
Light-mediated, palladium-catalyzed cyclizations of unactivated 1,6-dienes†
Weiyuan
Du
,
Gangfeng
Tang
,
Yiyong
Chen
,
Dawen
Xu
and
Hao
Guo
*
Department of chemistry, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China. E-mail: Hao_Guo@fudan.edu.cn; Fax: +86-21-55664361; Tel: +86-21-55664361
First published on 15th July 2014
Abstract
We report herein a light-mediated, palladium-catalyzed cyclization of unactivated 1,6-dienes under UV irradiation at rt. 1,5-Dimethylcyclopent-1-enes are formed as the only product in this transformation in moderate to excellent yields. This reaction offers a new insight into the realization of photochemical access to monocycles under ambient conditions.
Intramolecular photocyclization of dienes is an important research topic in organic chemistry,1 which has been studied for quite a long time and applied as a key step in total synthesis of many natural products.2 Continuous efforts devoted in this field have resulted in many methods to construct ring systems unavailable to thermal reactions. Copper salts were reported to be able to efficiently catalyze the [2 + 2]-photocycloaddition of both activated and unactivated 1,6-dienes, generating cis-bicyclo[3.2.0]heptanes in high yields highly stereoselectively.1c,e,3 Bach developed two protocols4,5 to achieve highly enantiopure bicyclo[3.2.0]heptanes or bicyclo[4.2.0]octanes from 1,6- or 1,7-dienes using chiral hydrogen-bonding template4 or Lewis acid5 catalysts. Yoon reported the visible light induced cyclizations of activated 1,6-dienes which led to bicyclo[3.2.0]heptanes highly regio- and stereoselectively.6,7 Meanwhile, photoinduced intramolecular transformations of dienes into monocycles have also received much attention. One example is the photocyclization of N-vinylacrylamides into six-membered lactams, which has been used as a powerful tool in organic synthesis.2a,8 Electron acceptor promoted electron transfer photoreaction of geraniol derivatives provided another way.9 Yoon reported a visible light induced reductive photocyclization of enones affording cyclopentanes efficiently.10 However, to the best of our knowledge, transition metal-catalyzed photochemical formation of monocyclic ring systems from unactivated dienes remained unexplored. In this regard, more efficient and ambient methods in this field are still in urgent need, since photochemistry is playing a very important role in modern chemistry. On the other hand, transition metal-catalyzed non-photochemical 1,6-diene cycloisomerizations provide efficient methods for the synthesis of cyclopentane