
Copper(I)-catalyzed enantioselective hydroboration of cyclopropenes: facile synthesis of optically active cyclopropylboronates†
Bing
Tian‡
ab,
Qiang
Liu‡
ab,
Xiaofeng
Tong
b,
Ping
Tian
*a and
Guo-Qiang
Lin
*a
aKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: tianping@sioc.ac.cn; lingq@sioc.ac.cn; Tel: (+86) 21-54925081
bShanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
First published on 10th September 2014
Abstract
Copper(I)-catalyzed enantioselective hydroboration of 3-aryl substituted cyclopropene-3-carboxylate is described, providing chiral cyclopropylboronates with excellent enantioselectivities (89–95% ee) in moderate to high yields (55–86%). The non-directing effect of the ester group was observed, and the reaction proceeded with solely trans-selectivity. The chiral boronates could be conveniently converted into chiral 1,2-diaryl substituted cyclopropane derivatives.
Introduction
The chiral cyclopropane framework represents the smallest carbocycles existing in a wide range of naturally-occurring compounds,1 chiral drugs, and insecticides, for instance, (+)-Coronatine,2 Saxagliptin (Onglyza®),3 EBC-219,4 Milnacipran,5 Deltamethrin,6 and (+)-Tranylcypromine7 (Fig. 1). These three-membered carbocycles, due to their unique structural and electronic properties, serve as extremely significant versatile building blocks in organic synthesis.8 Thus, a few interesting and characteristic transformations have continually emerged.9 Owing to their important biological activities and wide applications in organic chemistry, much attention has been paid to their efficient enantioselective syntheses.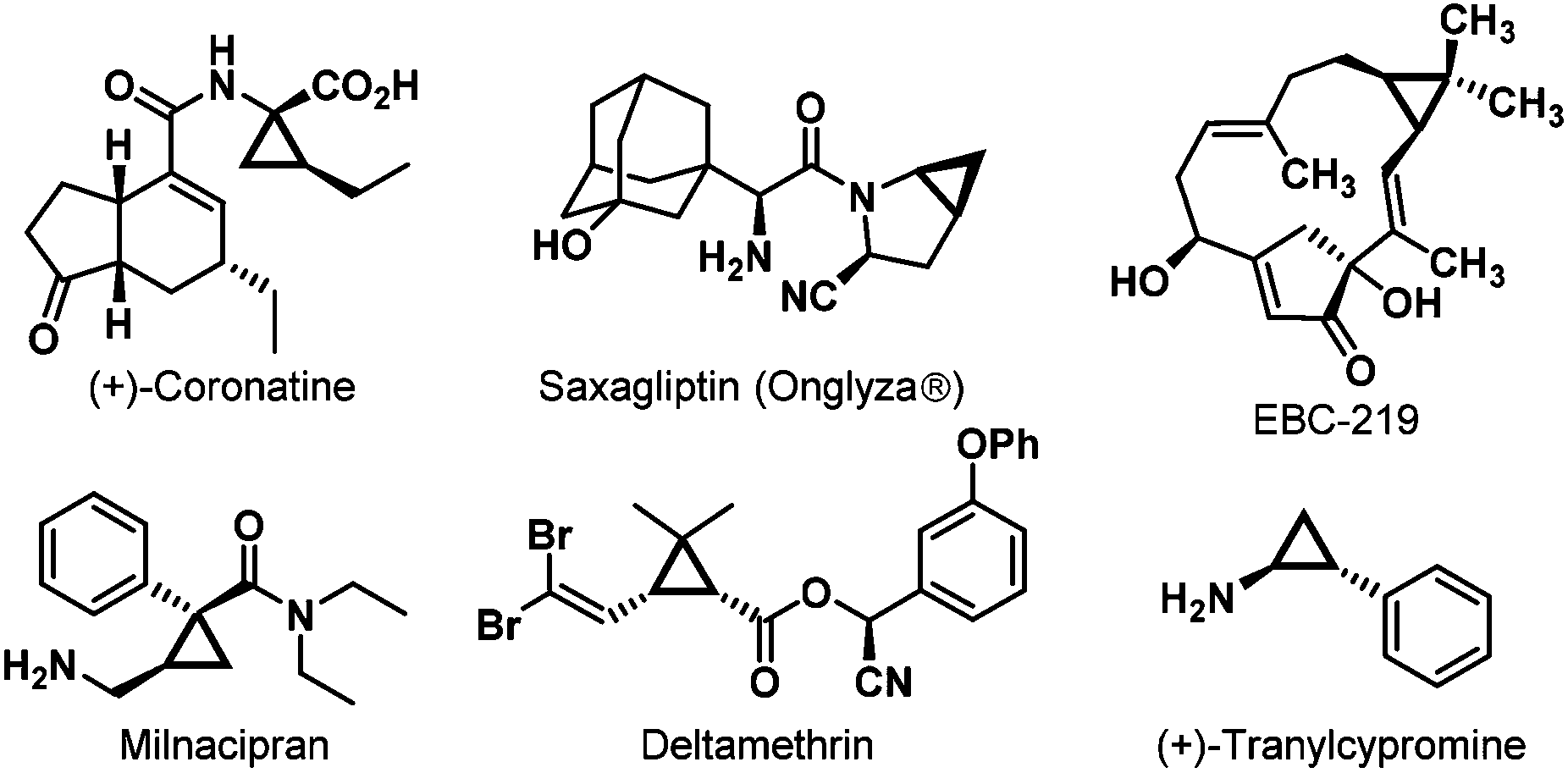 | ||
| Fig. 1 Cyclopropane-containing natural products, chiral drugs and insecticides. | ||
