
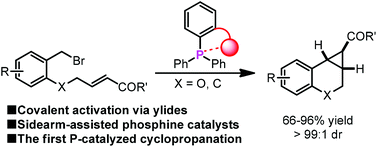
A sidearm-assisted phosphine for catalytic ylide intramolecular cyclopropanation†
Abstract
The first phosphine-catalyzed cyclopropanation reaction via covalent ylide catalysis has been realized with high efficiency in the presence of sidearm-assisted phosphine catalysts. An ether sidearm group is found critical to turn on the catalytic activity. Crystal structures of the catalyst-derived phosphonium salts indicated a significant O⋯P+ interaction between the pendant ether oxygen and the phosphonium center, which is believed to favor the catalyst regeneration. DFT calculations rationalize the insight of the sidearm effects.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