derivatives (Scheme 1).11–16 For example, RajanBabu16 reported Ni- or Pd-catalyzed cyclization of α,ω-dienes affording the kinetic product 2 in most cases. However, only one example showed that product 3 could be obtained at a moderate regioselectivity. Lloyd-Jones developed efficient methods for regiospecific formation of 2 or 3 using different palladium precatalysts and carefully studied the corresponding mechanisms.12b,c Besides the rich history of this reaction, photochemical processes of this structural motif are less explored. Thus, we showed great interest in developing new light-mediated cyclization of 1,6-dienes in this field. Herein, we wish to report our recent observation on the light-mediated palladium-catalyzed cyclizations of unactivated 1,6-dienes at ambient temperature, forming 1,5-dimethylcyclopent-1-ene derivatives highly regiospecifically.
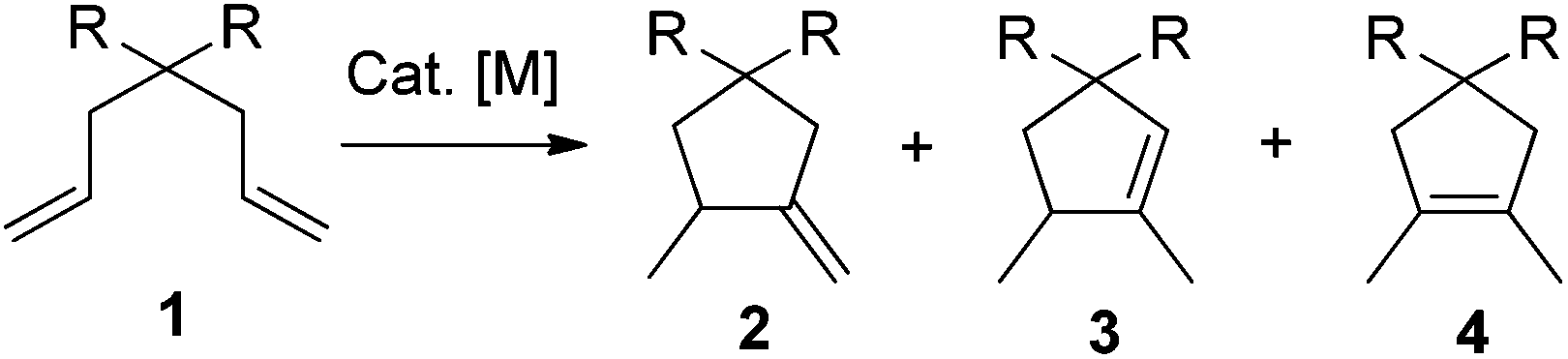 | ||
| Scheme 1 Transition metal-catalyzed non-photochemical 1,6-diene cycloisomerizations. | ||
At the beginning of our study, dimethyl 2,2-diallylmalonate 1a was chosen as the model substrate to explore the feasibility of the desired photoreaction. First of all, a CH2Cl2 solution of 1a was irradiated at λ = 185&254 nm at rt without any catalyst. After full conversion, no cyclic product was formed (Table 1, entry 1). Then a series of palladium catalysts were tested under the same irradiation conditions (Table 1, entries 2–7). The reactions using Pd(0) as the catalyst did not proceed (Table 1, entries 2–4). To our delight, the cyclization reaction occurred readily when Pd(OAc)2 was applied, affording dimethyl 3,4-dimethylcyclopent-2-ene-1,1-dicarboxylate 3a as the only product in 47% isolated yield (Table 1, entry 5). Further studies with other Pd(II) catalysts led to better results (Table 1, entries 6 and 7). The reaction under the catalysis of [Pd(allyl)Cl]2 gave the sole product 3a in 76% isolated yield (Table 1, entry 7). Thus, [Pd(allyl)Cl]2 was chosen as the standard catalyst for this reaction. Subsequently, the solvent effect was examined. The photoreaction proceeded to generate 3a in all the tested cases (Table 1, entries 8–10); however, no better results were observed. On the basis of these results, we set to explore the influence of the amount of the catalyst. When 5 mol% of [Pd(allyl)Cl]2 was applied, the yield significantly increased to 93% (Table 1, entry 11). Further reducing the amount of the catalyst resulted in a much lower yield (Table 1, entry 12). The following examination of the influence of the irradiation energy showed that with