Enhanced-electrocatalytic activity of Ni1−xFex alloy supported on polyethyleneimine functionalized MoS2 nanosheets for hydrazine oxidation†
Jing
Li
,
Weijie
Tang
,
Haidong
Yang
,
Zhengping
Dong
,
Jingwei
Huang
,
Shuwen
Li
,
Jia
Wang
,
Jun
Jin
* and
Jiantai
Ma
*
College of Chemistry, and Chemical Engineering, Lanzhou University, Lanzhou, China. E-mail: majiantai@lzu.edu.cn
First published on 14th October 2013
Abstract
A high-performance Ni1−xFex on polyethyleneimine (PEI)-functionalized molybdenum disulfide (MoS2) electrocatalyst has been synthesized by an electroplating in situ growth approach. Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy confirm the successful functionalization of MoS2 with PEI. The empty orbitals of Ni2+ and Fe2+ (Ni and Fe precursors) coordinated with the donated lone pairs of nitrogen atoms in PEI-mediated MoS2 and then the Ni2+ and Fe2+ were in situ reduced at a negative potential. Transmission electron microscope images and X-ray diffraction reveal that Ni85Fe15 nanoparticles with an average size of 2.25 nm are uniformly dispersed on the PEI–MoS2 sheets. The Ni1−xFex/PEI–MoS2 catalyst exhibits unexpectedly high activity towards the hydrazine oxidation reaction, which can be attributed to highly homogeneous dispersed Ni1−xFex alloy. It also shows enhanced electrochemical stability due to the structural integrity of PEI–MoS2. Finally, the Ni85Fe15/PEI–MoS2 catalyst is proved to be very valuable for applications in hydrazine fuel cells, as compared with Ni90Fe10/PANi–MoS2 and Ni85Fe15/MoS2 catalysts.
Introduction
Since 2004,1 graphene, a 2D single layer of carbon atoms with an hexagonal packed structure, has evoked wide interest due to its excellent physicochemical properties such as mechanical strength, electrical conductivity and ease of functionalization.1–3 Considerable efforts have been successfully devoted to modifying graphene with metal nanoparticles (NPs) and to measure the properties of the graphene-based composites.4–7 Besides well-known graphene, graphene-like species formed by layered inorganic materials such as molybdenum disulfide (MoS2) have also sparked increasing attention in recent years.8–18 In particular, the unique physical, optical, electrical properties of single-layered MoS2, and its potential for applications (e.g., in topological insulators, transistors, and lithium ion batteries)15–17 has generated an accelerating pace of research relative to that on bulk MoS2.11,12 Unfortunately, limited studies are available on the preparation of an alloy supported on graphene-like MoS2 as a synergistic catalyst in fuel cells.Hydrazine (N2H4), as a high-energy fuel molecule, has remarkable advantages including a superior theoretical standard equilibrium potential and a high power density.19 Numerous researchers have focused on studying the high power density of direct hydrazine–air fuel cells.20 Since the 1960s, the electrooxidation of hydrazine in alkaline media has been investigated extensively at platinum, palladium, gold, nickel, and silver electrodes.21–25 However, the high cost limits the use of noble metals as catalysts for direct alkaline hydrazine fuel cell anodes. Previous studies indicated that Ni-based alloy exhibits excellent electrocatalytic activity for hydrazine electrooxidation.19 Importantly, the catalytic activity depends not only on the metal NPs, but also on the nature of the support.26,27 For supported metal catalysts, the activity and selectivity are strongly influenced by the amount of metal, the size of the dispersed metal particles, the preparation method, and the support composition.28 With regard to support materials, single-layered MoS2 has abundant active edges13,14 and a large specific surface area10,15 so that it may be a good catalytic support to load metal NPs. Zhong et al. have electroplated Ni–Fe alloy on graphene-like MoS2 and applied the material in hydrazine oxidation. However, an obvious weakness exists in MoS2, which has poor conductivity.18 In order to enhance the electrical conductivity of single-layered MoS2 and to obtain a better dispersed active phase in the catalyst, the introduction of conducting polymers (CPs) to single-layered MoS2 is a novel idea. As a matter of fact, CPs are at present studied in view of their high electrical conductivity and they have extensive technological applications such as in electronic devices and sensors.29,30 Single-layered MoS2 functionalized with CPs exhibit a large surface-to-volume ratio, together with good chemical stability and a broad electrochemical window, rendering them attractive electrode materials.31,32 Meanwhile, CPs have also been explored for the dispersal of metallic particles.33
CPs include various types, mainly electron-conducting and ion-conducting. These polymers have different cross-linked structures, which may affect the electrocatalytic performance. One of the most traditional electro-conducting polymers, polyaniline (PANi), has been widely reported in the fields of nanotechnology.34–36 Nowdays, there has been much attention on PANi-based materials because of their good environmental stability and ease of synthesis.35,36 Among the ion-conducting polymers, polyethyleneimine (PEI) has also attracted extensive attention from experimental-scientific communities because of its relatively high conductivity and high-water dispersibility.37,38 Many studies on the advantages of PEI have focused on the physical adsorption of PEI on multiwalled carbon nanotubes (CNTs) in order to promote the solubility, biological compatibility and electrical conductance of CNTs.37,39,40 Moreover, PEI contains one of the highest density of amino groups among all the polymers, donating electrons that help to anchor metal ions.41,42
On the basis of these observations, in this work, we report a novel work on the synthesis of Ni1−xFex/PEI–MoS2 catalyst by using PEI as a functionalization and anchoring agent (Scheme 1). The template single-layered MoS2 was functionalized in a synergistic coupling method using PEI, to form PEI–MoS2.43 Composites based on MoS2 and PEI form a “sandwich” organization. We then electroplated Ni1−xFex alloy NPs on the novel support material to produce the electrocatalyst, this was a facile and rapid process, and proceeded at low temperature. Owing to the amino-rich structure of PEI, the enhanced nucleation was favorable to anchor Ni1−xFex NPs. And their interaction was suggested to originate from the special nickel/iron–nitride (Ni/Fe–N) with PEI that bridged between the Ni1−xFex NPs and MoS2 sheets. To the best of our knowledge, there have been few reports on using PEI for the preparation of MoS2-based hybrid nanomaterials. The present report is the first on the electrodeposition of Ni1−xFex alloy on PEI–MoS2 sheets with different mass ratios of Ni and Fe, and the study of their use for the electrocatalysis of hydrazine oxidation. In order to vary the composition of bimetallic NPs, the mass concentration ratio of NiSO4·6H2O and FeSO4·7H2O was changed as some test examples. In addition, the electrocatalytic activities of the Ni1−xFex/PEI–MoS2 catalyst were further compared with the Ni1−xFex/PANi–MoS2 catalyst in which MoS2 was wrapped physically by PANi. The advantage of this work is that PEI plays a role in mediated functionalization. It not only improves the catalytic performance of single-layered MoS2 for hydrazine oxidation, but also leads to homogeneous distribution of Ni1−xFex NPs.