Through Suzuki–Miyaura coupling, C–N coupling, Tamao oxidation reaction, etc., cyclopropylboronates could be readily converted into structurally and functionally diverse cyclopropanes.10 Thus, efficient enantioselective synthesis of optically active cyclopropylboronates has gradually become a spotlight. Recently, Ito and co-workers successfully established copper(I)-catalyzed asymmetric cyclopropanation reactions of allylic phosphates and carbonates with bis(pinacolato)diboron (B2pin2), affording optically active trans-silyl- and trans-aryl-substituted cyclopropylboronates (Scheme 1a).11,12 Gevorgyan and co-workers described rhodium-catalyzed asymmetric hydroboration of 3,3-disubstituted cyclopropenes, directly constructing enantiopure 2,2-disubstituted cyclopropylboronates. The directing effect of the ester group was found to be necessary for achieving cis-selectivity and high enantioselectivity (Scheme 1b).13,14 Herein, we present our findings in copper(I)-catalyzed asymmetric hydroboration of 3,3-disubstituted cyclopropenes.15 Interestingly, the non-directing effect of the ester group was observed in this case, and the reaction proceeded with solely trans-selectivity (Scheme 1c).
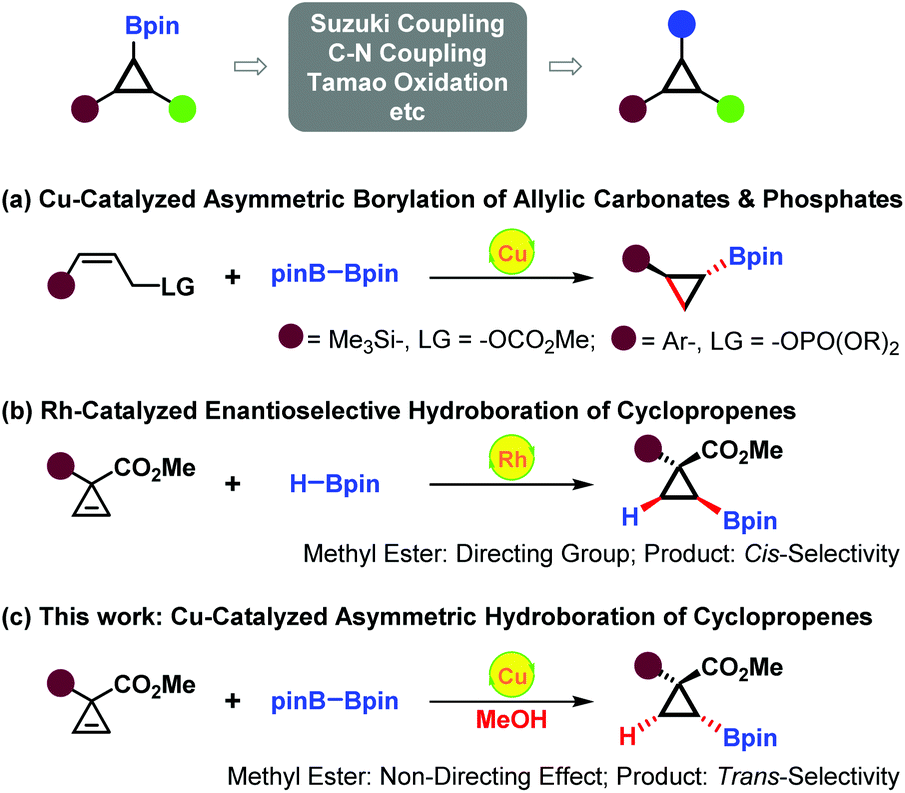 | ||
| Scheme 1 Enantioselective synthesis of optically active cyclopropylboronates. | ||
Results and discussion
At the outset, a set of representative chiral phosphorus ligands were investigated for the Cu-catalyzed asymmetric hydroboration of the cyclopropene substrate 1f, and the screening results are summarized in Table 1. The chiral bisphosphine ligand, (R,Sp)-Josiphos (L1), has been successfully employed in the Cu-catalyzed asymmetric conjugate hydroboration reaction of α,β-unsaturated compounds.16 However, only 62% yield and 40% ee were observed in our hydroboration (Table 1, entry 1). Phosphoramidite ((R)-MonoPhos, L2)17 and (R)-MOP (L3) ligands were subsequently subjected to this reaction, but