the decrease of the energy, the yield of 3a became lower until no reaction took place at λ = 365 nm (Table 1, entries 13–15), indicating that a high irradiation energy is necessary for this transformation. The control reactions without light at rt (Table 1, entry 16) or under reflux (Table 1, entry 17) did not give any conversion of starting diene 1a, showing that [Pd(allyl)Cl]2 itself cannot catalyze this cyclization under non-photochemical reaction conditions, which is consistent with the literature reports.17,18 Next, the reaction mixture was irradiated at λ = 185&254 nm for 1 hour, after which it was allowed to react for 11 hours without light. Only 5% of 3a was yielded (Table 1, entry 18), indicating that the photoreaction only proceeded in the first 1 hour when it was irradiated. To check whether [Pd(allyl)Cl]2 was the pre-catalyst which turned into the real catalytic species under photo-irradiation, a solution of [Pd(allyl)Cl]2 in CH2Cl2 was irradiated at λ = 185&254 nm for 1 hour, which led to the formation of palladium black quickly. Then 1a was added. After reacting for another 11 hours without light, 2% of 3a was formed (Table 1, entry 19). This result may suggest that [Pd(allyl)Cl]2 is a pre-catalyst in this system. High energy irradiation is necessary for the in situ generation of the real catalytic species whose lifetime is short in the absence of dienes. Thus, conditions A (λ = 185&254 nm, 5 mol% of [Pd(allyl)Cl]2, CH2Cl2, and rt) were applied for this cyclization.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. [Pd] (mol%) | Solvent | λ (nm) | Time (h) | Yield (%) |
| a A solution of 1a (0.2 mmol) in deaerated solvent (10 mL) in a quartz reaction tube was irradiated by a Matrix-10 photoreactor with sixteen ultraviolet lamps (10 W per lamp) at rt under an argon atmosphere. b Some unidentified products were formed. c Recovered yield of 1a. d The reaction was carried out without light. e The reaction was carried out under reflux. f The solution of 1a (0.2 mmol) and [Pd(allyl)Cl]2 (5 mol%) in deaerated CH2Cl2 (10 mL) was irradiated at λ = 185&254 nm for 1 hour, after which the reaction mixture was allowed to react for 11 hours without light. g The solution of [Pd(allyl)Cl]2 (5 mol%) in deaerated CH2Cl2 (10 mL) was irradiated at λ = 185&254 nm for 1 hour before 1a (0.2 mmol) was added. | |||||
| 1 | — | CH2Cl2 | 185&254 | 36 | 0b |
| 2 | Pd/C (5) | CH2Cl2 | 185&254 | 11 | NR |
| 3 | Pd(PPh3)4 (20) | THF | 185&254 | 5.5 | NR |
| 4 | Pd(dba)2 (20) | CH2Cl2 | 185&254 | 22 | NR |
| 5 | Pd(OAc)2 (20) | CH2Cl2 | 185&254 | 9.5 | 47 |
