
A sidearm-assisted phosphine for catalytic ylide intramolecular cyclopropanation†
Jian-Bo
Zhu
,
Hao
Chen
,
Saihu
Liao
,
Yu-Xue
Li
* and
Yong
Tang
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: tangy@mail.sioc.ac.cn; liyuxue@ sioc.ac.cn
First published on 10th September 2014
Abstract
The first phosphine-catalyzed cyclopropanation reaction via covalent ylide catalysis has been realized with high efficiency in the presence of sidearm-assisted phosphine catalysts. An ether sidearm group is found critical to turn on the catalytic activity. Crystal structures of the catalyst-derived phosphonium salts indicated a significant O⋯P+ interaction between the pendant ether oxygen and the phosphonium center, which is believed to favor the catalyst regeneration. DFT calculations rationalize the insight of the sidearm effects.
Ylide cyclopropanation provides a convenient and attractive method for the synthesis of cyclopropanes, an important motif that frequently occurs in natural products as well as in synthetically useful intermediates.1,2 Although both phosphorus ylide reactions and phosphine-catalyzed reactions have been intensively studied in the past few decades,3,4 phosphine-catalyzed cyclopropanation remains elusive.5–9 Rational theoretical studies by Aggarwal and Harvey on the leaving group ability of onium ylides show that compared with other ylides, such as ammonium, oxonium, sulfonium ylides, the phosphonium ylide needs to overcome a much higher energy barrier to act as a leaving group in a SN2 substitution reaction (Scheme 1).10 Accordingly, the key step is to regenerate phosphine in the ylide cyclopropanation to complete the catalytic cycle.5 As the leaving group ability depends on the nature of phosphine, it could be envisioned that a rational design of phosphine would provide a solution for the catalyst turnover in such a reaction. Based on this idea, recently, we found that the introduction of a sidearm, an ether group at the ortho-position of triphenylphosphine (Ph3P), could substantially alter the phosphine reactivity to enable its regeneration via a SN2 substitution, allowing the realization of the first phosphine-catalyzed cyclopropanation reaction (eqn (2) in Scheme 1). In this communication, we wish to report our efforts on this subject.
 | ||
| Scheme 1 Leaving group ability of phosphine and phosphine-mediated annulation reactions. | ||
Recently, we reported an intramolecular conjugation for the synthesis of chromans and their relevant analogues in the presence of 150 mol% PPh3.11 The mechanism of this reaction is proposed as shown in Scheme 2. According to this mechanism, two factors are crucial to change the pathway to the cyclopropane: reducing the hydrolysis rate and/or improving the leaving ability of phosphine. We envisioned that tuning the steric hindrance around the phosphorus in the phosphine might balance the nucleophilicity and leaving group ability, switching ylide hydrolysis into SN2 substitution and giving cyclopropanes as the desired products.
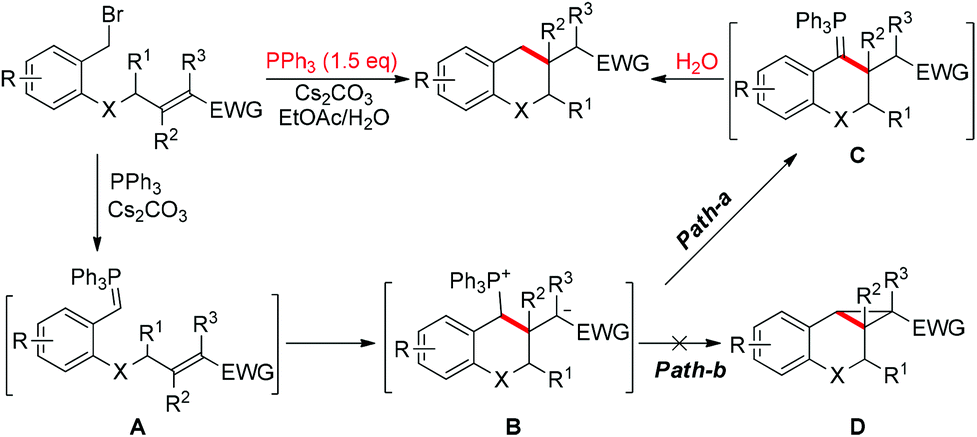 | ||
| Scheme 2 Reaction pathway to the conjugation product. | ||