 | ||
| Scheme 1 The process for the preparation of the Ni1−xFex/PEI–MoS2. | ||
Experimental
Materials
Molybdenum disulfide (99%), polyethyleneimine (PEI, Mn = 600) were purchased from Aladdin-reagent Co. Ltd. Aniline, hydrogen chloride, ammonium persulfate ((NH4)2S2O8), de-ionized water (18.2 MΩ) were commercially available and used throughout the experiments. All the other commercial chemicals were used without further purification.MoS2 preparation
The preparation of MoS2 has been reported previously.8 The details of the process are described as follows: 30 mg of MoS2 powder was added to 25 mL flasks. 10 mL of ethanol–water with an EtOH volume of 45% was added as the dispersion solvent. The sealed flask was sonicated for 16 h, and then the dispersion was centrifuged at 5000 rpm for 10 min to remove the aggregates. The supernatant was collected and the concentration of MoS2 in 45% ethanol–water was estimated to be 0.02 mg·mL−1. The prepared dispersion of graphene-like MoS2 was centrifuged to 0.2 mg mL−1.Preparation of PEI–MoS2
The 0.1 wt% PEI was dispersed in the prepared MoS2 (100 mg) aqueous solution. The suspension was sonicated in an ultrasonic bath and refluxed at 60 °C for 24 h and washed with deionized water twice. Then PEI–MoS2 was obtained after drying the powder at 80 °C.Preparation of PANi–MoS2
PANi–MoS2 composites were synthesized by the polymerization of aniline in the presence of MoS2 suspension. An HCl solution (2 M, 150 mL) containing a prepared MoS2 (100 mg) aqueous solution was sonicated at room temperature for about half an hour and then mixed with 0.1 mL aniline monomer. Followed by the addition of 0.26 g of (NH4)2S2O8 (dissolved in 10 mL of deionized water) that served as the oxidant. The reaction mixture was constantly stirred in an ice bath (temperature range of 0–5 °C) for 1 h and then reacted at room temperature for 12 h. The mixing was filtered and washed with distilled water–ethanol until the filtrate became colorless. PANi–MoS2 was obtained after drying the powder at 80 °C.Details of the direct current (DC) electroplating
An electrochemical cell with a two-electrode configuration was used for the experiments. A copper plate (50 mm × 10 mm × 1 mm) was used as the cathode and a glass carbon electrode (GCE) to which the materials (PEI–MoS2 and PANi–MoS2) were loaded was used as the anode. The two electrodes were placed into a cell containing NiSO4·6H2O and FeSO4·7H2O as the plating solution at 60 °C. The mass ratio of Ni NPs to Fe NPs was controlled by varying the mass concentration of NiSO4·6H2O (x = 250 g L−1) and FeSO4·7H2O (y = 2.7, 9.7, 25.6, 30.5, 32, 39 g L−1). This kind of electroplating is an anomalous type of codeposition. The nickel iron electroplating formula and DC electroplating experimental factors are given in Table S1 and S2 (see ESI†).Characterization
The morphologies of Ni1−xFex/PEI–MoS2 and Ni1−xFex/PANi–MoS2 were observed using a TECNAI G2Tf20 transmission electron microscope, TEM (FEI American). The Fourier transform infrared (FT-IR) spectra were obtained by the KBr disc technique and recorded on a NEXUS 670 instrument (Nicolet American). X-Ray photoelectron spectroscopy (XPS) was recorded on a PHI-5702 instrument. X-ray diffraction (XRD) measurements were performed on a Rigaku D/max-2400 diffractometer, using Cu-Kα radiation as the X-ray source in the range of 10–90 °C. The amount of PEI and PANi loaded on MoS2 were determined by thermogravimetric analysis (TGA) (Linseis German). The content of Ni1−xFex alloy in the Ni1−xFex/PEI–MoS2 and Ni1−xFex/PANi–MoS2 was analyzed by inductively coupled plasma (ICP) spectroscopy using an IRIS Advantage analyzer.Electrochemical measurement
Electrochemical measurements of Ni1−xFex/PEI–MoS2 (PANi–MoS2) hybrid were performed with a CHI660C electrochemical analyzer (CHI Instrument Corp. Shanghai) using a three-electrode system at 60 °C throughout this work. The concentration of the Ni1−xFex/PEI–MoS2 (Ni1−xFex/PANi–MoS2) in water–ethanol–Nafion (volume ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) solution was 1 mg mL−1. A GCE electrode loaded with 10 μL of the prepared Ni1−xFex/PEI–MoS2 (PANi–MoS2) solution served as the working electrode, a platinum wire and a saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode, respectively. The electrolyte was 0.1 M hydrazine hydrate–0.015 M NaOH which had been purged with N2 for 10 min prior to the experiment.
:1:1) solution was 1 mg mL−1. A GCE electrode loaded with 10 μL of the prepared Ni1−xFex/PEI–MoS2 (PANi–MoS2) solution served as the working electrode, a platinum wire and a saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode, respectively. The electrolyte was 0.1 M hydrazine hydrate–0.015 M NaOH which had been purged with N2 for 10 min prior to the experiment.
Results and discussion
TEM and XRD analysis of PEI–MoS2 and PANi–MoS2
Fig. 1 shows the dispersion and morphology together with the particle size distribution of the Ni85Fe15/PEI–MoS2 and Ni90Fe10/PANi–MoS2 hybrids . It can be seen clearly that the PEI–MoS2 sheets were successfully decorated with many well dispersed Ni1−xFex NPs (Fig. 1a). Electrochemical deposition of bimetallic Ni1−xFex NPs resulted in the appearance of small particles on the surface of PEI–MoS2. Systematic counting revealed that the size of about 60% of the NPs was 2.25 nm. Further analysis of the Ni1−xFex NPs by ICP spectroscopy showed a composition of 85%:15% (mass ratio). Fig. 1c reveals that the Ni1−xFex alloy NPs were also stabilized on PANi–MoS2. However, the dispersed metal particles on the PANi–MoS2 support were not uniform in size. Therefore, it can be confirmed that PEI played an important role in the formation of the Ni1−xFex/PEI–MoS2 composite materials. The PEI coupling on the MoS2 sheets produced a uniform distribution of the amino groups which served as condense centers for the immobilization of Ni and Fe NPs.
 | ||
| Fig. 1 TEM images of (a) Ni85Fe15/PEI–MoS2 and (c) Ni90Fe10/PANi–MoS2; HRTEM images of (b) Ni85Fe15/PEI–MoS2 and (d) Ni90Fe10/PANi–MoS2. | ||
As a comparison, the TEM image of the graphene like MoS2 and the Ni85Fe15/MoS2 hybrid are illustrated (see ESI,† Fig. S1). Fig. S1b† shows the TEM image of the Ni85Fe15 NPs directly deposited on the surface of MoS2, it is obvious that the metal NPs aggregate readily and the size of the metal NPs isn't remarkably uniform.