| 6 | Pd(acac)2 (20) | CH2Cl2 | 185&254 | 11 | 68 |
| 7 | [Pd(allyl)Cl]2 (20) | CH2Cl2 | 185&254 | 23 | 76 |
| 8 | [Pd(allyl)Cl]2 (20) | CHCl3 | 185&254 | 35 | 58 (10)c |
| 9 | [Pd(allyl)Cl]2 (20) | Dioxane | 185&254 | 33 | 65 |
| 10 | [Pd(allyl)Cl]2 (20) | DCE | 185&254 | 35 | 71 |
| 11 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | 185&254 | 11 | 93 |
| 12 | [Pd(allyl)Cl]2 (1) | CH2Cl2 | 185&254 | 11 | 71 |
| 13 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | 254 | 22 | 78 |
| 14 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | 313 | 22 | 60 (40)c |
| 15 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | 365 | 22 | NR |
| 16 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | Darkd | 72 | NR |
| 17 | [Pd(allyl)Cl]2 (5) | CH2Cl2 | Darkd,e | 24 | NR |
| 18 | [Pd(allyl)Cl]2 (5)f | CH2Cl2 | Darkd | 11 | 5 (88)c |
| 19 | [Pd(allyl)Cl]2 (5)g | CH2Cl2 | Darkd | 11 | 2 (89)c |
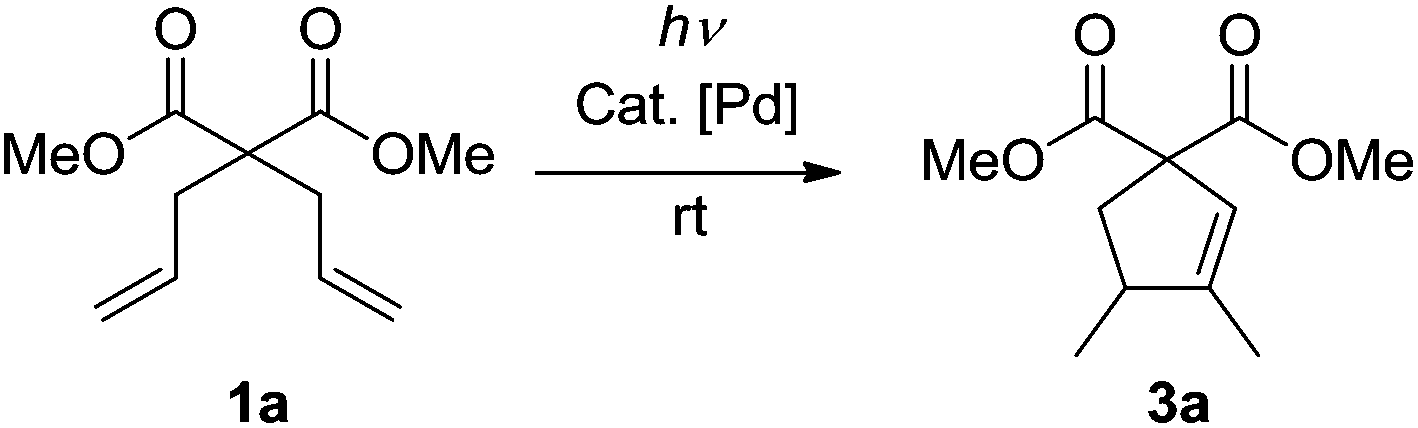
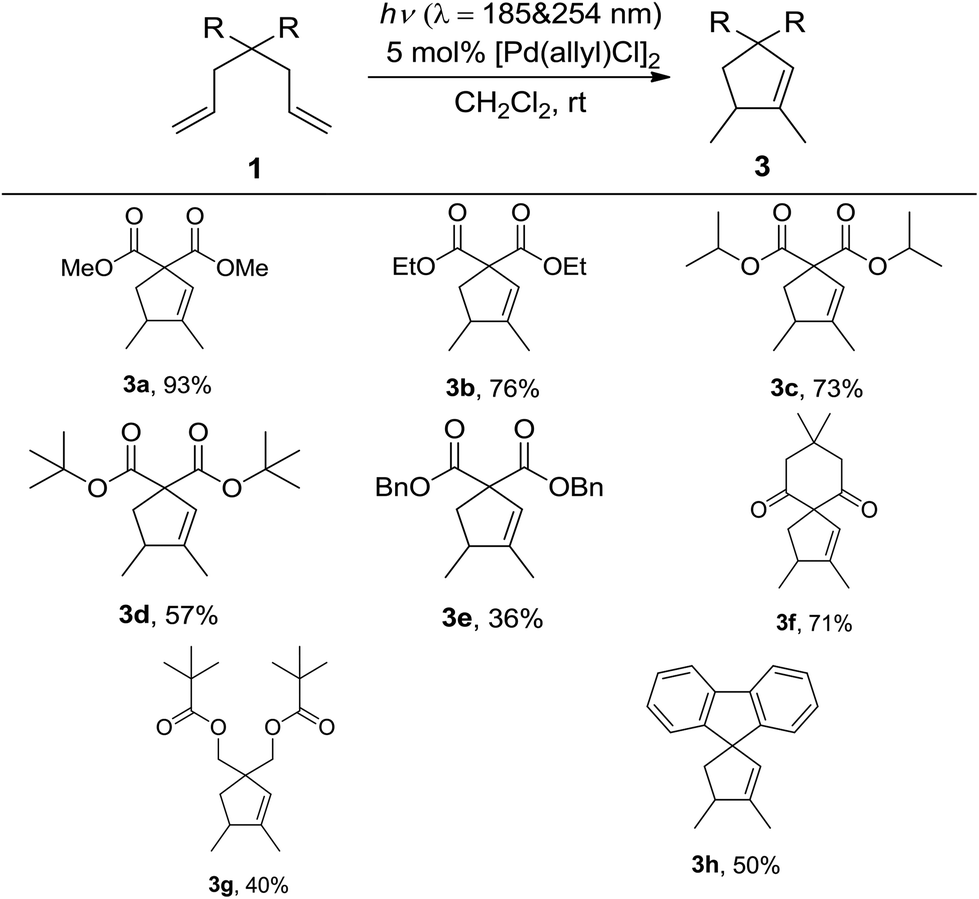
With the optimized reaction conditions in hand, the scope of this reaction with different 1,6-dienes under conditions A was investigated (Table 2). The reactions of 4,4-dialkoxycarbonyl substituted 1,6-dienes proceeded smoothly to form the corresponding five-membered cyclic products (3a, 3b, 3c, 3d, and 3e). The steric effect of the alkyl group in the alkoxycarbonyl group showed clear influence in this reaction, that is, the bulkier the alkyl group is, the lower yield the reaction gives (3a, 3b, 3c, and 3d). The low yield of 3e might not only be due to the steric effect of the benzyl group, but also its instability under UV-irradiation. When the 4,4-dialkylcarbonyl substituted substrate was applied under conditions A, the desired product 3f was afforded in 71% isolated yield. Further studies with other 4,4-dialkyl or aryl substituted reactants also led to satisfactory results (3g and 3h). Notably, in all the above cases, excellent chemo- and regioselectivities were observed, with 1,5-dimethylcyclopent-1-ene derivatives formed as the only product. And palladium black was formed after full conversion.
| a A solution of 1 (0.2 mmol) and [Pd(allyl)Cl]2 (5 mol%) in deaerated CH2Cl2 (10 mL) in a quartz reaction tube was irradiated by a Matrix185-10 photo reactor with sixteen ultraviolet lamps (10 W per lamp) at rt under an argon atmosphere. |
|---|
|
