A problem solving approach for the diastereoselective synthesis of (5′S)- and (5′R)-5′,8-cyclopurine lesions†‡
Chryssostomos
Chatgilialoglu
*ab,
Carla
Ferreri
a,
Annalisa
Masi
a,
Anna
Sansone
a,
Michael A.
Terzidis
a and
Michail
Tsakos
ac
aIstituto per la Sintesi Organica e la Fotoreattività, Consiglio Nazionale delle Ricerche, Via P.Gobetti 101, 40129 Bologna, Italy. E-mail: chrys@isof.cnr.it
bInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, Patriarchon Grigorion and Neapoleos Str. GR15310, Athens, Greece
cLaboratory of Organic Chemistry, Department of Chemistry, University of Athens, Panepistimiopolis 15771, Athens, Greece
First published on 4th June 2014
Abstract
A short, efficient and high-yielding approach for the diastereoselective syntheses of the four (5′S)- and (5′R)-5′,8-cyclopurine lesions and their phosphoramidites has been developed. The intermediates prepared by these synthetic pathways provide an easy access to those modified nucleosides that constitute an important molecular library for recognition of DNA damage. With respect to previous synthesis, the key methodologies for cyclization steps and phosphoramidite synthesis were ameliorated.
Introduction
5′,8-Cyclopurines, both A and G, have been identified as lesions in human cellular DNA.1 These lesions have been proven to be highly mutagenic2–4 both in mammalian cells and in vitro and are known to accumulate with aging in a tissue specific manner, in the order: liver > kidney > brain.5 Such lesions are formed by the action of ionizing radiation generating HO˙ radicals and are named tandem lesions since the damage occurs at two sites, 2-deoxyribose and the purine base.6 It has been shown that the formation of the lesion is initiated by abstraction of the H5′ atom of the 2-deoxyribose moiety by HO˙ followed by intramolecular cyclization between C5′ and C8 and subsequent oxidation of the resulting N7-radical (Scheme 1).6,7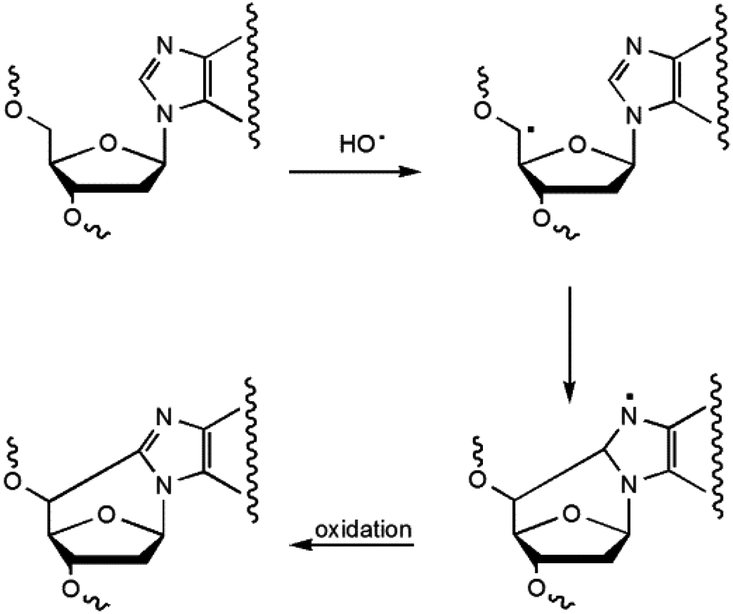 | ||
| Scheme 1 Formation of 5′,8-cyclopurines in DNA. | ||
5′,8-Cyclo-2′-deoxyadenosine (cdA) and 5′,8-cyclo-2′-deoxyguanosine (cdG) are formed both as 5′R- and 5′S-diastereoisomers (Fig. 1). The 5′,8-cyclopurine lesions are specifically repaired by nucleotide excision repair (NER), with different efficiency for the R and S forms.8,9 In order to undertake epidemiological and comparative studies on the occurrence of such lesions and to examine the effect of the above-mentioned diastereoisomers on the efficiency of known enzymatic repair systems, it is extremely important to establish effective synthetic and analytical methods, as well as to improve the design and use of biomimetic systems for modelling the biological occurrence of the lesions and performing mechanistic studies.