In addition, energy dispersive X-ray (EDX) analysis was utilized to determine the chemical composition of the Ni85Fe15/PEI–MoS2 and Ni90Fe10/PANi–MoS2 catalysts. Peaks for Ni, Fe, Mo, S and N can be observed in Fig. 2 and indicate the composition of the catalysts. The peak of Cu arises from the copper grid in TEM analysis.
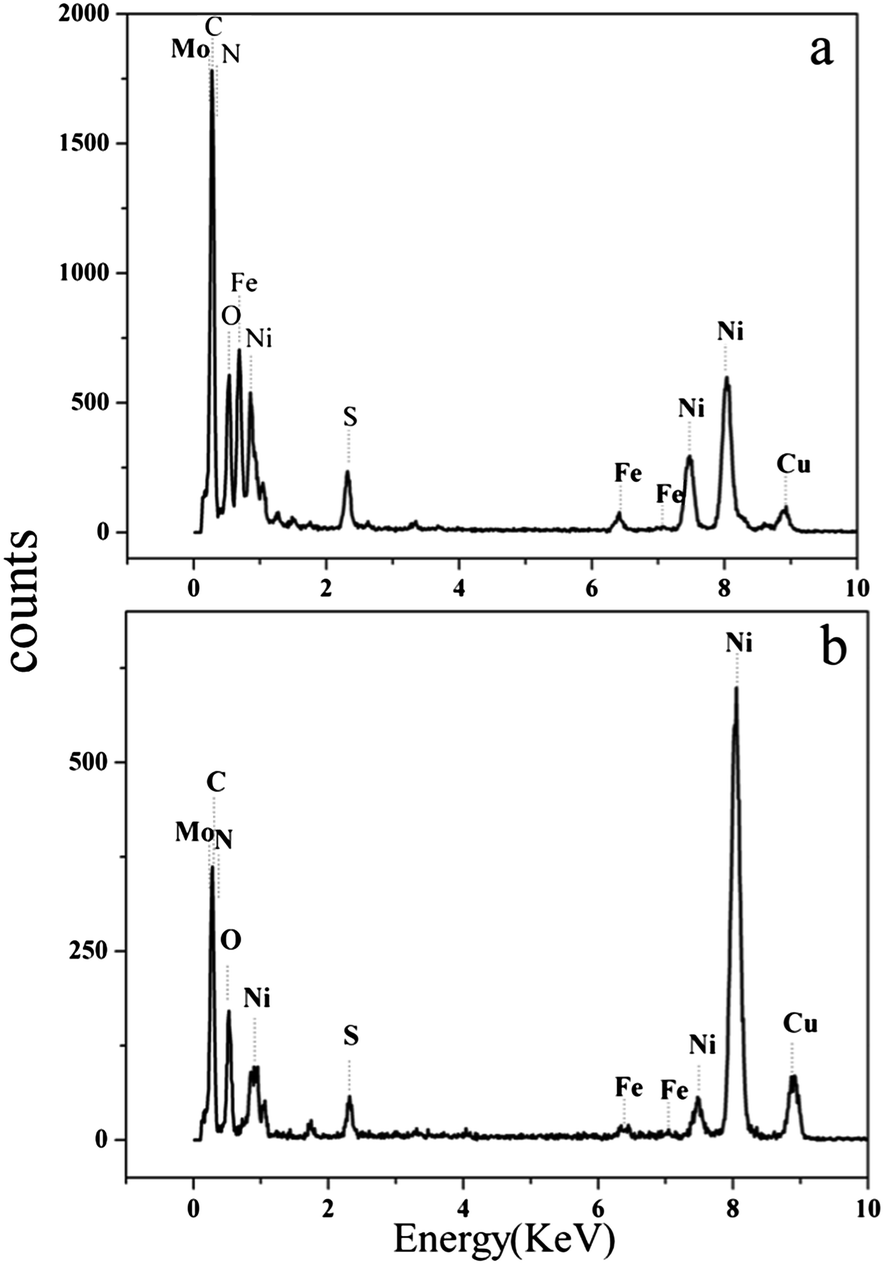 | ||
| Fig. 2 EDX images of (a) Ni85Fe15/PEI–MoS2 and (b) Ni90Fe10/PANi–MoS2. | ||
Fig. 3 shows the XRD patterns of the Ni85Fe15/PEI–MoS2 and Ni85Fe15/PEI–MoS2 hybrids to further investigate the crystalline structure. The primary (002) diffraction peak can be observed at 2θ = 14.3°, which is in good agreement with the hexagonal structure of MoS2.44 Moreover, it is seen that the crystal structure of Ni1−xFex is face-centered cubic (fcc), diffraction peaks at 2θ values of 44.5°, 51.9° and 76.4° can be assigned to the (111), (200), and (220) crystal planes of the Ni1−xFex alloy, respectively.45 Thus, the successful electroplating of Ni–Fe ions to Ni–Fe NPs can be confirmed. In addition, it is shown that the Ni90Fe10/PANi–MoS2 hybrid has a crystalline structure similar to that of Ni85Fe15/PEI–MoS2.