When considering the mechanism of this reaction, we first contemplated the palladium hydride initiated reaction pathway (Scheme 2), which was proposed by Lloyd-Jones based on careful studies of similar Pd-catalyzed but non-photochemical cyclizations.12b,c Under thermal conditions, [Pd(allyl)Cl]2 is quite ineffective for this cycloisomerization, since the catalyst generation requires a free ligand site through loss of chloride. Upon irradiation by high energy UV light, the heterolytic Pd–Cl bond cleavage occurs readily to afford intermediate 5 which reacts with 1 rapidly to form the carbopalladation product 6. β-H elimination of 6 generates the monochloro-bearing palladium hydride species which otherwise would turn into Pd black easily without alkene ligands. Coordination of this active palladium hydride intermediate with reactant 1 leads to the stabilized palladium hydride catalyst 7. The subsequent hydropalladation, carbopalladation, β-H elimination, hydropalladation, and β-H elimination, finally result in the highly regioselective formation of product 3.
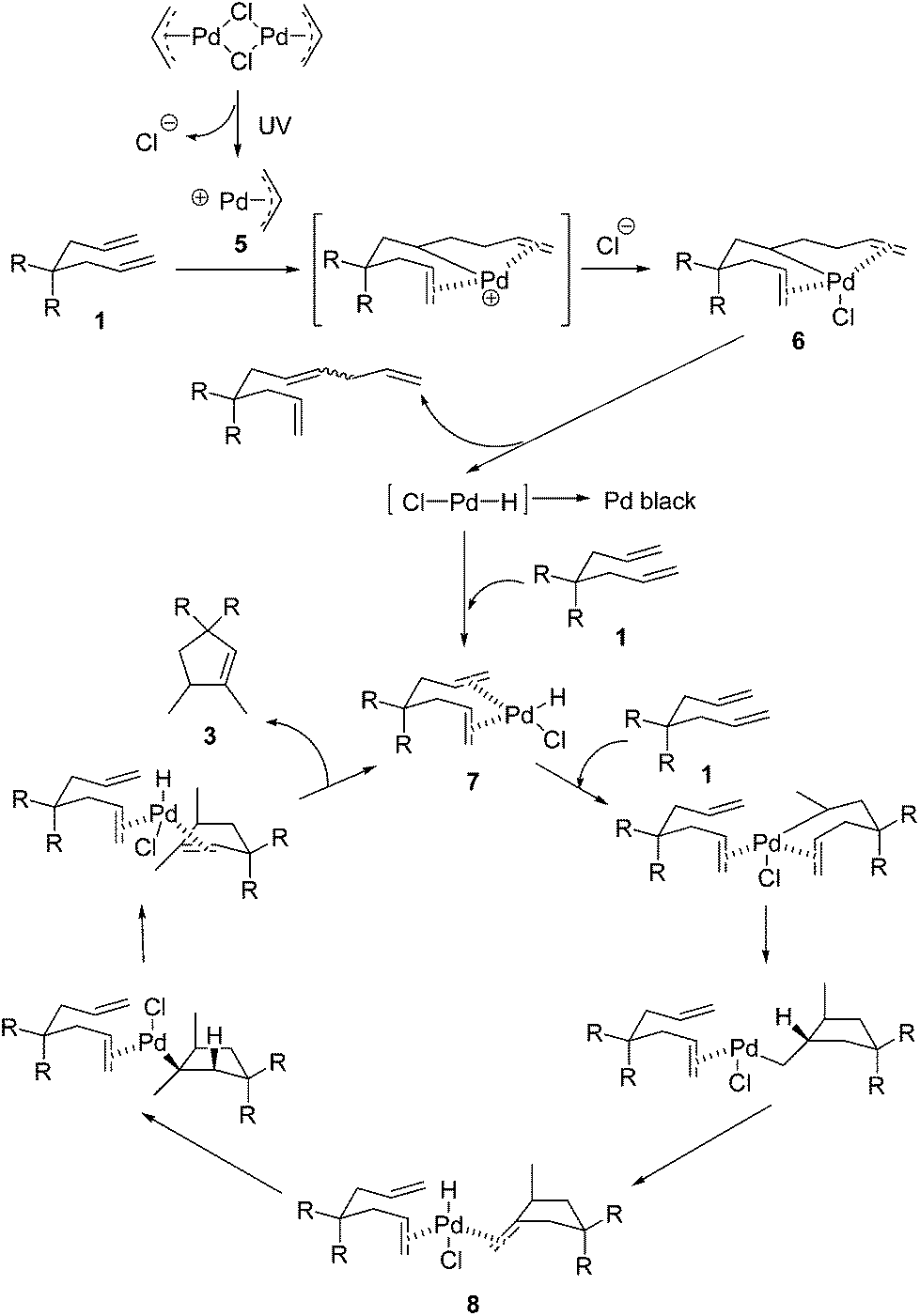 | ||
| Scheme 2 The proposed mechanism. | ||
In Lloyd-Jones’ mechanism12b,c it is clear that product 3 is formed directly from the reactant 1 in a continuous process, not via the isomerization of the kinetic product 2 step by step. Although 2 is afforded as an intermediate in the whole reaction procedure, it is not free but coordinated to the palladium centre to generate a (1)Pd(H)(Cl)(2) complex 8. The following key step of hydropalladation rather than ligand dissociation leads to the regiospecific formation of 3. Indeed, 2 was not detected in the reaction mixture. To check whether this was due to a fast isomerization from 2 to 3 under UV light via a different mechanism, irradiation of 2a under conditions A was investigated (Scheme 3). Without the addition of 1a, the conversion was not complete and the yield was significantly low (15%). To check whether the possible in situ transformation of 2a to 3a occurred only with the assistance of 1a, 1 equivalent of 1a was added to this reaction system. After full conversion of both 1a and 2a, the total yield was 50% (based on 1a + 2a). Subtracting the 90% yield of 3a generated from 1a alone, the estimated yield of 3a from the conversion of 2a is less than 30%. These results indicated that isomerization from free 2 to 3 is not a possible reaction pathway in this case.
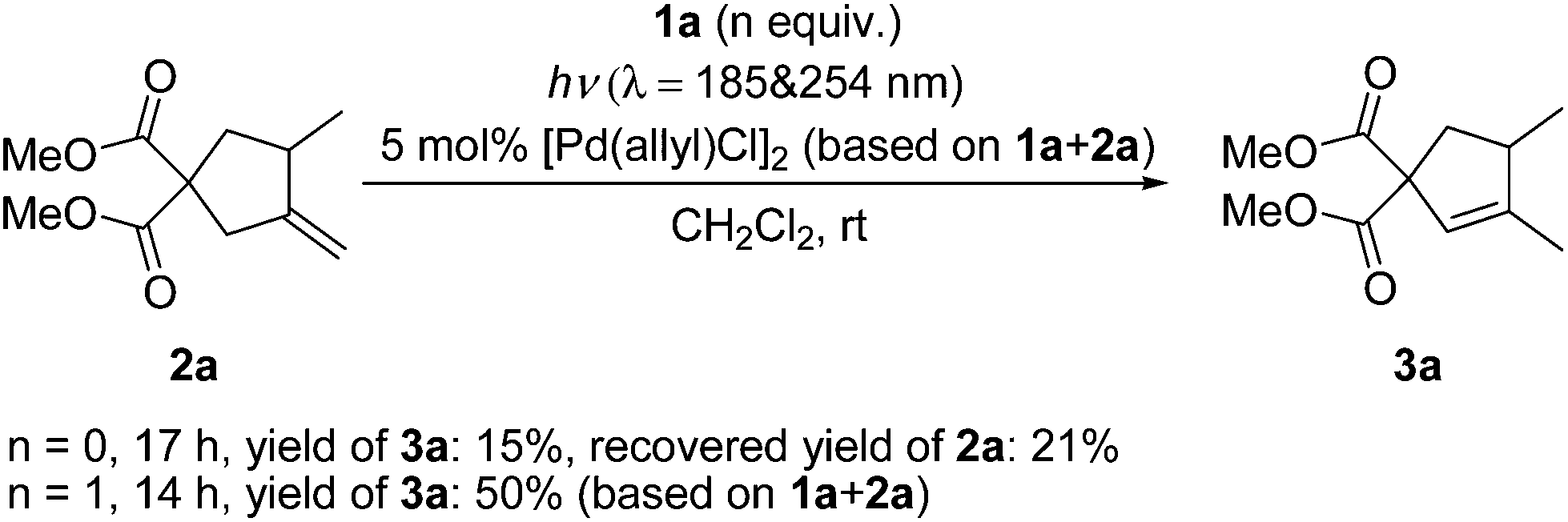 | ||
| Scheme 3 Irradiation of 2a under conditions A with or without 1a. | ||
Diene 9 was shown to exhibit catalyst inhibition in Lloyd-Jones’ mechanism.12b,c Similarly, 9 was not observed in the reaction mixture and the results of test reactions with 9 (Scheme 4) gave the same observation as Lloyd-Jones made under thermal conditions.