no promising outcomes were obtained (Table 1, entries 2 and 3). To our delight, the ligand (R)-BINAP (L4) could dramatically improve the yield and ee of hydroboration product 3f to 75% and 94%, respectively (Table 1, entry 4). Several electronically different bisphosphine ligands (L5–L8) were applied in this reaction, but no better results were achieved (Table 1, entries 5–8).|
|
|||||
|---|---|---|---|---|---|
| Entry | L* | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a The reaction was carried out with 1f (0.15 mmol), B2Pin2 (2, 0.3 mmol), CuCl (10 mol%), chiral ligand (L*, 12 mol%) and NaOtBu (11 mol%) in anhydrous toluene (1.0 mL) at room temperature under a N2 atmosphere, unless otherwise noted. b Yield of the isolated product. c Determined by HPLC analysis using a chiral stationary phase. d At 0 °C. e L4 (15 mol%) was used. f L4 (20 mol %) was used. B2Pin2 = bis(pinacolato)diboron. | |||||
| 1 | L1 | Toluene | 6 | 62 | 40 |
| 2 | L2 | Toluene | 8 | 16 | 22 |
| 3 | L3 | Toluene | 8 | 30 | 51 |
| 4 | L4 | Toluene | 6 | 75 | 94 |
| 5 | L5 | Toluene | 8 | 58 | 93 |
| 6 | L6 | Toluene | 10 | 85 | −89 |
| 7 | L7 | Toluene | 12 | 60 | 93 |
| 8 | L8 | Toluene | 16 | 40 | 79 |
| 9d | L4 | Toluene | 24 | 46 | 90 |
| 10 | L4 | THF | 6 | 44 | 84 |
| 11 | L4 | DCM | 6 | 32 | 94 |
| 12e | L4 | Toluene | 6 | 80 | 95 |
| 13f | L4 | Toluene | 6 | 78 | 94 |
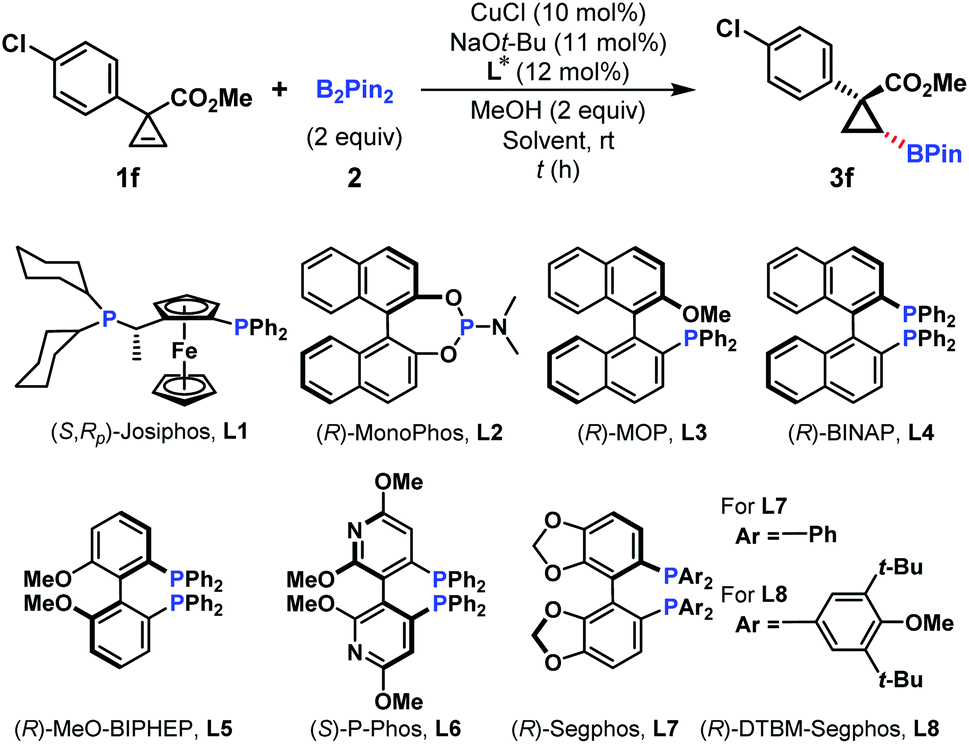
Next, the reaction temperature and the solvent were investigated to further improve the enantioselectivity. Unfortunately, they led to different levels of erosion in yields and ee values (Table 1, entries 9–11). Increasing the ligand loading to 15 mol% resulted in a slight improvement of both yield and ee values (Table 1, entry 12). However, further increasing the ligand loading failed to give better results (Table 1, entry 13).