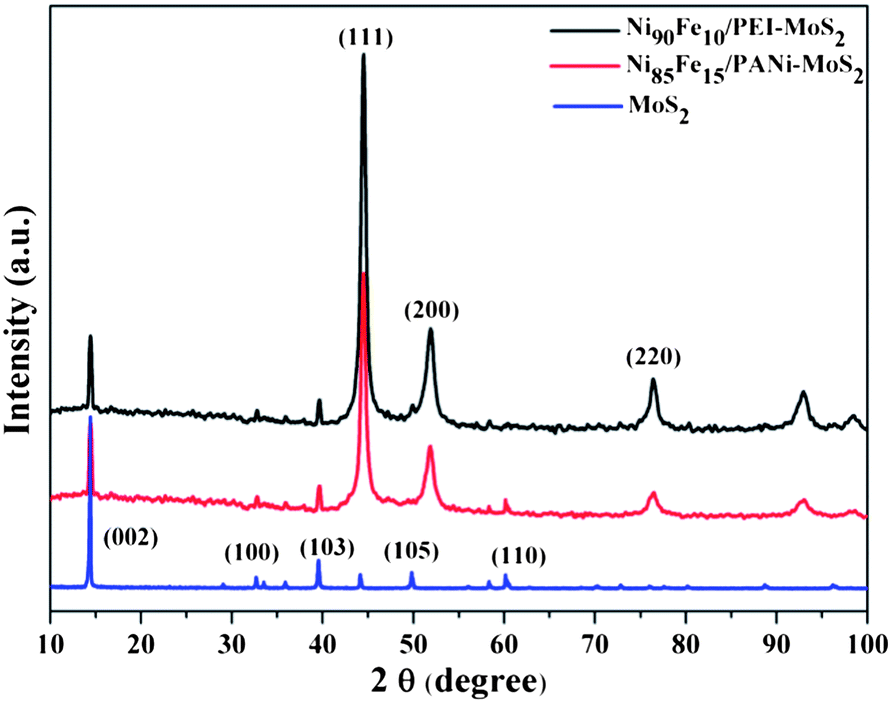 | ||
| Fig. 3 XRD patterns of the Ni90Fe10/PANi–MoS2 hybrid, the Ni85Fe15/PEI–MoS2 hybrid and MoS2. | ||
FT-IR analysis of PEI–MoS2 and PANi–MoS2
FT-IR spectra of MoS2, pure PEI, PEI–MoS2, Ni85Fe15/PEI–MoS2, PANi–MoS2, and Ni90Fe10/PANi–MoS2 are illustrated in Fig. 4. As can be seen from the spectrum of MoS2, the band at 467 cm−1 could be assigned to stretching of the Mo–S vibrations.46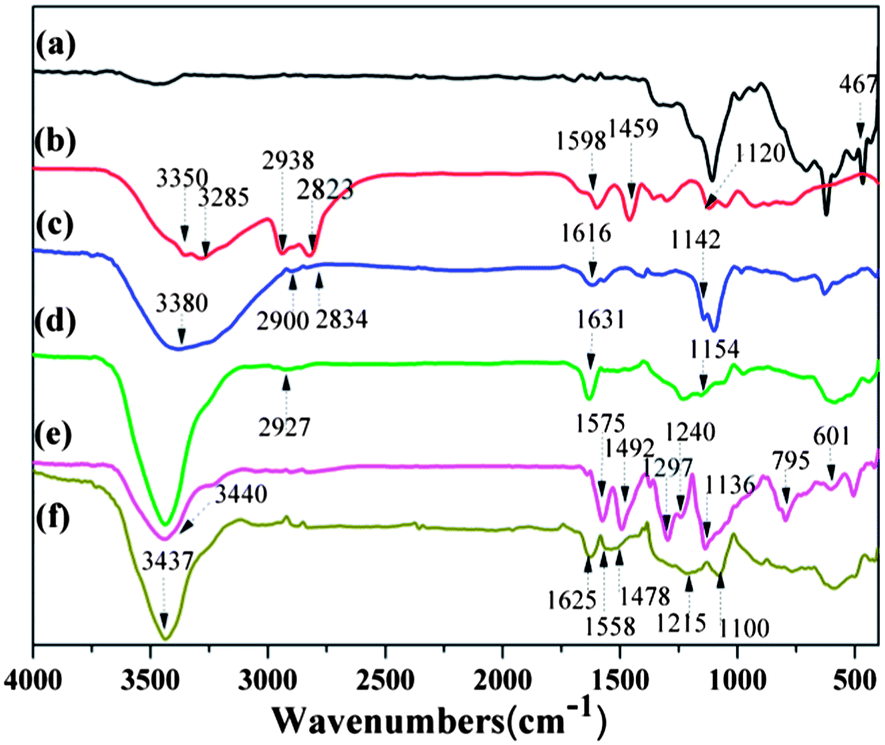 | ||
| Fig. 4 FT-IR spectra of (a) MoS2, (b) PEI, (c) PEI–MoS2, (d) Ni85Fe15/PEI–MoS2, (e) PANi–MoS2, (f) Ni90Fe10/PANi–MoS2. | ||
In the spectrum of PEI, two bands at 3350 and 3285 cm−1 arise from NH2 stretching vibrations, the strong peaks at 2938 and 2823 cm−1 that can be assigned to asymmetric and symmetric vibrations of the CH2 group, respectively, and the peak at 1459 cm−1 which corresponds to inplane bending of CH2. Peaks for the bending vibration of the NH group and the stretching vibration of the C–N groups of PEI can be seen at 1598 cm−1 and 1120 cm−1, respectively.47 However, there are subtle differences at the same positions between the IR spectrum of the PEI–MoS2 complex and that of MoS2 or PEI alone. The stretching vibrations of NH2 groups, NH groups and C–N groups shift to higher frequencies (3380 cm−1, 1616 cm−1, 1142 cm−1) for the PEI–MoS2 hybrid material, which clearly proves our hypothesis that synergistic coupling occurs between the amino-rich PEI and MoS2 sheets. Overall, the IR spectrum of PEI–MoS2 shows peaks that are characteristic of their main functional groups. When depositing Ni1−xFex alloy on the PEI–MoS2 sheets, the stretching vibrations of the main functional groups also shift to higher frequencies, which is thought to originate from the interaction of nickel/iron–nitride (Ni/Fe–N).
For the IR spectrum of PANi–MoS2,48,49 the band at 3440 cm−1 is attributable to N–H stretching vibration. The main peaks at 1575 and 1492 cm−1 can be assigned to the stretching vibrations of quinone and benzene rings, respectively. The bands at 1297 and 1240 cm−1 are attributed to C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode, while the band at 1136 cm−1 is assigned to the in-plane bending of C–H. A band at about 800 cm−1 could be attributed to the out-of-plane bending of C–H, while the many low-intensity peaks ranging from 780 to 580 cm−1 can be assigned to the vibrations of the C–H bonds in the benzene rings. The spectrum clearly indicates that PANi has been wrapped onto the surface of MoS2. Moreover, the spectrum of Ni90Fe10/PANi–MoS2 is shown in Fig. 4f. The stretching vibrations of the main characteristic groups shifting to lower frequency indicate the formation of Ni1−xFex alloy on PANi–MoS2.
N stretching mode, while the band at 1136 cm−1 is assigned to the in-plane bending of C–H. A band at about 800 cm−1 could be attributed to the out-of-plane bending of C–H, while the many low-intensity peaks ranging from 780 to 580 cm−1 can be assigned to the vibrations of the C–H bonds in the benzene rings. The spectrum clearly indicates that PANi has been wrapped onto the surface of MoS2. Moreover, the spectrum of Ni90Fe10/PANi–MoS2 is shown in Fig. 4f. The stretching vibrations of the main characteristic groups shifting to lower frequency indicate the formation of Ni1−xFex alloy on PANi–MoS2.