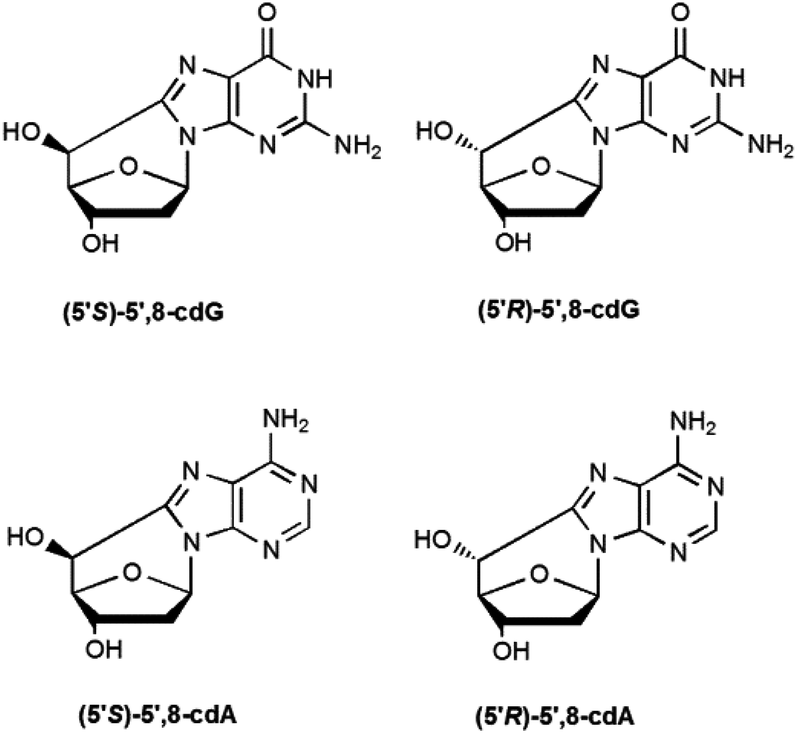 | ||
| Fig. 1 Chemical structures of the cdA and cdG stereoisomeric lesions. | ||
However, the synthetic routes developed so far to obtain the four 5′,8-cyclopurines display some limitations due to several steps and low yields. Furthermore, the R and S diastereoisomers are difficult to separate from each other. The huge interest raised in the study of these lesions during the last few years has then prompted our research toward a simplified access to these intermediates offering shorter synthetic pathways with higher yields. To this aim we have developed synthetic routes for the four 5′,8-cyclopurine nucleosides,10,11 an important molecular library that can be used for the production of analytical standards for the recognition of DNA damage. We report herein the optimization of the synthetic pathways for the diastereoselective syntheses of the four (5′S)- and (5′R)-5′,8-cyclopurine lesions and their phosphoramidites used for the automated synthesis of model oligonucleotides (ODNs).
Results and discussion
Problem solving approach for the synthesis of (5′R)- and (5′S)-5′,8-cyclo-2′-deoxyguanosine diastereoisomers
Previously reported protocols for the preparation of the (5′R)- and (5′S)-cdG phosphoramidites started from the N2-isobutyroyl-2′-deoxyguanosine.12 This methodology had some limitations due to the low yield and several steps that were necessary to obtain both diastereoisomers. Furthermore, the synthesis of the unprotected nucleosides, useful also as analytical standard for HPLC/MS analysis, offers some challenges. Among the strategies described in the literature,13,14 one begins with the photolysis of 8-bromo-2′-deoxyguanosine in aqueous solutions with a 26% yield and a 5′S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5′R ratio of 1:8.14 Following the latter methodology apart from the problem of the low yield, several attempts to selectively protect one of the two secondary OH groups were unsuccessful. More recently, we reported a new methodology based on a radical cascade protocol that afforded the key derivatives that can be used for the preparation of the cyclopurine phosphoramidites, compatible with the automated synthesis of oligonucleotides (Scheme 2).11 This route represented a good approach to obtain the intermediate (5′S)-6 with high yield, while the yield of compound (5′R)-7 needed to be further improved. To do this, on the basis of the above direct synthesis, we performed the Mitsunobu reaction to invert the C5′ configuration, going from compound 6 to 7 (Scheme 2), but all attempts failed, probably due to the basic character of the double bond C–N in the dimethyl formamido group (DMF) preventing the reaction to occur.