With the optimal reaction conditions identified, various aryl-substituted cyclopropenes were investigated, and the results are summarized in Table 2. All 4-substituted phenyl substrates, regardless of the electron-donating or electron-withdrawing properties of the substituent at the phenyl ring, afforded the hydroboration products in moderate to high yields (55–86%) and with high to excellent enantioselectivities (89–95% ee, Table 2, entries 1–7). Interestingly, p-, m-, and o-bromophenyl substituted cyclopropene substrates (1g, 1h, and 1i) gave almost the same yields with high to excellent levels of enantioselectivities (Table 2, entries 7–9). As for 2-naphthyl and disubstituted phenyl substrates, the hydroboration reaction also proceeded smoothly with high yields and excellent enantioselectivities (Table 2, entries 10–12). In general, cyclopropene substrates bearing electron-withdrawing phenyl substituents provided better yields (Table 2, entries 1–3 vs. 4–6, 12).
| a The reaction was carried out with 1 (0.15 mmol), B2Pin2 (2, 0.3 mmol), CuCl (10 mol%), (R)-BINAP (L4, 15 mol%) and NaOtBu (11 mol%) in anhydrous toluene (1.0 mL) at room temperature under a N2 atmosphere. b Reaction time. c Yield of the isolated product. d Determined by HPLC analysis using a chiral stationary phase. |
|---|
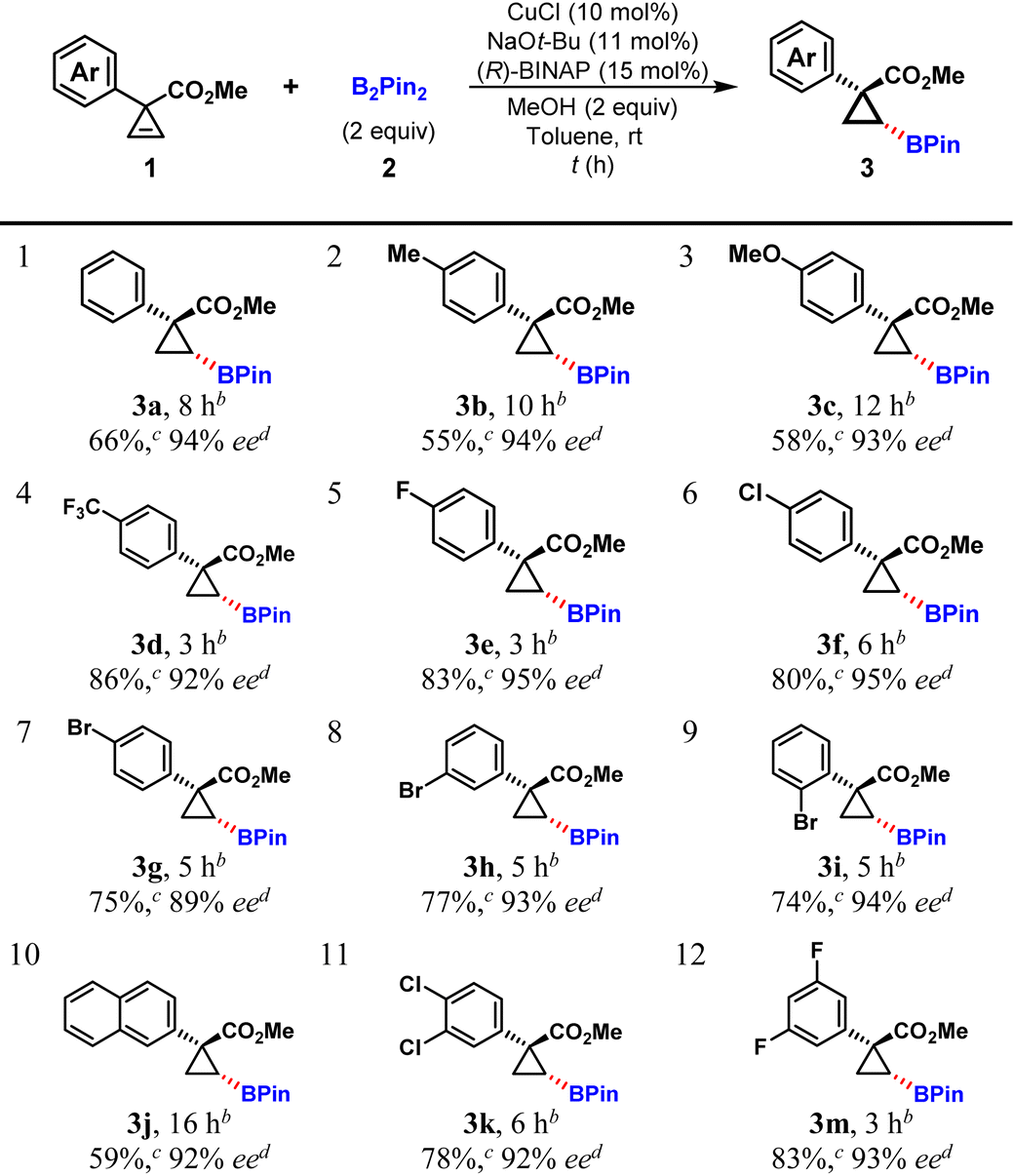
|
Given the highly enantioselective nature of this hydroboration reaction, the methyl substituted substrates 1n and 1o were tested under the standard conditions. Unfortunately, no desired products were observed, indicating that the α-substituent played an important role in the cyclopropene reactivity (Scheme 2, eqn (1) and (2)). As for the diester substrate 1p, the hydroboration reaction readily occurred with excellent enantioselectivity, albeit in a lower yield. This was partially attributed to the decomposition of the starting material (Scheme 2, eqn (3)).