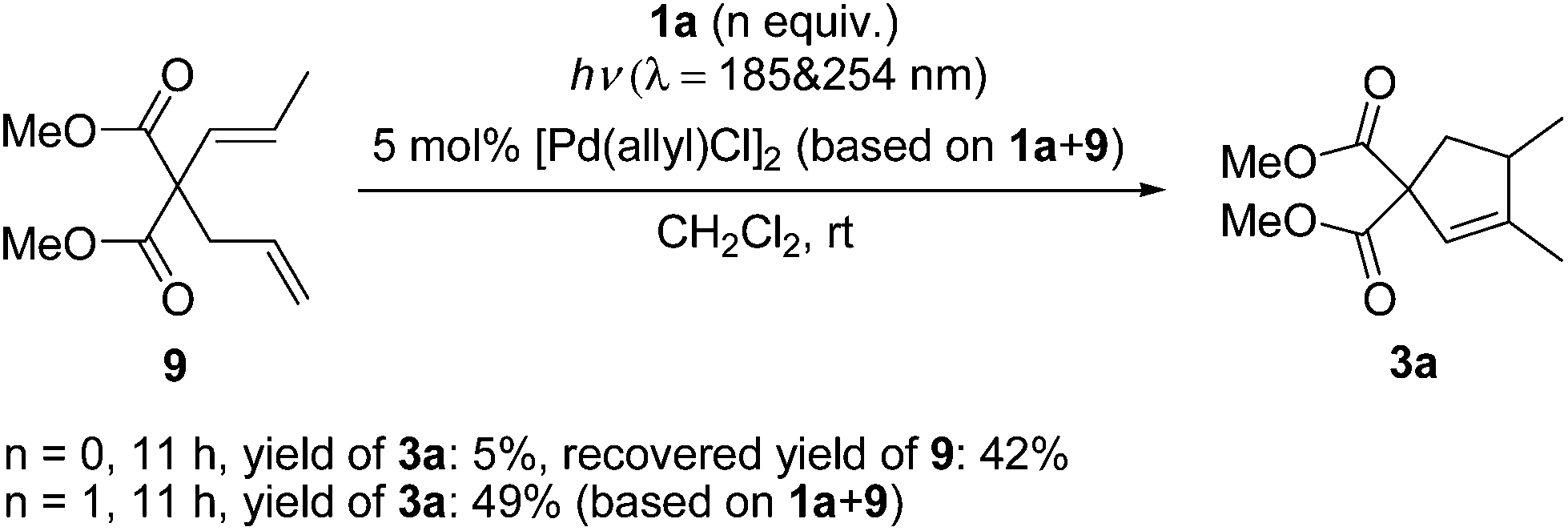 | ||
| Scheme 4 Irradiation of 9 under conditions A with or without 1a. | ||
To obtain more mechanistic information, deuterium-labeled substrates 1019 and 1120 were synthesized and applied under conditions A. After cyclizations, two labeled products 3a-d2 and 3a-d4 were formed respectively (Scheme 5). In the cyclization of 10, 3a-d2 was formed with clear label distribution which showed that the deuterium at C(2) in 10 was transferred to C(1) in 3a-d2 without any deuterium loss. However, for the reaction of 11, one of the allylic deuteriums at C(3) was transferred to C(7) in 3a-d4 with a significant loss of deuterium.
 | ||
| Scheme 5 Labeling experiments. | ||
These D-labelling experiments offer useful insights into the photochemical process. To draw a clear picture of events that happened in this process, the catalytic cycles of 10 (Scheme 6A) and 11 (Scheme 6B) are depicted in Scheme 6. For the cyclization of 10, the only Pd-D species is intermediate 8. This (1)Pd(D)(Cl)(2) complex has a very short lifetime and undergoes deuteropalladation rapidly.12c Thus, no H/D exchange occurs with 8. While for the cyclization of 11, the key Pd-D species is intermediate 7. This alkene ligand stabilized complex has a relatively long lifetime,12c which we suspect might allow H/D exchange with the solvent or a trace amount of water. The detailed mechanism for this deuterium loss is not clear and warrants further studies. Overall, the above observations fit well with the proposed mechanism.