XPS analysis of N1s spectrum of PEI and PEI–MoS2
In conjunction with FT-IR spectroscopy, XPS has increasingly been used to study the interactions between metal ions and electron-donating ligands.50,51 As is shown in Fig. 5a and b, the N1s spectrum was used to determine the nitrogen configurations. In the research work on PEI, the N1s spectrum can be deconvoluted to several individual peaks which are assigned toN– (398.51 eV), –NH– (399.23 eV), and –NH2 (399.84 eV). However, the peak position of these nitrogen types varies as shown in Fig. 5b after MoS2 was modified by PEI. The binding energies were 398.71, 399.85, and 400.88 eV for N–, –NH–, and –NH2, respectively. The peak shift of –NH2 is as large as 1 eV, which is explained by the synergistic effect of Mo ions and the donating electrons of –NH2.42 The charge of nitrogen and its neighbouring atoms and the electron redistribution all influence the precise position of different nitrogen types. Thus, the binding energy of N– and –NH– results in little peak shift.
 | ||
| Fig. 5 N1s spectra of (a) PEI and (b) PEI–MoS2. | ||
TGA analysis of PEI–MoS2 and PANi–MoS2
TGA analysis (Fig. 6) was also used to characterize the surface modification of the MoS2. The weight loss stage below 200 °C is a result of the evaporation of physically adsorbed water and residual solvent. As is shown in Fig. 6b, there is a sharp decrease at about 400 °C and this could be due to the decomposition of PEI. Comparing the TGA curves of MoS2 and PEI–MoS2, the weight loss of PEI in PEI–MoS2 is 20.91%. In Fig. 6c, a rapid decrease begins at about 200 °C, and there is a 18.74% weight loss of PANi. It is worth mentioning that the PEI–MoS2 hybrid has a much better thermal stability than that of PANi–MoS2.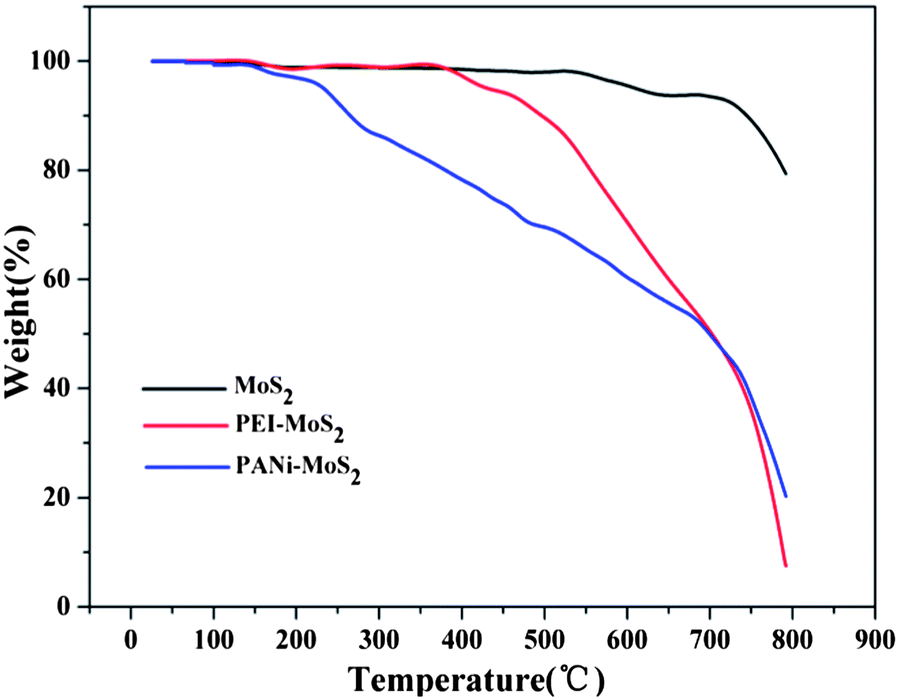 | ||
| Fig. 6 TGA curves of MoS2, PEI–MoS2 and PANi–MoS2. | ||
Hydrazine oxidation on the hybrid electrodes in the 0.1 M hydrazine hydrate/0.015 M NaOH solution
The electrochemical activity of the hydrazine oxidation reaction has been evaluated using cyclic voltammetry. Cyclic voltammograms of hydrazine on a GCE (in Fig. 7) and on a MoS2/GCE were first recorded. On both electrodes, no oxidation peaks were seen. When PEI–MoS2/GCE and PANi–MoS2/GCE were applied, a similar oxidation peak appeared at −0.2 V, while on a PEI–MoS2/GCE the oxidation current was higher and the capacitive current was bigger than that on a PANi–MoS2/GCE, confirming that the PEI–MoS2 material has a higher effective surface area and a better synergistic catalytic performance for hydrazine oxidation.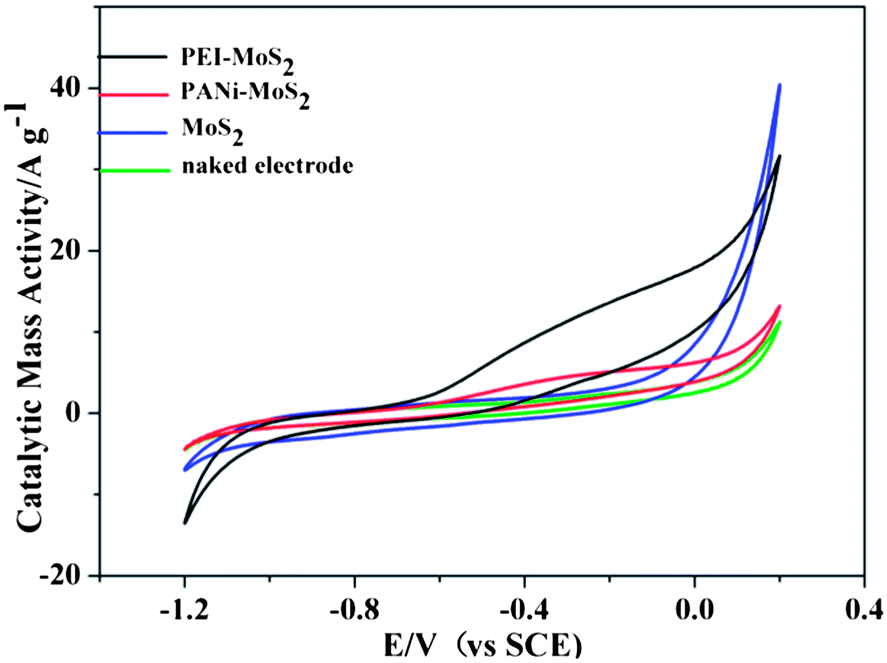 | ||
| Fig. 7 Cyclic voltammetric measurements of the PEI–MoS2 hybrid, the PANi–MoS2 hybrid, the MoS2 and naked electrode at a scan rate of 50 mV s−1. | ||
Then the electrocatalytic oxidation of hydrazine with bimetallic Ni1−xFex NPs on the PEI–MoS2/GCE and PANi–MoS2/GCE were studied. The mass ratios of Ni to Fe were varied in order to obtain the optimum content for hydrazine electrocatalysis. These NPs were produced by varying the mass concentration ratios of [NiSO4·6H2O]/[FeSO4·7H2O] during the electrodeposition process. The final mass ratio of Ni to Fe was analyzed by ICP spectroscopy (see ESI,† Table S3). Fig. 8 records a series of Ni1−xFex mass ratios ranging from 95:5 to 70:30 on the PEI–MoS2/GCE and PANi–MoS2/GCE, respectively. In Fig. 8a, the Ni1−xFex/PEI–MoS2 catalyst presents a high peak of anodic oxidation current. With an increase of the amount of Ni NPs, it leads to a positive shift of the oxidation potential as well as an enhancement of the anodic current. Nevertheless, the amount of Ni NPs doesn't solely influence the electrocatalytic activity. Our previous studies reported that nickel is the major active catalyst in the oxidation of hydrazine, while iron is the synergistic one.52 It is interesting to note that different metal ratios give a minor variation in the oxidation potential. Analyzed from a theoretical view (Fig. 8a), a Ni85Fe15/PEI–MoS2 is better for voltammetric detection of hydrazine since a higher oxidation current is recorded. This results in better sensitivity