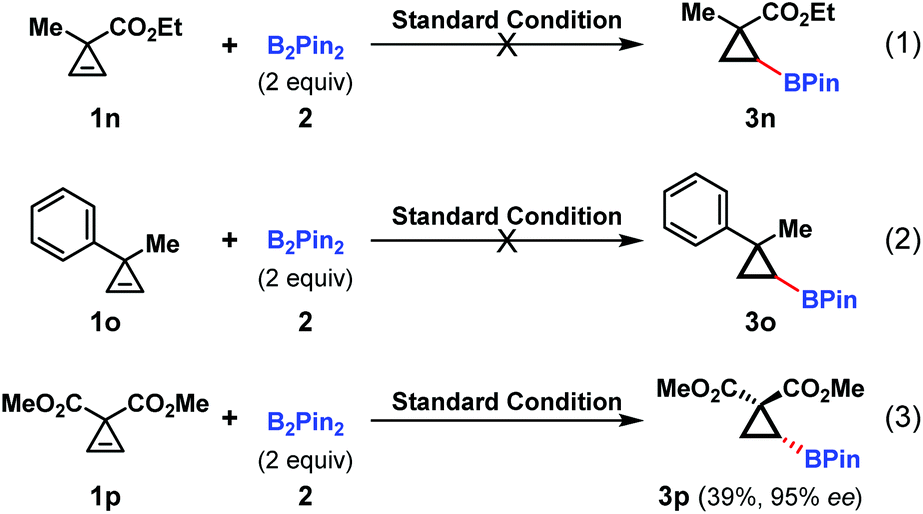 | ||
| Scheme 2 Cu-catalyzed asymmetric hydroboration of cyclopropenes 1n, 1o and 1p. | ||
The relative configuration of hydroboration products 3 was determined using NOE interactions; for example, the NOE interactions between the aryl group and the boronate group in 3g and 3j clearly revealed that both of them were on the same side of the cyclopropane plane (Fig. 2). Thus trans-cyclopropylboronates were achieved in this Cu(I)-catalyzed asymmetric hydroboration of cyclopropenes.
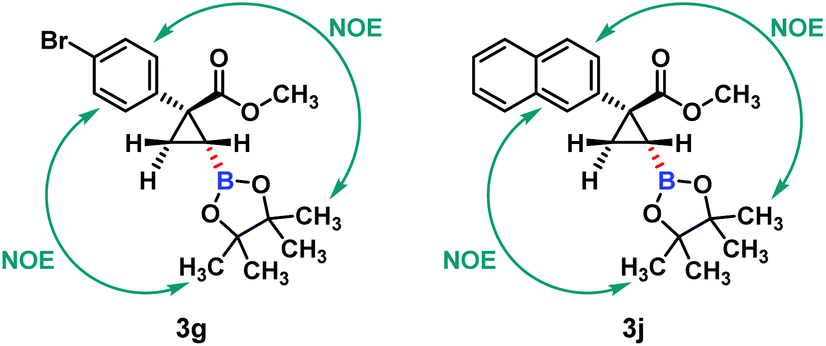 | ||
| Fig. 2 The NOE interactions between the aryl group and the boronate group in 3g and 3j. | ||
To determine the absolute configurations of the hydroboration product 3a in Table 2,18 we converted cyclopropylboronate 3a, through Suzuki–Miyaura coupling with iodobenzene (4), into a known compound (1R,2S)-5 in almost quantitative yield with no loss of the enantiomeric excess.19 Thus, the absolute configuration of cyclopropylboronate 3a was unambiguously assigned as 1R,2R. The absolute configurations of other hydroboration products in Table 2 were assigned on the basis of their chemical correlation with (1R,2R)-3a (Scheme 3).
 | ||
| Scheme 3 Determining the absolute configuration of cyclopropylboronate 3a. | ||
To probe the ‘hydrogen’ source of this hydroboration reaction, [D4]-methanol experiment was investigated. cis-Deuterated product 3a (50%) was observed, suggesting that the proton partially came from methanol and this hydroboration reaction was a syn-addition process (Scheme 4).