:5′R ratio of 1:8.14 Following the latter methodology apart from the problem of the low yield, several attempts to selectively protect one of the two secondary OH groups were unsuccessful. More recently, we reported a new methodology based on a radical cascade protocol that afforded the key derivatives that can be used for the preparation of the cyclopurine phosphoramidites, compatible with the automated synthesis of oligonucleotides (Scheme 2).11 This route represented a good approach to obtain the intermediate (5′S)-6 with high yield, while the yield of compound (5′R)-7 needed to be further improved. To do this, on the basis of the above direct synthesis, we performed the Mitsunobu reaction to invert the C5′ configuration, going from compound 6 to 7 (Scheme 2), but all attempts failed, probably due to the basic character of the double bond C–N in the dimethyl formamido group (DMF) preventing the reaction to occur.
 | ||
| Scheme 2 Synthesis of (5′S)-cdG (6) and (5′R)-cdG (7) by the radical cascade protocol. Steps: (a) Bu3SnH, AIBN ACN, 2.5 h, reflux, 48%, (5′S)/(5′R) 6.5:1; (b) (CH3)2NCH(OCH3)2, THF, 2.5 h, rt; (c) TBAF, THF, 30 min, −18 °C, 77% (5′S)-6 and 83% (5′R)-7. | ||
Therefore, the problem solving approach was to design a different synthetic strategy by changing the amino group protection from dimethyl formamido to isobutyroyl, thus forming an amidic bond with a lower electron donating character.
Compound 2 was protected using isobutyroyl chloride, followed by a selective cleavage of the triethylsilyl ether after treatment with TBAF at low temperature affording the epimer 9 with a 83% yield. The Mitsunobu reaction, using 4-nitrobenzoic acid and diisopropylazodicarboxylate (DIAD), afforded 10 and upon hydrolysis of the p-nitrobenzoate group, the (5′R)- epimer 11 was isolated pure with a 91% yield (Scheme 3).
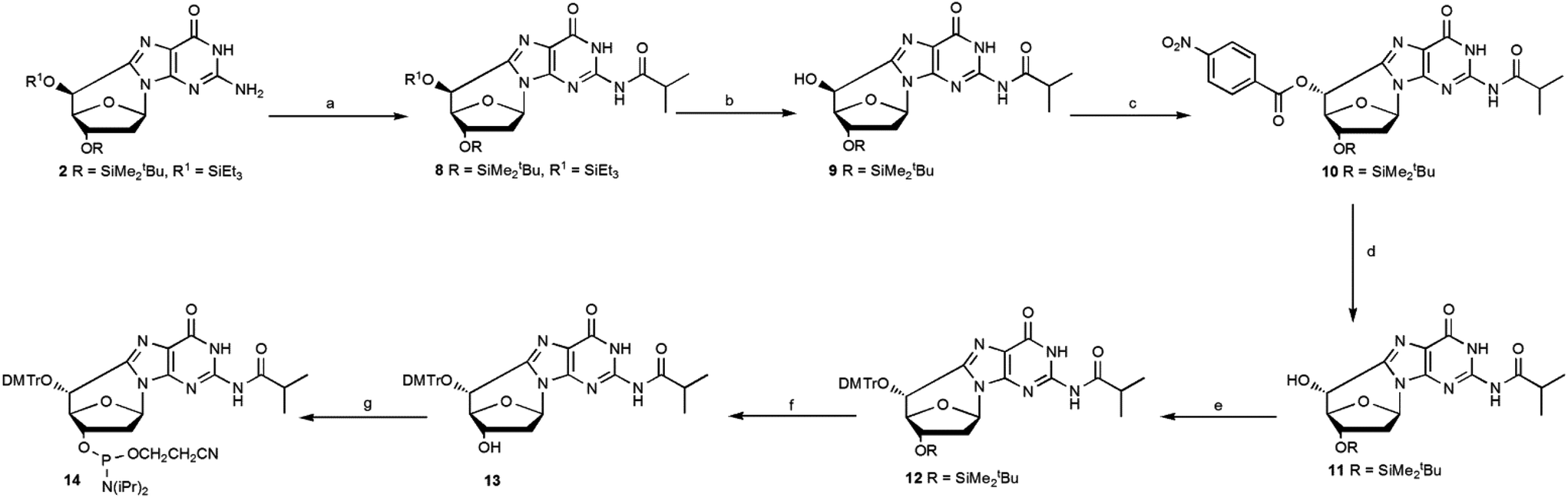 | ||
