Abe, Org. Lett., 2010, 12, 4152–4155 CrossRef PubMed.
- D. M. White and J. Sonnenberg, J. Am. Chem. Soc., 1966, 88, 3825–3829 CrossRef.
- 13 structures of 2C–1′N HABIs, excluding redeterminations and additional solvate structures, have been deposited in the CSD (WebCSD ver. 1.1.1, January 2013 update).
- M. Kawano, T. Sano, J. Abe and Y. Ohashi, Chem. Lett., 2000, 1372–1373 CrossRef.
- S. Hatano, T. Horino, A. Tokita, T. Oshima and J. Abe, J. Am. Chem. Soc., 2013, 135, 3164–3172 CrossRef PubMed.
- S. Delbaere, M. Orio, J. Berthet, M. Sliwa, S. Hatano and J. Abe, Chem. Commun., 2013, 49, 5841–5843 RSC.
- A metal organic framework comprised of 2C–2′C dimers stabilised by coordination of all four nitrogen atoms to CuBr4 units has also been reported: X.-F. Huang, D.-W. Fu and R.-G. Xiong, Cryst. Growth Des., 2008, 8, 1795–1797 Search PubMed.
- S. E. Wolkenberg, D. D. Wisnoski, W. H. Leister, Y. Wang, Z. Zhao and C. W. Lindsley, Org. Lett., 2004, 6, 1453–1456 CrossRef PubMed.
- As a result of poor diffraction quality and the inclusion of highly disordered solvent, the structure is used only to confirm connectivity.
- G. R. Coraor, L. A. Cescon, R. Dessauer, E. F. Silversmith and E. J. Urban, J. Org. Chem., 1971, 36, 2262–2267 CrossRef.
- A. Kikuchi, T. Iyoda and J. Abe, Chem. Commun., 2002, 1484–1485 RSC.
- A. Kikuchi, F. Iwahori and J. Abe, J. Am. Chem. Soc., 2004, 126, 6526–6527 CrossRef PubMed.
- A. Kikuchi and J. Abe, Chem. Lett., 2005, 34, 1552–1553 CrossRef.
- P. Liao, M. E. Itkis, R. T. Oakley, F. S. Tham and R. C. Haddon, J. Am. Chem. Soc., 2004, 126, 14297–14302 CrossRef PubMed.
- As this method produces the desired isomer only at the focus of the irradiation, it is not appropriate to give a yield; furthermore, the intention was only to produce high-quality single crystals for diffraction experiments, rather than to provide a large-scale preparative method for this isomer.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- C. Rüchardt and H. D. Beckhaus, Angew. Chem., Int. Ed. Engl., 1980, 19, 429–440 CrossRef.
- G. Gunbas, N. Hafezi, W. L. Sheppard, M. M. Olmstead, I. V. Stoyanova, F. S. Tham, M. P. Meyer and M. Mascal, Nat. Chem., 2012, 4, 1018–1023 CrossRef PubMed.
- R. Winiker, H. D. Beckhaus and C. Rüchardt, Chem. Ber., 1980, 113, 3456–3476 CrossRef.
- M. A. Flammtermeer, H. D. Beckhaus, K. Peters, H. G. Vonschering and C. Rüchardt, Chem. Ber., 1985, 118, 4665–4673 CrossRef.
- G. Kaupp and J. Boy, Angew. Chem., Int. Ed. Engl., 1997, 36, 48–49 CrossRef.
- F. Effenberger, K. E. Mack, R. Niess, F. Reisinger, A. Steinbach, W. D. Stohrer, J. J. Stezowski, I. Rommel and A. Maier, J. Org. Chem., 1988, 53, 4379–4386 CrossRef.
- A. A. Leitch, C. E. McKenzie, R. T. Oakley, R. W. Reed, J. F. Richardson and L. D. Sawyer, Chem. Commun., 2006, 1088–1090 RSC.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef PubMed.
- A. A. Fokin, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, J. Am. Chem. Soc., 2012, 134, 13641–13650 CrossRef PubMed.
- X. Chen, X. Wang, Z. Zhou, Y. Li, Y. Sui, J. Ma, X. Wang and P. P. Power, Angew. Chem., Int. Ed., 2013, 52, 589–592 CrossRef PubMed.
- The M06-2X functional provides a good description of non-covalent interactions, important in large systems such as these, and has been used recently to model effectively systems with long C–C bonds (ref. 42): Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef.
- A. Tam, G. Moe and W. Happer, Phys. Rev. Lett., 1975, 35, 1630–1633 CrossRef.
- T. Okutsu, K. Nakamura, H. Haneda and H. Hiratsuka, Cryst. Growth Des., 2003, 4, 113–115 Search PubMed.
- H. Adachi, K. Takano, Y. Hosokawa, T. Inoue, Y. Mori, H. Matsumura, M. Yoshimura, Y. Tsunaka, M. Morikawa, S. Kanaya, H. Masuhara, Y. Kai and T. Sasaki, Jpn. J. Appl. Phys., 2003, 42, L798–L800 CrossRef.