As PPh3 is inert to this reaction (entry 1, Table 1), initially, we synthesized bulky phosphines 3a and 3b with an ortho-propyl group and an ortho-methoxymethyl group. With 10 mol% of 3a, no cyclopropanation product was obtained at room temperature; however, with 10 mol% of 3b as a catalyst, we found that the desired cyclopropane was afforded with 33% yield (entries 2 & 3). In contrast, several other common phosphines with varied electronic nature were also tested in this reaction, but no product could be isolated in all these cases (entries 4–6). Then we employed 3b as the catalyst for further study. In the presence of 10 mol% 3b, optimization of the reaction parameters (temperature, solvent, and base) increased the yield to 64% (entries 7–13). As the activity was improved by the replacement of an ortho-propyl group in 3a with the ortho ether group in 3b (sidearm effect), it intrigued us to prepare several phosphine catalysts that also have this ether functional group for enhancing the efficiency. Though decreased yields were obtained with 3c and 3d (entries 14 & 15), possibly caused by the steric hindrance of the ether group, to our delight, the less hindered acetal phosphine 3e was found very effective for this cyclopropanation, improving the yield to 90% (entries 20 & 21). In contrast, thioether 3f gave lower yield, and ester 3g was even inactive (entries 17 & 18). With 3h as a catalyst, 53% yield was obtained. Noticeably, neither a sulfide nor an amine catalyst afforded the desired cyclopropanation product even under the optimal literature conditions, although tetrahydrothiophene is very efficient for the phenol derived α,β-unsaturated ester (entries 22 and 23).6c,7a
|
|
|||||
|---|---|---|---|---|---|
| Entry | PR3 | Solvent | T (°C) | t (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), catalyst (10 mol%), base (0.6 mmol), solvent (4 mL). DABCO = 1,4-diazobicyclo[2.2.2]octane. b Isolated yield. c With 20 mol% catalyst. | |||||
| 1 | PPh3 | DCE | rt | 21 | — |
| 2 | 3a | DCE | rt | 21 | — |
| 3 | 3b | DCE | rt | 21 | 33 |
| 4 | PCy3 | DCE | rt | 21 | — |
| 5 | (4-ClC6H4)3P | DCE | rt | 21 | — |
| 6 | (4-MeOC6H4)3P | DCE | rt | 21 | — |
| 7 | 3b | DCE | 80 | 20 | 52 |
| 8 | 3b | DME | 80 | 16 | 15 |
| 9 | 3b | PhCH3 | 80 | 16 | 7 |
| 10 | 3b | CH3CN | 80 | 14 | 24 |
| 11 | 3b | CHCl3 | 65 | 24 | 64 |
| 12 | 3b/Na2CO3 | CHCl3 | 65 | 24 | — |
| 13 | 3b/K2CO3 | CHCl3 | 65 | 24 | 54 |
| 14 | 3c | CHCl3 | 65 | 24 | 46 |
| 15 | 3d | CHCl3 | 65 | 24 | 41 |
| 16 | 3e | CHCl3 | 65 | 24 | 77 |
| 17 | 3f | CHCl3 | 65 | 24 | 25 |
| 18 | 3g | CHCl3 | 65 | 24 | — |
| 19 | 3h | CHCl3 | 65 | 24 | 53 |
| 20 | 3e | CHCl3 | 65 | 48 | 80 |
| 21c | 3e | CHCl3 | 65 | 48 | 90 |
| 22 | Tetrahydrothiophene | DCE | 80 | 34 | — |
| 23 | DABCO/Na2CO3 | CH3CN | 80 | 24 | — |

The scope of this cyclopropanation reaction was investigated under the optimized reaction conditions, and the results are tabulated in Table 2. Various α,β-unsaturated carbonyl compounds with different structures worked well, giving the desired [n.1.0] bicycloalkanes 2a––w in good to high yields. Notably, in all examples, only a single diastereoisomer was observed. The electronic nature of the substituent on the phenol ring slightly influences the yields (83–93%, entries 1–6). The substituent effect is more obvious on the benzoyl part, in which the electron-donating groups gave in general higher yields (entries 8–14), probably because the electron-donating group would increase the nucleophilicity of the enolate in the key intramolecular SN2-substitution step to regenerate the catalyst. Remarkably, aliphatic enone is also suitable for this reaction (94%, entry 17). And the all carbon [n.1.0] bicycles can also be obtained in even higher yields (91–95%, entries 18–21). It is worth mentioning that the enal substrate 1v was also compatible with the current reaction conditions; and the product 2v is a key intermediate for the synthesis of a potential antidepressant.12 The α,β-unsaturated ester 1w was also examined as a substrate, using 20 mol% of 3e, after 45 hours, 71% yield was obtained in toluene at 110 °C (entry 23).