and a lower detection limit. While for energy and environmental related applications, a Ni70Fe30/PEI–MoS2 is more promising since the oxidation potential is reduced, leading to higher efficiencies.53,54 The oxidation of Ni85Fe15 in Ni85Fe15/PEI–MoS2 occurs at more positive potentials compared to Ni70Fe30/PEI–MoS2, suggesting that there may be some relation between hydrazine oxidation reason (HOR) and Ni1−xFex alloy.55 How to select a preferable ratio of Ni to Fe in the Ni1−xFex/PEI–MoS2 for hydrazine oxidation is an important issue. Ye et al.56 electrodeposited Au–Pt alloy particles with various Pt/Au molar ratios (including 4:1, 1:1, 1:4) on indium tin oxide (ITO) substrates. Then they applied the catalyst to investigate the reaction of methanol oxidation. Eventually, Ye et al. concluded that the electrocatalytic performance of Pt4Au1 was better than that of Pt although the oxidation peak potential shifted positively by about 160 mV with Pt4Au1 compared to that with Pt. In our study, the oxidation potential of Ni85Fe15/PEI–MoS2 shifted positively by just about 100 mV compared to that with Ni70Fe30/PEI–MoS2 and the oxidation current of Ni85Fe15/PEI–MoS2 was obviously 2 times higher than that of Ni70Fe30/PEI–MoS2. Thus, Ni85Fe15/PEI–MoS2 is considered to be a preferable electrocatalyst for hydrazine oxidation. A similar finding is observed in Fig. 8b, where it can be seen that the electrocatalytic performance of Ni90Fe10/PANi–MoS2 is better for hydrazine oxidation. The maximum oxidation current peaks for Ni1−xFex/PEI–MoS2 and Ni1−xFex/PANi–MoS2 correspond to different mass ratios of Ni to Fe (85:15 and 90:10), suggesting that the interactions between PEI–MoS2 (PANi–MoS2) and Ni1−xFex alloy are distinguishable.
 | ||
| Fig. 8 Cyclic voltammetric measurements of Ni1−xFex/PEI–MoS2 (a) and Ni1−xFex/PANi–MoS2 (b) electrode with different Ni/Fe mass ratios at a scan rate of 50 mV s−1. Insets: mass activities at respective peaks. | ||
As a comparison, the Ni1−xFex alloy NPs were also deposited on MoS2 with different Ni1−xFex mass ratios at the same mass loading under the same conditions (see ESI,† Fig. S2).
Fig. 9 compares the cyclic voltammograms for the oxidation of hydrazine on a Ni85Fe15/PEI–MoS2/GCE, a Ni90Fe10/PANi–MoS2/GCE, a Ni85Fe15/MoS2/GCE, a Ni85Fe15/GCE, a PEI–MoS2/GCE, a PANi–MoS2/GCE, a MoS2/GCE. The maximum current of the hydrazine hydrate oxidation peaks for Ni85Fe15/PEI–MoS2 and Ni90Fe10/PANi–MoS2 hybrids were higher than that of the Ni85Fe15/MoS2 catalyst, which indicates the obvious advantage of modifying the single-layer MoS2 sheets (e.g. larger active area) with CPs. Remarkably, the oxidation current on a Ni85Fe15/PEI–MoS2/GCE is higher than that on a Ni90Fe10/PANi–MoS2/GCE and on a Ni85Fe15/GCE, confirming the enhancement effect of PEI–MoS2 as the support. On a Ni85Fe15/PEI–MoS2/GCE, Ni90Fe10/PANi–MoS2/GCE and Ni85Fe15/MoS2/GCE the oxidation occurs at quite similar potentials. According to these results, we found that PEI–MoS2 supported metal NPs are promising platforms for electrochemical oxidation of hydrazine.
 | ||
| Fig. 9 Cyclic voltammetric measurements of the Ni85Fe15/PEI–MoS2 hybrid, the Ni90Fe10/PANi–MoS2 hybrid, the Ni85Fe15/MoS2 hybrid, the Ni85Fe15 alloy hybrid, the PEI–MoS2 hybrid, the PANi–MoS2 hybrid, and MoS2 at a scan rate of 50 mV s−1. Inset: mass activities at respective peaks. | ||
The stability of catalysts is important for the application of fuel cells. Chronoamperometric measurements were carried out to appraise the durability of the catalysts. Fig. 10 shows the i–t curves of the Ni85Fe15/PEI–MoS2 hybrid and the Ni90Fe10/PANi–MoS2 at a working potential of −0.2 V. In the initial period, the decay current density decreased rapidly for all the catalysts. This may be due to the formation of the intermediate species (such as N2ads, NH3ads) during the hydrazine oxidation reaction.19 However, during the whole time, the larger residual currents for the Ni85Fe15/PEI–MoS2 hybrid compared with those of the Ni90Fe10/PANi–MoS2 hybrid indicate that PEI–MoS2 offers ideal support for the stability of the catalyst.