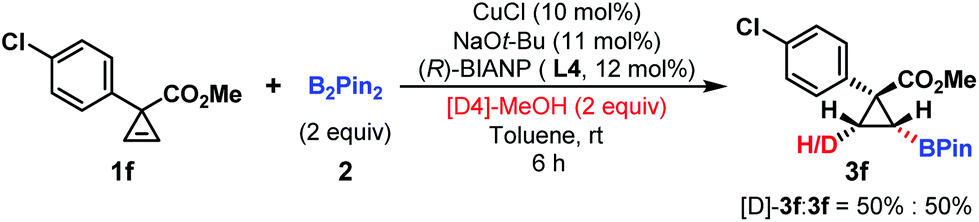 | ||
| Scheme 4 [D4]-Methanol experiment. | ||
Piecing together the above details and preceding results,20 a plausible reaction mechanism is proposed in Fig. 3. Initiation of the reaction through the transmetallation of a (pinacolato)boron group (BPin) from boron to copper species A generated the borylated copper B, which subsequently underwent syn-addition from the aryl group side21 to the double bond of the cyclopropene substrate 1 to afford the borylated cyclopropyl-copper intermediate D. The intermediate D was readily protonated by trace water or methanol to regenerate A and liberate the trans-product 3. Due to the bigger steric hindrance of the methyl ester group (Cvs.E), the weak coordination between copper and carboxyl groups could not overcome this energy barrier. Therefore, the cis-product was not observed.
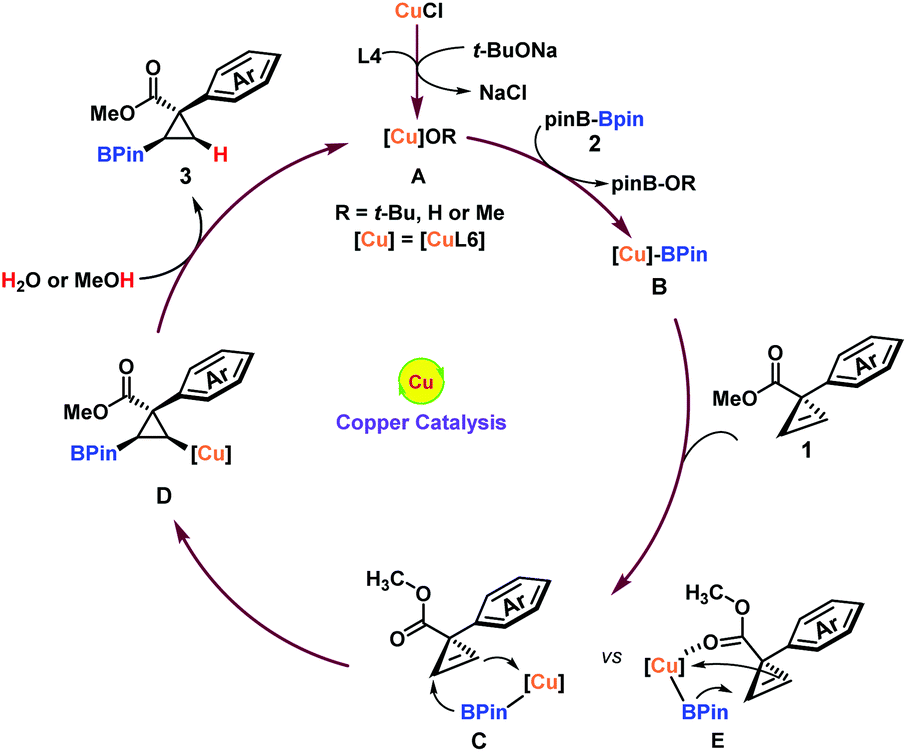 | ||
| Fig. 3 Proposed mechanism. | ||
Conclusions
In summary, copper-catalyzed asymmetric hydroboration reaction of 3-aryl, 3-methylester substituted cyclopropenes has been successfully established. This reaction proceeded smoothly at room temperature, affording optically active trans-cyclopropylboronates with excellent enantioselectivities (89–95% ee) in moderate to high yields (55–86%). The non-directing effect of the methylester group was observed and this method was actually complimentary to the earlier reported cis-borylated cyclopropane products through rhodium catalysis. The chiral boronates could be readily transformed to chiral 1,2-diaryl substituted cyclopropanes through Suzuki–Miyaura coupling reaction. Further studies on the applications of cyclopropylboronates are in progress in our laboratories.Experimental section
General information
All solvents were dried before use by following the standard procedures. Unless otherwise indicated, all starting materials purchased from commercial suppliers were used without further purification. The 1H and 13C NMR spectra were recorded on a Bruker AV 400 MHz in the indicated solvents. Chemical shifts are reported in δ (ppm) referenced to the internal standard TMS for 1H NMR and to CDCl3 (δ = 77.10 ppm) for 13C NMR. Coupling constants (J) are quoted in Hz. Optical rotations were measured on a JASCO P-1030 polarimeter. IR spectra were recorded on a Nicolet iN 10 MX. ESI mass spectra were recorded on an Agilent 1200/G6100A. HRMS of boron-containing compounds is based on 10B. For the preparation of substrates 4a, see the ESI.†General procedure for Cu-catalyzed hydroboration of 3,3-disubstituted cyclopropenes
A dried Schlenk flask was charged with CuCl (1.5 mg, 0.015 mmol, 10 mol%), (R)-(+)-BINAP (14 mg, 0.0225 mmol, 15 mol%), B2pin2 (2, 76.2 mg, 0.3 mmol, 2.0 equiv.), NaOtBu (1.6 mg, 0.0165 mmol, 11 mol%) and anhydrous toluene (1.0 mL) under a nitrogen atmosphere. After the mixture was stirred at room temperature for 40 min, a solution of cyclopropene 1 (0.15 mmol) in anhydrous toluene (0.5 mL) was added, followed by anhydrous MeOH (12.2 μL, 0.30 mmol, 2.0 equiv.). The resulting mixture was stirred at room temperature for the time indicated in Table 2, then filtered through Celite®, and concentrated in vacuo. The residue was purified by silica gel (300–400 mesh) column chromatography using hexane–ethyl acetate (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent to afford the desired product 3.