|
|
||||
|---|---|---|---|---|
| Entry | Product | t (h) | Yieldb (%) | |
| a Reaction conditions: 1a (0.3 mmol), 3e (10 mol%), base (0.6 mmol), CHCl3 (4 mL). b Isolated yield. c With 20 mol% 3e. d With 20 mol% 3e, in toluene, at 110 °C. | ||||
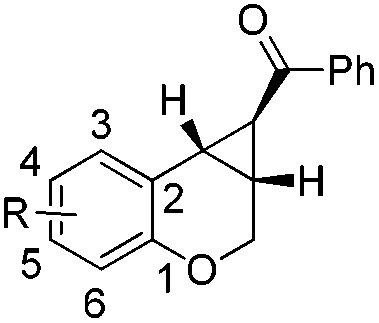
|
||||
| 1c | R = H | 2a | 25 | 90 |
| 2 | 4-Me | 2b | 46 | 92 |
| 3 | 4-OMe | 2c | 37 | 84 |
| 4c | 4-Br | 2d | 26 | 83 |
| 5 | 6-Me | 2e | 37 | 9314 |
| 6 | 6-F | 2f | 40 | 86 |
| 7 | 5,6-Benzo | 2g | 40 | 91 |

|
||||
| 8c | Y = 2-OMe | 2h | 40 | 96 |
| 9c | 3-OMe | 2i | 21 | 94 |
| 10c | 4-OMe | 2j | 26 | 91 |
| 11c | 4-F | 2k | 26 | 80 |
| 12c | 4-Br | 2l | 25 | 66 |
| 13c | 4-Ph | 2m | 40 | 84 |
| 14c | 2,3-Benzo | 2n | 63 | 80 |
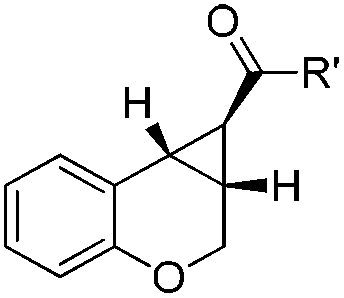
|
||||
| 15c | R′ = 2-furyl | 2o | 18 | 72 |
| 16c | 2-Thienyl | 2p | 18 | 84 |
| 17c | Cy | 2q | 24 | 94 |
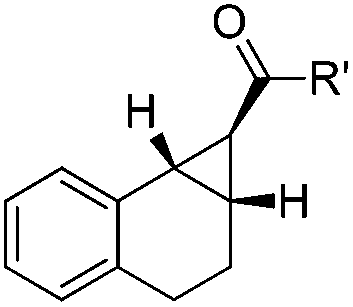
|
||||
| 18 | R′ = 2-furyl | 2r | 45 | 92 |
| 19c | 2-Thienyl | 2s | 45 | 91 |
| 20 | Ph | 2t | 68 | 95 |
| 21c | 4-BrC6H4 | 2u | 63 | 93 |
| 22c | H | 2v | 18 | 81 |
| 23d | OCH2CF3 | 2w | 45 | 71 |
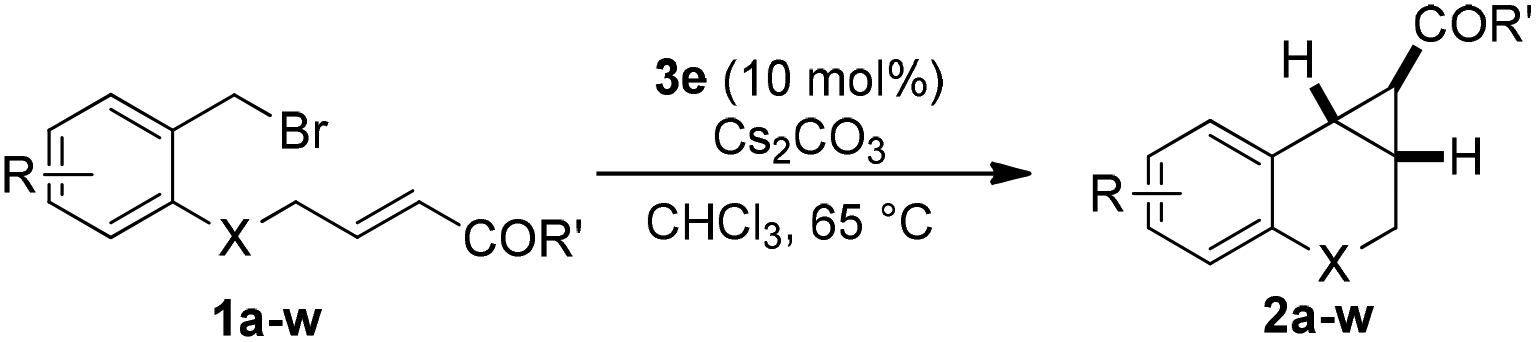