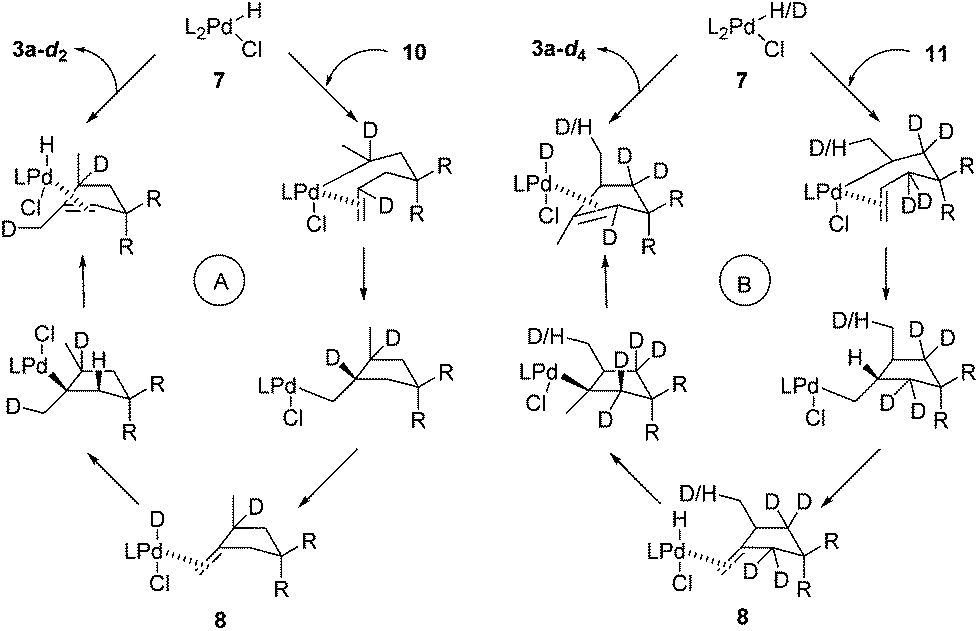 | ||
| Scheme 6 The catalytic circles of 10 and 11. | ||
Based on the above results which correlate quite well with Lloyd-Jones’ work,12c we conclude that this reaction proceeds via the proposed reaction pathway as shown in Scheme 2.
Conclusions
In conclusion, we have developed a light-mediated palladium-catalyzed cyclization of unactivated 1,6-dienes, providing a novel catalytic protocol for the synthesis of 1,5-dimethylcyclopent-1-ene derivatives. This method offers a new avenue for the realization of photochemical access to monocycles. It is noteworthy that the precatalyst [Pd(allyl)Cl]2, which is usually ineffective in terms of thermal diene cycloisomerization, is rendered effective through continuous irradiation. Further studies on the mechanism and the applications of this system to new catalytic reactions for organic synthesis are being carried out in our laboratory.Acknowledgements
We greatly acknowledge the financial support from Shanghai Rising-Star Program (14QA1400500), National Basic Research Program of China (973 Program, 2012CB720300), National Nature Science Foundation of China (21102016), Shanghai Scientific and Technological Innovation Project (13520711500), Shanghai Pujiang Program (11PJD004), and Specialized Research Fund for the Doctoral Program of Higher Education of China (20110071120004).Notes and references
- (a) D. W. C. MacMillan, C. K. Prier and D. A. Rankic, Chem. Rev., 2013, 113, 5322 CrossRef PubMed; (b) Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Dekker, New York, 2005 Search PubMed; (c) Handbook of Organic Photochemistry and Photobiology, ed. W. Horspool and F. Lenci, CRC Press, Florida, 2004 Search PubMed; (d) M. T. Crimmins, Chem. Rev., 1988, 88, 1453 CrossRef CAS; (e) R. G. Salomon, Tetrahedron, 1983, 39, 485 CrossRef CAS.
- (a) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000 CrossRef CAS PubMed; (b) N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed; (c) M. F. Greaney and J. Iriondo-Alberdi, Eur. J. Org. Chem., 2007, 4801 Search PubMed; (d) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449 CrossRef CAS PubMed; (e) J. D. Winkler, C. M. Bowen and F. Liotta, Chem. Rev., 1995, 95, 2003 CrossRef CAS.
- N. Hoffmann, ChemSuschem, 2012, 5, 352 CrossRef CAS PubMed.
- (a) T. Bach, C. Müller, A. Bauer, M. M. Maturi, M. C. Cuquerella and M. A. Miranda, J. Am. Chem. Soc., 2011, 133, 16689 CrossRef PubMed; (b) T. Bach, K. A. B. Austin and E. Herdtweck, Angew. Chem., Int. Ed., 2011, 50, 8416 CrossRef PubMed; (c) T. Bach, C. Müller and A. Bauer, Angew. Chem., Int. Ed., 2009, 48, 6640 CrossRef PubMed; (d) T. Bach and S. Breitenlechner, Angew. Chem., Int. Ed., 2008, 47, 7957 CrossRef PubMed; (e) T. Bach, H. Bergmann, B. Grosch and K. Harms, J. Am. Chem. Soc., 2002, 124, 7982 CrossRef CAS PubMed; (f) T. Bach, H. Bergmann and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 2302 CrossRef CAS.