:1) as an eluent to afford the desired product 5 (25 mg, 99% yield). [α]26D −38.9 (c 1.0, CHCl3) for 94% ee; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.05–6.94 (m, 8H), 6.70–6.68 (m, 2H), 3.58 (s, 3H), 3.06–3.01 (m, 1H), 2.06 (dd, J = 9.2 Hz, 4.8 Hz, 1H), 1.82–1.78 (m, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm) 174.42, 136.42, 134.80, 131.99, 128.40, 128.10, 127.76, 127.09, 126.37, 52.68, 37.45, 33.19, 20.54; ESI-MS: [M + Na]⊕ 275.1; HRMS (FTMS-ESI): [M + Na]⊕ calcd for C17H16O2Na⊕ 275.1043, found 275.1037; IR (KBr) ν (cm−1) 3648, 3412, 3061, 3086, 3029, 2953, 1966, 1897, 1720, 1602, 1496, 1456, 1447, 1428, 1376, 1342, 1255, 1205, 1189, 1104, 1050, 989, 865, 788, 760, 742, 702, 650, 545; HPLC: OJ-H Column; detected at 214 nm; n-hexane–i-propanol = 95/5; flow rate = 0.7 mL min−1; retention time: 11.5 min (R,S-isomer), 16.3 min (S,R-isomer).
:1) as an eluent to afford the desired product 3.
:1) as an eluent to afford the desired product 5 (25 mg, 99% yield). [α]26D −38.9 (c 1.0, CHCl3) for 94% ee; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.05–6.94 (m, 8H), 6.70–6.68 (m, 2H), 3.58 (s, 3H), 3.06–3.01 (m, 1H), 2.06 (dd, J = 9.2 Hz, 4.8 Hz, 1H), 1.82–1.78 (m, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm) 174.42, 136.42, 134.80, 131.99, 128.40, 128.10, 127.76, 127.09, 126.37, 52.68, 37.45, 33.19, 20.54; ESI-MS: [M + Na]⊕ 275.1; HRMS (FTMS-ESI): [M + Na]⊕ calcd for C17H16O2Na⊕ 275.1043, found 275.1037; IR (KBr) ν (cm−1) 3648, 3412, 3061, 3086, 3029, 2953, 1966, 1897, 1720, 1602, 1496, 1456, 1447, 1428, 1376, 1342, 1255, 1205, 1189, 1104, 1050, 989, 865, 788, 760, 742, 702, 650, 545; HPLC: OJ-H Column; detected at 214 nm; n-hexane–i-propanol = 95/5; flow rate = 0.7 mL min−1; retention time: 11.5 min (R,S-isomer), 16.3 min (S,R-isomer).
Acknowledgements
Financial support for this work was generously provided by the National Natural Science Foundation of China (NSFC 21372243, 21232009, 21102161), the Shanghai Municipal Committee of Science and Technology (13JC1406900), and the State Key Laboratory of Bioorganic and Natural Products Chemistry. We thank Dr Hanqing Dong (Arvinas Inc.) for his help in the preparation of this manuscript.Notes and references
- For selected recent reviews, see: (a) D. Y.-K. Chen, R. H. Pouwerb and J.-A. Richardc, Chem. Soc. Rev., 2012, 41, 4631 RSC; (b) P. Tang and Y. Qin, Synthesis, 2012, 2969 CAS.
- B. J. F. Feys, C. E. Benedetti, C. N. Penfold and J. G. Turner, Plant Cell, 1994, 6, 751 CrossRef CAS PubMed.
- D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S.-P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, J. Med. Chem., 2005, 48, 5025 CrossRef CAS PubMed.
- L. A. Maslovskaya, A. I. Savchenko, E. H. Krenske, C. J. Pierce, V. A. Gordon, P. W. Reddell, P. G. Parsons and C. M. Williams, Angew. Chem., Int. Ed., 2014, 53, 7006 CrossRef CAS PubMed.
- S. N. Vaishnavi, C. B. Nemeroff, S. J. Plott, S. G. Rao, J. Kranzler and M. J. Owens, Biol. Psychiatry, 2004, 55, 320 CrossRef CAS PubMed.