To further understand the role of the sidearm effect, we developed the single crystals of phosphonium salts 4a, 4b and 4e, which are derived from phosphines 3a, 3b and 3e.13 Interestingly, the approaching of the oxygen atom to the phosphonium was observed, with an O⋯P distance of 3.22 Å and 3.38 Å respectively, which are quite close to the sum of the van der Waals radii (3.32 Å) (Fig. 1). In a stark contrast, the C atom is apart from the P atom in the ortho-propyl group substituted phosphine. This interesting phenomenon indicated that interactions between the O atom and the P atom might account for the positive effect of the ether sidearm. A similar interaction of Te+⋯O was also observed in the crystal structure of a telluronium salt containing an ether group.15
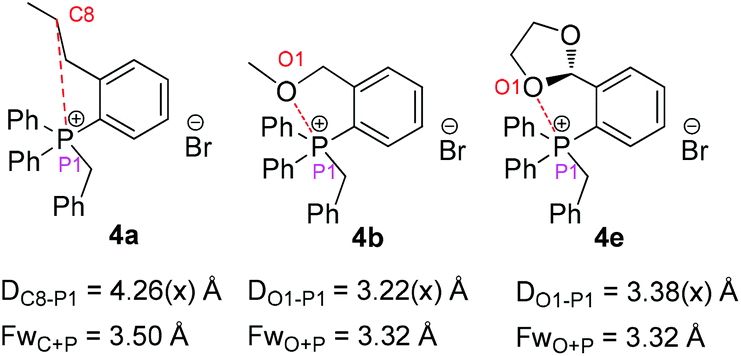 | ||
| Fig. 1 X-ray crystal structure data of the phosphonium salts. | ||
To rationalize the sidearm effect, density functional theory (DFT)16 studies have been performed with the GAUSSIAN09 program17 using the M0618 method. For the Cs atom the SDD basis set with an effective core potential (ECP)19 was used; for the rest of the atoms, the 6-31+G** basis set was used. Geometry optimization was performed in chloroform (ε = 4.711) using the SMD20 method. Harmonic vibration frequency calculations were carried out at 338.15 K, the optimized structures are all shown to be either minima (with no imaginary frequency) or transition states (with one imaginary frequency).