- T. Okutsu, K. Furuta, M. Terao, H. Hiratsuka, A. Yamano, N. Ferté and S. Veesler, Cryst. Growth Des., 2005, 5, 1393–1398 Search PubMed.
- A. P. O'Mullane, N. Fay, A. Nafady and A. M. Bond, J. Am. Chem. Soc., 2007, 129, 2066–2073 CrossRef PubMed.
- C. Zhao and A. M. Bond, J. Am. Chem. Soc., 2009, 131, 4279–4287 CrossRef PubMed.
- A. J. Alexander and P. J. Camp, Cryst. Growth Des., 2008, 9, 958–963 Search PubMed.
- M. R. Ward, I. Ballingall, M. L. Costen, K. G. McKendrick and A. J. Alexander, Chem. Phys. Lett., 2009, 481, 25–28 CrossRef PubMed.
- C. Duffus, P. J. Camp and A. J. Alexander, J. Am. Chem. Soc., 2009, 131, 11676–11677 CrossRef PubMed.
- M. R. Ward and A. J. Alexander, Cryst. Growth Des., 2012, 12, 4554–4561 Search PubMed.
- B. A. Garetz, J. E. Aber, N. L. Goddard, R. G. Young and A. S. Myerson, Phys. Rev. Lett., 1996, 77, 3475–3476 CrossRef CAS.
- J. Zaccaro, J. Matic, A. S. Myerson and B. A. Garetz, Cryst. Growth Des., 2000, 1, 5–8 CrossRef.
- X. Sun, B. A. Garetz and A. S. Myerson, Cryst. Growth Des., 2006, 6, 684–689 CAS.
- X. Sun, B. A. Garetz and A. S. Myerson, Cryst. Growth Des., 2008, 8, 1720–1722 CAS.
- M. R. Ward, S. McHugh and A. J. Alexander, Phys. Chem. Chem. Phys., 2012, 14, 90–93 RSC.
- In this method, clusters are aligned into a nucleation seed by the electric field of a focused, high intensity laser using the optical Kerr effect.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental detail, synthetic procedures, SC-XRD structure of ETPI-H, copies of all NMRs, DFT calculated orbital plots and Cartesian coordinates of all optimised structures. CCDC 920449–920451. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra43892a |
| ‡ Current address: Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. |
| § Current address: School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| ¶ Current address: Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 72D, UK. |
|| Crystal data forETPI-H·2EtOH: C33H28N2·2(C2H6O), λ = 0.71073 Å, monoclinic, P21/c, Z = 4, a = 11.0885(12) Å, b = 29.010(3) Å, c = 9.9651(10) Å, β = 102.424(3)°, V = 3130.4(6) Å3, T = 120 K, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 707 reflections measured, 6420 independent, final R indices: wR2 = 0.1339 (all data), R1 = 0.0523 (I > 2σ(I)). Crystal data forETPI-CN: C66H54N4, λ = 0.6689 Å, triclinic, P 707 reflections measured, 6420 independent, final R indices: wR2 = 0.1339 (all data), R1 = 0.0523 (I > 2σ(I)). Crystal data forETPI-CN: C66H54N4, λ = 0.6689 Å, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, a = 10.503(4) Å, b = 14.511(6) Å, c = 19.926(8) Å, α = 86.646(6)°, β = 82.585(6)°, γ = 70.912(5)°, V = 2845.6(19) Å3, T = 100 K, 4788 reflections measured, 4782 independent, wR2 = 0.2519 (all data), R1 = 0.0905 (I > 2σ(I)). Crystal data forETPI-CC: C66H54N4, λ = 0.6689 Å, triclinic, P, Z = 1, a = 10.017(3) Å, b = 11.112(3) Å, c = 11.721(3) Å, α = 69.889(3)°, β = 87.557(3)°, γ = 83.195(3)°, V = 1216.5(6) Å3, T = 150 K, 10462 reflections measured 3641 independent, wR2 = 0.1725 (all data), R1 = 0.0576 (I > 2σ(I)). , Z = 2, a = 10.503(4) Å, b = 14.511(6) Å, c = 19.926(8) Å, α = 86.646(6)°, β = 82.585(6)°, γ = 70.912(5)°, V = 2845.6(19) Å3, T = 100 K, 4788 reflections measured, 4782 independent, wR2 = 0.2519 (all data), R1 = 0.0905 (I > 2σ(I)). Crystal data forETPI-CC: C66H54N4, λ = 0.6689 Å, triclinic, P, Z = 1, a = 10.017(3) Å, b = 11.112(3) Å, c = 11.721(3) Å, α = 69.889(3)°, β = 87.557(3)°, γ = 83.195(3)°, V = 1216.5(6) Å3, T = 150 K, 10462 reflections measured 3641 independent, wR2 = 0.1725 (all data), R1 = 0.0576 (I > 2σ(I)). |
| This journal is © The Royal Society of Chemistry 2014 |