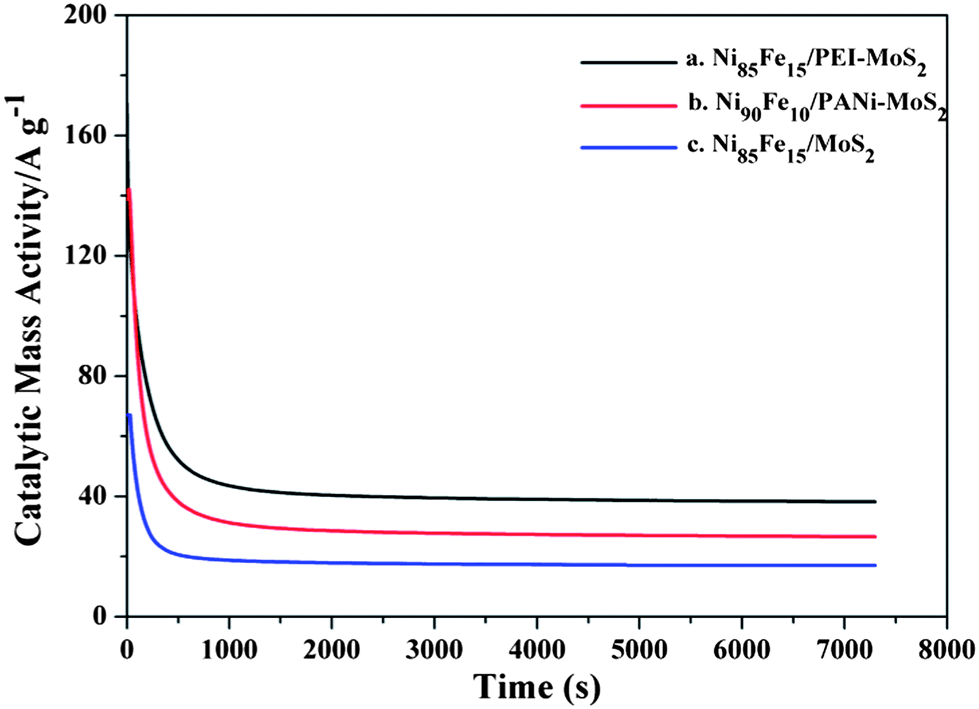 | ||
| Fig. 10 Chronoamperometric measurement in 0.1 M hydrazine hydrate and 0.015 M NaOH at −0.2 V. (a) The Ni85Fe15/PEI–MoS2 hybrid; (b) the Ni90Fe10/PANi–MoS2 hybrid; (c) the Ni85Fe15/MoS2 hybrid. | ||
Mechanism of formation of Ni1−xFex/PEI–MoS2 catalyst
The following mechanism for the Ni1−xFex/PEI–MoS2 hybrid which has superior electrocatalytic activity for hydrazine electrooxidation is proposed. It is well known that MoS2 is a good electron acceptor and PEI on the other hand is a very good electron donor.43 PEI has an amino-rich structure with polar groups, and the donated lone pairs of nitrogen atoms in the –NH2 polar groups of a PEI unit may attach to Mo atoms. Meanwhile, other functional groups, such as –NH– andN–, which can act as nucleation centers for Ni1−xFex NPs. In addition, the exposed sulfur edges can play an important role in anchoring Ni1−xFex NPs as well. Given such a background, the surface of the Ni1−xFex NPs becomes electron-rich when the potential gradually changes from positive to negative, which can promote the dissociation of N2H4 and adsorbed hydrogen atom on the metal surface.12,57 The PEI–MoS2 hybrid presents more homogeneous active sites to anchor more Ni1−xFex NPs. The increasing number of activated regions can enhance the catalytic reaction directly. Therefore, the Ni1−xFex/PEI–MoS2 catalyst shows distinguished electrocatalytic activity for hydrazine oxidation.
Conclusion
In this paper, we have demonstrated a novel and simple in situ growth method to prepare the Ni1−xFex/PEI–MoS2 composite material. This synthetic route involved a mild heat-treatment process, which induced the synergistic coupling between PEI and MoS2. The introduction of PEI to MoS2 enhanced the interaction between Ni1−xFex alloy and MoS2, so as to improve the stability of the catalyst. More importantly, PEI-functionalized MoS2 provided more homogeneous active sites to attach Ni1−xFex NPs. By electrochemical testing, the Ni85Fe15/PEI–MoS2 catalyst for hydrazine oxidation exhibits unexpected, surprisingly pronounced electrocatalytical activity in alkaline solutions, comparable to Ni90Fe10/PANi–MoS2 and Ni85Fe15/MoS2 catalyst but far exceeding in durability. Various methods for the characterization of the composites were presented, which prove that the Ni85Fe15/PEI–MoS2 catalyst is very promising for portable applications in hydrazine fuel cells.Acknowledgements
This research was supported by the National Science Foundation of China (No. 21345003), the Key Laboratory of Catalytic engineering of Gansu Province and Resources Utilization, Gansu Province for financial support, the Fundamental Research Funds for the Central Universities (Lzujbky-2013-67).Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- S. Anandan, A. Manivel and M. Ashokkumar, Fuel Cells, 2012, 12, 956–962 CrossRef CAS.
- U. Lange, T. Hirsch, V. M. Mirsky and O. S. Wolfbeis, Electrochim. Acta, 2011, 56, 3707–3712 CrossRef CAS PubMed.
- C. Nethravathi, M. Rajamathi, N. Ravishankar, L. Basit and C. Felser, Carbon, 2010, 48, 4343–4350 CrossRef CAS PubMed.
- S. Bhaviripudi, X. T. Jia, M. S. Dresselhaus and J. Kong, Nano Lett., 2010, 10, 4128–4133 CrossRef CAS PubMed.
- K.-G. Zhou, N.-N. Mao, H.-X. Wang, Y. Peng and H.-L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed.
- X. S. Zhou, L. J. Wan and Y. G. Guo, Chem. Commun., 2013, 49, 1838–1840 RSC.
- C. N. R. Rao and A. Nag, Eur. J. Inorg. Chem., 2010, 4244–4250 CrossRef CAS.
- Z. Y. Zeng, Z. Y. Yin, X. Huang, H. Li, Q. Y. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- Z. Y. Yin, H. Li, L. Jiang, Y. M. Shi, Y. H. Sun, G. Lu, Q. Zhang, X. D. Chen and H. Zhang, ACS Nano, 2012, 6, 74–80 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- K. K. Liu, W. J. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Su, C. S. Chang, H. Li, Y. M. Shi, H. Zhang, C. S. Lai and L. J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS PubMed.
- H. Li, G. Lu, Z. Y. Yin, Q. Y. He, Q. Zhang and H. Zhang, Small, 2012, 8, 682–686 CrossRef CAS PubMed.