Both the poor and the optimal phosphines, PPh3 and 3e, were used to explore the reaction pathways Path-a and Path-b starting from the intermediate B. As shown in Fig. 2, for PPh3, the hydrogen shift pathway (10.5 kcal mol−1) is more favorable than the cyclopropanation pathway (11.6 kcal mol−1), which is well consistent with the experimental observations that only a trace amount of the cyclopropanation product was obtained. For the optimal phosphine 3e, the barrier of cyclopropanation (11.8 kcal mol−1) is almost the same as that of PPh3. However, the barriers of the hydrogen shift pathways increase to 12.9 kcal mol−1 (3e-TS-H-Shift) and 11.7 kcal mol−1 (3e-TS-H-Shift2), respectively. Thus, the cyclopropanation pathway becomes favorable for 3e.
 | ||
| Fig. 2 The calculated reaction pathways for phosphine PPh3. The energy barriers for phosphine 3e are also given (in italics). The selected bond lengths are in angstrom, and the relative free energies in chloroform (Gsol) are in kcal mol−1. Calculated at the M06/6-31+G**/SDD level. | ||
These results indicate that the sidearm on the phosphine changes the selectivity of the reaction mainly by suppressing the hydrogen shift process. A detailed structural analysis21 shows that in 3e-B, the acetal group increases the repulsion among the three phenyl rings on the P atom, leading to a more flat “umbrella”, which is more sensitive to the steric effect. Compared with PPh3-TS-H-Shift, the attacking of CsHCO3 on the phosphonium group in 3e-TS-H-Shift causes larger repulsion, resulting in a more rigid structure with an entropy loss of 4.759 cal mol−1 K−1, which is equivalent to a barrier increase of 1.6 kcal mol−1. Based on these understandings, it is natural that the phosphonium group has a much less effect on the “backside-attack” cyclopropanation transition state than the “side-attack” protonation transition state. For phosphines 3a, 3b and 3f, the rate determining steps of Path-a and Path-b were also calculated, and the results are in good agreement with the experiments.21
Conclusions
In summary, a phosphine-catalyzed cyclopropanation reaction has been successfully realized for the first time with an ether sidearm-assisted phosphine catalyst. X-ray crystal analysis indicated an interaction between the ether oxygen and the phosphonium atom, which was believed to be crucial for switching the phosphine reactivity from a typical ylide route to an unprecedented catalytic cyclopropanation path. DFT calculations rationalize the insight of the sidearm effects. This beneficial effect of a sidearm group would provide a brightening idea for the design and modification of phosphines and other sulfide and amine catalysts. Further investigation on the mechanism and the development of an asymmetric version are in progress in our laboratory.Acknowledgements
We are grateful for the financial support from the National Natural Sciences Foundation of China (no. 21121062 and 21272248), the Chinese Academy of Sciences. We thank Dr Xue-Bing Leng (SIOC) and Mr Jie Sun (SIOC) for X-ray crystal analysis.Notes and references
- (a) A. H. Li, L. X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS PubMed; (b) L. X. Dai, X. L. Hou and Y. G. Zhou, Pure Appl. Chem., 1999, 71, 369 CAS; (c) H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (d) Y. Tang, S. Ye and X. L. Sun, Synlett, 2005, 2720 CrossRef CAS PubMed; (e) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS PubMed.