| Scheme 3 Mitsunobu reaction for the inversion of the C5′ configuration of the (5′S)-cdG diastereoisomer and synthesis of the (5′R)-cdG phosphoramidite (14). Steps: (a) iPrC(O)Cl, DMAP, py, 85%; (b) TBAF, THF, −20 °C, 25 min, 98%; (c) PPh3, DIAD, 4-NBA, THF, r.t., 92%; (d) LiOH, THF, r.t., 99%; (e) DMTrCl, dry py, 70 °C, 6 h, 85%; (f) TBAF, THF, 15 min, r.t., 92%; (g) iPr2NP(Cl)O(CH2)2CN, DIEA, dry CH2Cl2, Ar, 2.5 h, 82% (see ESI‡ for details). | ||
Compound 11 was converted into the phosphoramidite building block 14, after optimization of the standard procedures (Table 1).15
| Synthesis of phosphoramidite (5′R)-cdG 14 | ||||
|---|---|---|---|---|
| Compound | Present work | Yield % | Romieu et al. conditions | Yield% |
| 12 | DMTrCl (4 eq.) t = 6 h, T = 70 °C | 85 | DMTrCl (3 eq.) t = 4 h, T = 80 °C | 65 |
| Solv. dry Py | Solv. dry Py | |||
| 13 | TBAF–THF 1 M (2.3 eq.) t = 15 min, T = r.t. | 92 | TBAF–THF 1 M (2 eq.) t = 4 h, T = r.t. | 75 |
| 14 | DIEA (3 eq.) | 82 | DIEA (1.9 eq.) | 78 |
| CEP-Cl (3 eq.) t = 2.5 h, T = r.t. | CEP-Cl (1.1 eq.) t = 30 min, T = r.t. | |||
| Solv. dry CH2Cl2 | Solv. dry CH2Cl2 | |||
The overall yield, from 2 to 14, according to the methodologies described herein is 30%, 4 times higher than the previous one reported.12 In particular, the synthetic Scheme 3 provides very useful solutions for an easy access to the R epimer as well as to obtain higher amounts of (5′R)-5′,8-cyclo-deoxyguanosine and its phosphoramidite.
The overall yield of the phosphoramidite (5′S)-cdG 17 was improved by optimizing the conditions of the steps from 6 to 17 (Scheme 4).
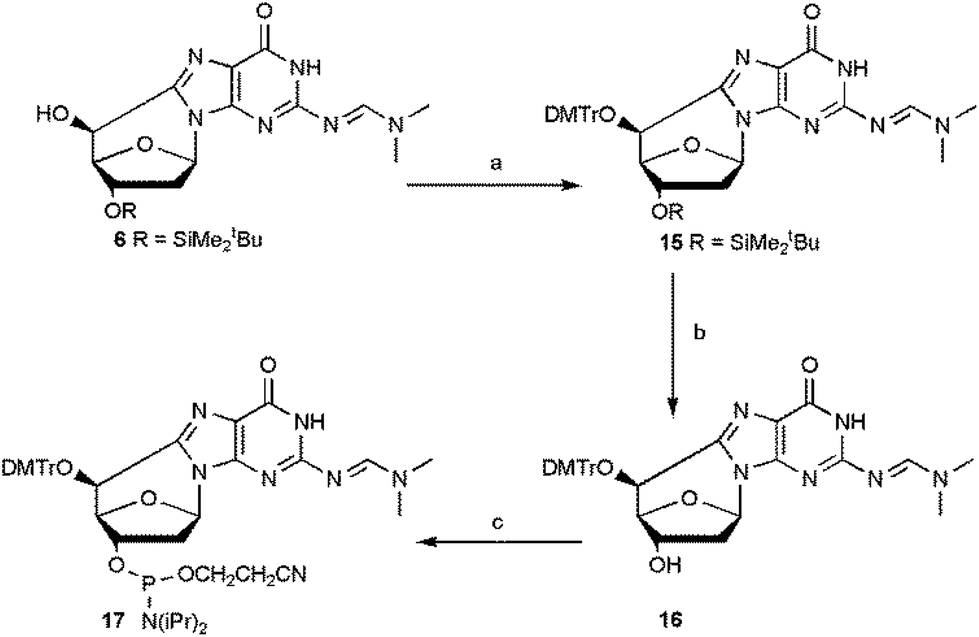 | ||
| Scheme 4 Synthesis of (5′S)-cdG (17) phosphoramidite. Steps: (a) DMTrCl, dry py, 70 °C, 6 h, 74%; (b) TBAF, THF, 15 min, 92%; (c) iPr2NP(Cl)O(CH2)2CN (CEP-Cl), DIEA, dry CH2Cl2, Ar, 1 h, 82% (see ESI‡ for details). | ||
Problem solving approach for the synthesis of (5′R)- and (5′S)-5′,8-cyclo-2′-deoxyadenosine diastereoisomers