- K. Chang, W. X. Chen, L. Ma, H. Li, F. H. Huang, Z. D. Xu, Q. B. Zhang and J. Y. Lee, J. Mater. Chem., 2011, 21, 6251–6257 RSC.
- X. Zhong, H. Yang, S. Guo, S. Li, G. Gou, Z. Niu, Z. Dong, Y. Lei, J. Jin, R. Li and J. Ma, J. Mater. Chem., 2012, 22, 13925 RSC.
- J. Sanabria-Chinchilla, K. Asazawa, T. Sakamoto, K. Yamada, H. Tanaka and P. Strasser, J. Am. Chem. Soc., 2011, 133, 5425–5431 CrossRef CAS PubMed.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255–1260 CAS.
- V. Rosca and M. T. M. Koper, Electrochim. Acta, 2008, 53, 5199–5205 CrossRef CAS PubMed.
- N. Maleki, A. Safavi, E. Farjami and F. Tajabadi, Anal. Chim. Acta, 2008, 611, 151–155 CrossRef CAS PubMed.
- M. Hosseini, M. M. Momeni and M. Faraji, J. Mol. Catal. A: Chem., 2011, 335, 199–204 CrossRef CAS PubMed.
- R. Wojcieszak, M. Zieliński, S. Monteverdi and M. M. Bettahar, J. Colloid Interface Sci., 2006, 299, 238–248 CrossRef CAS PubMed.
- M. Hosseini and M. M. Momeni, J. Mater. Sci., 2010, 45, 3304–3310 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- G. Budroni and A. Corma, Angew. Chem., Int. Ed., 2006, 45, 3328–3331 CrossRef CAS PubMed.
- D. G. Mustard and C. H. Bartholomew, J. Catal., 1981, 67, 186–206 CrossRef CAS.
- G. Inzelt, M. Pineri, J. W. Schultze and M. A. Vorotyntsev, Electrochim. Acta, 2000, 45, 2403–2421 CrossRef CAS.
- S. Nambiar and J. T. W. Yeow, Biosens. Bioelectron., 2011, 26, 1825–1832 CrossRef CAS PubMed.
- C. Y. Wang, D. Li, C. O. Too and G. G. Wallace, Chem. Mater., 2009, 21, 2604–2606 CrossRef CAS.
- S. R. C. Vivekchand, C. S. Rout, K. S. Subrahmanyam, A. Govindaraj and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9–13 CrossRef CAS PubMed.
- G. Wu, L. Li, J. H. Li and B. Q. Xu, Carbon, 2005, 43, 2579–2587 CrossRef CAS PubMed.
- X. F. Lu, W. J. Zhang, C. Wang, T. C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671–712 CrossRef CAS PubMed.
- J. Yan, T. Wei, B. Shao, Z. J. Fan, W. Z. Qian, M. L. Zhang and F. Wei, Carbon, 2010, 48, 487–493 CrossRef CAS PubMed.
- S. Chen, Z. Wei, X. Qi, L. Dong, Y.-G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
- X. G. Hu, T. Wang, X. H. Qu and S. J. Dong, J. Phys. Chem. B, 2006, 110, 853–857 CrossRef CAS PubMed.
- X. Fang, H. Ma, S. L. Xiao, M. W. Shen, R. Guo, X. Y. Cao and X. Y. Shi, J. Mater. Chem., 2011, 21, 4493–4501 RSC.
- X. Y. Cao, J. J. Chen, S. H. Wen, C. Peng, M. W. Shen and X. Y. Shi, Nanoscale, 2011, 3, 1741–1747 RSC.
- Y. Liu, D.-C. Wu, W.-D. Zhang, X. Jiang, C.-B. He, T. S. Chung, S. H. Goh and K. W. Leong, Angew. Chem., Int. Ed., 2005, 117, 4860–4863 CrossRef.
- L. Bai, H. Zhu, J. S. Thrasher and S. C. Street, ACS Appl. Mater. Interfaces, 2009, 1, 2304–2311 CAS.
- P. L. Kuo, W. F. Chen, H. Y. Huang, I. C. Chang and S. A. Dai, J. Phys. Chem. B, 2006, 110, 3071–3077 CrossRef CAS PubMed.
- H. Zhang, K. P. Loh, C. H. Sow, H. R. Gu, X. D. Su, C. Huang and Z. K. Chen, Langmuir, 2004, 20, 6914–6920 CrossRef CAS PubMed.
- K. Chang and W. Chen, Chem. Commun., 2011, 47, 4252 RSC.
- S. Bai, X. P. Shen, G. X. Zhu, Z. Xu and J. Yang, CrystEngComm, 2012, 14, 1432–1438 RSC.
- T. Weber, J. C. Muijsers, H. vanWolput, C. P. J. Verhagen and J. W. Niemantsverdriet, J. Phys. Chem., 1996, 100, 14144–14150 CrossRef CAS.
- T. Yang, A. Hussain, S. Bai, I. A. Khalil, H. Harashima and F. Ahsan, J. Controlled Release, 2006, 115, 289–297 CrossRef CAS PubMed.
- N. A. Kumar, H. J. Choi, Y. R. Shin, D. W. Chang, L. M. Dai and J. B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- X. M. Feng, G. Yang, Q. Xu, W. H. Hou and J. P. Zhu, Macromol. Rapid Commun., 2006, 27, 31–36 CrossRef CAS.
- S. Deng and Y.-P. Ting, Water Res., 2005, 39, 2167–2177 CrossRef CAS PubMed.
- S. B. Deng, R. B. Bai and J. P. Chen, Langmuir, 2003, 19, 5058–5064 CrossRef CAS.
- H. D. Yang, X. Zhong, Z. P. Dong, J. Wang, J. Jin and J. T. Ma, RSC Adv., 2012, 2, 5038–5040 RSC.
- K. Asazawa, K. Yamada, H. Tanaka, A. Oka, M. Taniguchi and T. Kobayashi, Angew. Chem., Int. Ed., 2007, 119, 8170–8173 CrossRef.
- Q. Wan, Y. Liu, Z. Wang, W. Wei, B. Li, J. Zou and N. Yang, Electrochem. Commun., 2013, 29, 29–32 CrossRef CAS PubMed.
- M. Khosravi and M. K. Amini, Int. J. Hydrogen Energy, 2010, 35, 10527–10538 CrossRef CAS PubMed.
- W. Ye, H. Kou, Q. Liu, J. Yan, F. Zhou and C. Wang, Int. J. Hydrogen Energy, 2012, 37, 4088–4097 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra42757a |
| This journal is © The Royal Society of Chemistry 2014 |