- For recent examples of cyclopropanation with ylide reagents, see: (a) J. Wang, X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, J. Org. Chem., 2013, 78, 6322 CrossRef CAS PubMed; (b) C. Q. Wang, Tetrahedron Lett., 2012, 53, 7003 CrossRef CAS PubMed; (c) L. Q. Lu, Z. H. Ming, J. An, C. Li, J. R. Chen and W. J. Xiao, J. Org. Chem., 2012, 77, 1072 CrossRef CAS PubMed; (d) A. Biswas, S. De Sarkar, L. Tebben and A. Studer, Chem. Commun., 2012, 48, 5190 RSC; (e) R. Zhou, X. M. Deng, J. C. Zheng, Q. Shen, X. L. Sun and Y. Tang, Chin. J. Chem., 2011, 29, 995 CrossRef CAS; (f) Y. Cheng, J. An, L. Q. Lu, L. Luo, Z. Y. Wang, J. R. Chen and W. J. Xiao, J. Org. Chem., 2011, 76, 281 CrossRef CAS PubMed; (g) B. H. Zhu, R. Zhou, J. C. Zheng, X. M. Deng, X. L. Sun, Q. Shen and Y. Tang, J. Org. Chem., 2010, 75, 3454 CrossRef CAS PubMed; (h) S. L. Riches, C. Saha, N. F. Filgueira, E. Grange, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 7626 CrossRef CAS PubMed; (i) R. Appel, N. Hartmann and H. Mayr, J. Am. Chem. Soc., 2010, 132, 17894 CrossRef CAS PubMed; (j) C. Y. Zhu, X. Y. Cao, B. H. Zhu, C. Deng, X. L. Sun, B. Q. Wang, Q. Shen and Y. Tang, Chem. – Eur. J., 2009, 15, 11465 CrossRef CAS PubMed; (k) Y. H. Zhao, G. Zhao and W. G. Cao, Tetrahedron: Asymmetry, 2007, 18, 2462 CrossRef CAS PubMed; (l) R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240 CrossRef CAS PubMed.
- For reviews on phosphine catalysis, see: (a) O. I. Kolodiazhnyi, Phosphorus Ylides: Chemistry and Application in Organic Synthesis, Wiley-VCH, New York, 1999 Search PubMed; (b) X. Y. Lu, C. M. Zhang and Z. R. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (d) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed; (e) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS PubMed; (f) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (g) X. L. Sun and Y. Tang, Acc. Chem. Res., 2008, 41, 937 CrossRef CAS PubMed; (h) L. W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC.
- Recent examples on phosphine catalysis: (a) F. R. Zhong, X. W. Dou, X. Y. Han, W. J. Yao, Q. Zhu, Y. Z. Meng and Y. X. Lu, Angew. Chem., Int. Ed., 2013, 52, 943 CrossRef CAS PubMed; (b) R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 2525 CrossRef CAS PubMed; (c) X. Y. Han, F. R. Zhong, Y. Q. Wang and Y. X. Lu, Angew. Chem., Int. Ed., 2012, 51, 767 CrossRef CAS PubMed; (d) F. R. Zhong, X. Y. Han, Y. Q. Wang and Y. X. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed; (e) B. Tan, N. R. Candeias and C. F. Barbas, J. Am. Chem. Soc., 2011, 133, 4672 CrossRef CAS PubMed; (f) H. Xiao, Z. Chai, C. W. Zheng, Y. Q. Yang, W. Liu, J. K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS PubMed; (g) J. E. Wilson, J. W. Sun and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 161 CrossRef CAS PubMed; (h) S. Takizawa, N. Inoue, S. Hirata and H. Sasai, Angew. Chem., Int. Ed., 2010, 49, 9725 CrossRef CAS PubMed; (i) Y. Q. Jiang, Y. L. Shi and M. Shi, J. Am. Chem. Soc., 2008, 130, 7202 CrossRef CAS PubMed; (j) L. W. Ye, X. L. Sun, Q. G. Wang and Y. Tang, Angew. Chem., Int. Ed., 2007, 46, 5951 CrossRef CAS PubMed.
- For reviews on catalytic ylide cyclopropanation, see: (a) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841 CrossRef CAS PubMed; (b) S.-H. Liao, P. Wang and Y. Tang, Covalent Activations: Ylides, in Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. Dalko, Wiley-VCH & Sons, Weinheim, Germany, 2013 Search PubMed.
- For examples of sulfide-catalyzed cyclopropanation, see: (a) V. K. Aggarwal, H. W. Smith, G. Hynd, R. V. H. Jones, R. Fieldhouse and S. E. Spey, J. Chem. Soc., Perkin Trans. 1, 2000, 3267 RSC; (b) X. M. Deng, P. Cai, S. Ye, X. L. Sun, W. W. Liao, K. Li, Y. Tang, Y. D. Wu and L. X. Dai, J. Am. Chem. Soc., 2006, 128, 9730 CrossRef CAS PubMed; (c) L. W. Ye, X. L. Sun, C. Y. Li and Y. Tang, J. Org. Chem., 2007, 72, 1335 CrossRef CAS PubMed.
