
From systems biology to systems chemistry: metabolomic procedures enable insight into complex chemical reaction networks in water†
Abstract
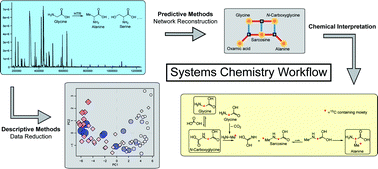
Metabolomics comprises of the monitoring of small molecules present in a biological system as a function of time and space. Coupled with emerging modeling approaches, it facilitates predictions of reaction sequences. Here, we explore the potential of metabolomic tools for analyzing the complex chemical systems in a model reaction, the hydrothermal reforming (HTR) of glycine. The profiles for more than 20 monitored compounds were used to reconstruct the glycine reaction network. The mechanism of glycine conversion into serine and alanine was validated, where new carbon–carbon (C–C) bonds are formed from the C2-position of glycine. We thus demonstrated that metabolomic methods are useful for the analysis of complex combinatorial problems in chemistry.
 Please wait while we load your content...
Please wait while we load your content...