Romieu et al.12 developed the synthesis of (5′S)-5′,8-cyclo-deoxyadenosine and its phosphoramidite, starting from N6-benzoyl-2′-deoxyadenosine following a multistep synthetic pathway with a low overall yield of 3.5%. The synthesis of the (5′R) diastereoisomer was obtained by inversion of the C5′ configuration in the (5′S)-cdA intermediate using the Corey method with an overall yield of 1.6%.16 The low yield and the length of the synthetic pathway are the limiting steps for accessing these lesions. In the literature some attempts are reported to optimize the synthetic pathways of cycloadenosine lesions,10,16 and we thought a route to improve the overall yield (Scheme 5).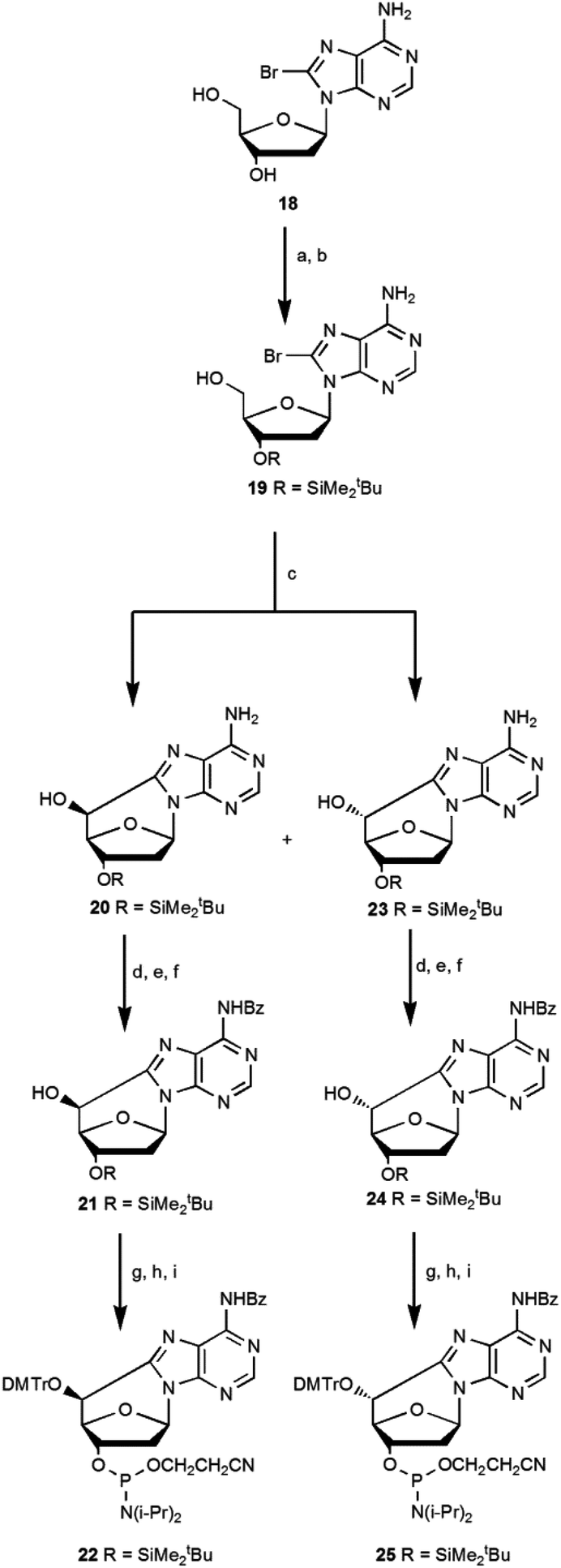 | ||
| Scheme 5 A new synthetic approach for the synthesis of (5′S)-cdA (22) and (5′R)-cdA (25) phosphoramidite. Steps: (a) TBDMSCl, imid, DMF, 1.5 h, 95%; (b) TFA–H2O 1:1, THF, 0 °C, 1.1 h, 65%; (c) UV (125 W medium pressure Hg lamp), ACN–H2O 1:1, Ar, 1.2 h, (5′R)/(5′S) 7:3 (39% 23, 16% 20); (d) TES-Cl, imid, dry CH2Cl2, Ar, 30 min, 90% (R) and 90% (S); (e) benzoyl chloride, py, Et3N, 0 °C, 3 h, 69% (R) and 70% (S); (f) TBAF, THF, 4 h, 95% (R) and 95% (S); (g) DMTrCl, Et3N, dry py, 70 °C, 18 h, 87% (R) and 74% (S); (h) TBAF, THF, 1 h, 89% (R) and 88% (S); (i) iPr2NP(Cl)O(CH2)2CN, DIEA, dry CH2Cl2, Ar, 40 min, 85% (R) and 88% (S) (see ESI‡ for details). | ||
In our current approach 2′-deoxyadenosine was brominated using Br2 in dioxane (18), then the 5′OH and 3′OH groups were protected with tert-butyldimethylsilyl chloride (TBDMSCl) and selectively deprotected at 5′OH using trifluoroacetic acid (TFA) to give the mono protected compound 19. Its conversion by UV photolysis generated the cyclopurines 5′,8-cdA 20 and 23 in a 5′S/5′R ratio of 3:7 with a respective yield, after purification, of 16% and 39%.10 The above described one-pot radical cascade process represents a very convenient