- For examples of amine-catalyzed ylide cyclopropanation, see: (a) C. D. Papageorgiou, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2003, 42, 828 CrossRef CAS PubMed; (b) N. Bremeyer, S. C. Smith, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 2681 CrossRef CAS; (c) C. D. Papageorgiou, M. A. C. de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef CAS PubMed; (d) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024 CrossRef CAS PubMed; (e) I. S. del Villar, A. Gradillas, G. Dominguez and J. Perez-Castells, Tetrahedron Lett., 2010, 51, 3095 CrossRef CAS PubMed.
- For Te, As-catalyzed cyclopropanation, see: (a) Y. Z. Huang, Y. Tang, Z. L. Zhou, W. Xia and L. P. Shi, J. Chem. Soc., Perkin Trans. 1, 1994, 893 RSC; (b) W. W. Liao, K. Li and Y. Tang, J. Am. Chem. Soc., 2003, 125, 13030 CrossRef CAS PubMed; (c) H. Jiang, X. L. Sun, C. Y. Zhu, L. X. Dai and Y. Tang, Tetrahedron, 2008, 64, 5032 CrossRef CAS PubMed.
- For examples of cyclopropanation using stoicheometric phosphonium ylides, see: (a) S. Redon, S. Leleu, X. Pannecoucke, X. Franck and F. Outurquin, Tetrahedron, 2008, 64, 9293 CrossRef CAS PubMed; (b) E. Bunuel, S. D. Bull, S. G. Davies, A. C. Garner, E. D. Savory, A. D. Smith, R. J. Vickers and D. J. Watkin, Org. Biomol. Chem., 2003, 1, 2531 RSC; (c) S. Ahmad, L. M. Doweyko, S. Dugar, N. Grazier, K. Ngu, S. C. Wu, K. J. Yost, B. C. Chen, J. Z. Gougoutas, J. D. DiMarco, S. J. Lan, B. J. Gavin, A. Y. Chen, C. R. Dorso, R. Serafino, M. Kirby and K. S. Atwal, J. Med. Chem., 2001, 44, 3302 CrossRef CAS PubMed; (d) A. Krief, L. Provins and A. Froidbise, Tetrahedron Lett., 1998, 39, 1437 CrossRef CAS . Also see ref. 3a.
- V. K. Aggarwal, J. N. Harvey and R. Robiette, Angew. Chem., Int. Ed., 2005, 44, 5468 CrossRef CAS PubMed.
- J. B. Zhu, P. Wang, S. H. Liao and Y. Tang, Chem. Commun., 2013, 49, 4570 RSC.
- A. A. Cordi, I. Berque-Bestel, T. Persigand, J. M. Lacoste, A. Newman-Tancredi, V. Audinot and M. J. Millan, J. Med. Chem., 2001, 44, 787 CrossRef CAS PubMed.
- CCDC 992105 (4a), CCDC 962019 (4b), CCDC 962015 (4e) contain the supplementary crystallographic data for this paper. For crystal structures of 4a, 4b and 4e, see ESI.†.
- When the Z-isomer of 1e was employed as a substrate in the catalytic ylide intramolecular cyclopropanation, the desired product 2e was produced in 21% yield with the same diastereoselectivity of the E-configuration substrate 1e, and 51% of the Z-isomer was recovered.
- Z. Z. Huang, S. Ye, W. Xia, Y. H. Yu and Y. Tang, J. Org. Chem., 2002, 67, 3096 CrossRef CAS PubMed.
- (a) P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef; (b) W. Kohn and L. Sham, J. Phys. Rev., 1965, 140, A1133 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 2009, 113, 6378 CrossRef CAS PubMed.
- For details, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and the characterization data of new compounds. CCDC 992105 (4a), 962019 (4b) and 962015 (4e). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00232f |
| This journal is © the Partner Organisations 2014 |