- D. A. Laskowski, Rev. Environ. Contam. Toxicol., 2002, 174, 49 CAS.
- W. Zhang, T. Kilicarslan, R. F. Tyndale and E. M. Sellers, Drug Metab. Dispos., 2001, 26, 897 Search PubMed.
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed.
- For selected recent examples, see: (a) H. Xiong, H. Xu, S.-H. Liao, Z.-W. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed; (b) S. M. Wales, M. M. Walker and J. S. Johnson, Org. Lett., 2013, 15, 2558 CrossRef CAS PubMed; (c) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed.
- (a) G.-H. Fang, Z.-J. Yan and M.-Z. Deng, Org. Lett., 2004, 6, 357 CrossRef CAS PubMed; (b) S. Bénard, L. Neuville and J. Zhu, Chem. Commun., 2010, 46, 3393 RSC; (c) P. B. Brondani, H. Dudek, J. S. Reis, M. W. Fraaije and L. H. Andrade, Tetrahedron: Asymmetry, 2012, 23, 703 CrossRef CAS PubMed.
- (a) H. Ito, Y. Kosaka, K. Nonoyama, Y. Sasaki and M. Sawamura, Angew. Chem., Int. Ed., 2008, 47, 7424 CrossRef CAS PubMed; (b) C. Zhong, S. Kunii, Y. Kosaka, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 11440 CrossRef CAS PubMed.
- For selected Cu-catalyzed asymmetric tandem borylation reactions, see: (a) H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990 CrossRef CAS PubMed; (b) A. R. Burns, J. S. González and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 10827 CrossRef CAS PubMed; (c) N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934 CrossRef CAS PubMed; (d) P. Liu, Y. Fukui, P. Tian, Z.-T. He, C.-Y. Sun, N.-Y. Wu and G.-Q. Lin, J. Am. Chem. Soc., 2013, 135, 11700 CrossRef CAS PubMed.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198 CrossRef CAS PubMed.
- For other catalyzed hydrometallations, see: (a) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2002, 124, 11566 CrossRef CAS PubMed; (b) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688 CrossRef CAS PubMed.
- For selected Cu-catalyzed asymmetric hydroborations of the aryl- and silyl-substituted alkenes, see: (a) Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3160 CrossRef CAS PubMed; (b) R. Corberán, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079 CrossRef PubMed; (c) F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204 CrossRef CAS PubMed.
- For selected examples of Cu-catalyzed conjugate hydroboration, see: (a) J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145 CrossRef CAS PubMed; (b) H.-S. Sim, X. Feng and J. Yun, Chem. – Eur. J., 2009, 15, 1939 CrossRef CAS PubMed; (c) I.-H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS PubMed; (d) J. M. O'Brien, K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10630 CrossRef PubMed; (e) I. Ibrahem, P. Breistein and A. Córdova, Angew. Chem., Int. Ed., 2011, 50, 12036 CrossRef CAS PubMed; (f) A. L. Moure, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2011, 47, 6701 RSC; (g) H. Wu, S. Radomkit, J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277 CrossRef CAS PubMed; (h) S. Kobayashi, P. Xu, T. Endo, M. Ueno and T. Kitanosono, Angew. Chem., Int. Ed., 2012, 51, 12763 CrossRef CAS PubMed; (i) Y. Luo, I. D. Roy, A. G. E. Madec and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 4186 CrossRef CAS PubMed; (j) Z.-T. He, Y.-S. Zhao, P. Tian, C.-C. Wang, H.-Q. Dong and G.-Q. Lin, Org. Lett., 2014, 16, 1426 CrossRef CAS PubMed.
- C. Sole, A. Bonet, A. H. M. de Vries, J. G. de Vries, L. Lefort, H. Gulyás and E. Fernández, Organometallics, 2012, 31, 7855 CrossRef CAS.
- Direct single crystal incubation of hydroboration product 3g resulted in the production of a dimer compound through deboration and the [2 + 2] reaction process. See ESI† for the details.
- (a) H. M. L. Davies and G. H. Lee, Org. Lett., 2004, 6, 1233 CrossRef CAS PubMed; (b) R. Sambasivan and Z. T. Ball, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef CAS PubMed.
- For selected Cu(I)-catalyzed asymmetric additions of cyclopropenes, see: (a) X. Liu and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 5600 CrossRef CAS PubMed; (b) N. Yan, X. Liu and J. M. Fox, J. Org. Chem., 2008, 73, 563 CrossRef CAS PubMed; (c) V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382 CrossRef CAS PubMed; (d) V. Tarwade, R. Selvaraj and J. M. Fox, J. Org. Chem., 2012, 77, 9900 CrossRef CAS PubMed.
- In the cyclopropene substrate 1, the aryl group was almost vertical to the cyclopropene plane. As a result, the aryl group side was supposed to be less hindered than the methyl ester side.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of substrates, characterization data, 1H, 13C NMR, MS and IR spectra. CCDC 1004894. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00157e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2014 |