- (a) T. Bach and R. Brimioulle, Science, 2013, 342, 840 CrossRef PubMed; (b) T. Bach, R. Brimioulle and H. Guo, Chem. – Eur. J., 2012, 18, 7552 CrossRef PubMed; (c) T. Bach, H. Guo and E. Herdtweck, Angew. Chem., Int. Ed., 2010, 49, 7782 CrossRef PubMed.
- (a) T. P. Yoon, ACS Catal., 2013, 3, 895 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- (a) T. P. Yoon and Z. Lu, Angew. Chem., Int. Ed., 2012, 51, 10329 CrossRef PubMed; (b) T. P. Yoon, E. L. Tyson and E. P. Farney, Org. Lett., 2012, 14, 1110 CrossRef PubMed; (c) T. P. Yoon, M. A. Ischay and Z. Lu, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef PubMed; (d) T. P. Yoon, M. A. Ischay, M. E. Anzovino and J. Du, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef PubMed.
- (a) T. Naito, Chem. Pharm. Bull., 2008, 56, 1367 CrossRef CAS; (b) I. Ninomiya and T. Naito, Heterocycles, 1981, 15, 1433 CrossRef CAS.
- (a) M. Demuth, K. D. Warzecha and X. Xing, Pure Appl. Chem., 1997, 69, 109 Search PubMed; (b) H. D. Roth, T. Herbertz, H. Weng and D. Zhou, Pure Appl. Chem., 1997, 69, 809 CAS; (c) H. D. Roth, H. Weng and C. Scarlata, J. Am. Chem. Soc., 1996, 118, 10947 CrossRef; (d) M. Demuth, U. Hoffmann, Y. Gao, B. Pandey, S. Klinge, K. D. Warzecha, C. Krüger and H. D. Roth, J. Am. Chem. Soc., 1993, 115, 10358 CrossRef.
- T. P. Yoon, J. Du, L. R. Espelt and I. A. Guzei, Chem. Sci., 2011, 2, 2115 RSC.
- For reviews, see: (a) B. M. Trost and M. J. Krische, Synlett, 1998, 1 CrossRef PubMed; (b) G. C. Lloyd-Jones, Org. Biomol. Chem., 2003, 1, 215 RSC.
- (a) R. Grigg, T. R. B. Mitchell and A. Ramasubbu, J. Chem. Soc., Chem. Commun., 1979, 669 RSC; (b) K. L. Bray, J. P. H. Charmant, I. J. S. Fairlamb and G. C. Lloyd-Jones, Chem. – Eur. J., 2001, 7, 4205 CrossRef CAS; (c) G. C. Lloyd-Jones, K. L. Bray, M. P. Muñoz, P. A. Slatford, E. H. P. Tan, A. R. Tyler-Mahon and P. A. Worthington, Chem. – Eur. J., 2006, 12, 8650 CrossRef PubMed.
- (a) W. Leitner, C. Böing, J. Hahne and G. Franciò, Adv. Synth. Catal., 2008, 350, 1073 CrossRef; (b) M. Kotora, D. Nečas, M. Turský and I. Tišlerová, New J. Chem., 2006, 30, 671 RSC; (c) W. Leitner, C. Böing, J. Hahne and G. Franciò, Chem. Commun., 2005, 1456 Search PubMed; (d) W. Leitner, C. Böing, J. Hahne and G. Franciò, Adv. Synth. Catal., 2005, 347, 1537 CrossRef.
- (a) I. J. S. Fairlamb, G. P. McGlacken and F. Weissberger, Chem. Commun., 2006, 988 RSC; (b) K. Itoh, Y. Yamanoto, Y. I. Nakagai and N. Ohkoshi, J. Am. Chem. Soc., 2001, 123, 6372 CrossRef PubMed; (c) K. Itoh, Y. Yamanoto, N. Ohkoshi and M. Kameda, J. Org. Chem., 1999, 64, 2178 CrossRef PubMed.
- T. Livinghouse and S. Okamoto, J. Am. Chem. Soc., 2000, 122, 1223 CrossRef.
- T. V. RajanBabu and B. Radetich, J. Am. Chem. Soc., 1998, 120, 8007 CrossRef.
- See ref. 18 in G. C. Lloyd-Jones, K. L. Bray, J. P. H. Charmant and I. J. S. Fairlamb, Chem. – Eur. J., 2001, 7, 4205 CrossRef.
- See entry 5, Table 1 in G. C. Lloyd-Jones, K. L. Bray, I. J. S. Fairlamb, J. P. Kaiser and P. A. Slatford, Top. Catal., 2002, 19, 49 CrossRef.
- G. C. Lloyd-Jones and K. L. Bray, Eur. J. Org. Chem., 2001, 1635 Search PubMed.
- G. C. Lloyd-Jones, L. A. Evans, N. Fey, M. P. Muñoz and P. A. Slatford, Angew. Chem., Int. Ed., 2009, 48, 6262 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, spectral distribution of irradiance density for the UV lamps, experimental procedures and the characterization data of compounds. See DOI: 10.1039/c4qo00145a |
| This journal is © the Partner Organisations 2014 |