way to have the two epimers R and S more readily accessible, with shorter synthetic pathways endowed with higher yields, and importantly the mono protected derivatives (20, 23) found to be easily separable on silica. It is worth noting that the unprotected exocyclic amino group facilitated the cyclization and subsequent separation of diastereoisomers.
The free hydroxyl group at C5′ of the compounds 20 and 23 was protected with triethylsilyl chloride, and after the coupling of the exocyclic amino group with benzoyl chloride, the selective deprotection of the Et3SiO moiety was achieved for each diastereomer. Compounds 21 and 24 were converted into phosphoramidite building blocks 22 and 25 after optimization of the standard procedures15 and resulted in an overall yield of 9.3% for the (5′R)-25 diastereoisomer and 3.3% for the (5′S)-22 diastereoisomer.
The new approach exploiting alternative synthetic strategies allowed us to improve the overall yield of the (5′R)-cdA lesion 5 times compared to the previously reported procedures.12,16 The key methodologies were ameliorated for the cyclization step and the phosphoramidite synthesis. In order to improve the yields of the final steps, we tuned appropriately the reaction conditions, as summarized in Table 2.
| Synthesis of phosphoramidite (5′S)-cdA (22) | ||||
|---|---|---|---|---|
| Step | Present work | Yield% | Romieu et al. conditions | Yield% |
| g | DMTrCl (3 eq.) t = 18 h, T = 70 °C | 74 | DMTrCl (2 eq.) t = 4 h, T = 70 °C | 60 |
| Solv. dry Py/Et3N | Solv. dry Py | |||
| h | TBAF–THF 1 M (2.5 eq.) t = 3 h, T = r.t. | 88 | TBAF–THF 1 M (2 eq.) t = 4 h, T = r.t. | 72 |
| i | DIEA (3 eq.) | 88 | DIEA (0.5 eq.) | 62 |
| CEP-Cl (3 eq.) t = 30 min, T = r.t. | CEP-Cl (1.3 eq.) t = 4 h, T = r.t. | |||
| Solv. dry CH2Cl2 | Solv. dry CH2Cl2 | |||
| Synthesis of phosphoramidite (5′R)-cdA (25) | ||||
|---|---|---|---|---|
| Step | Present work | Yield% | Romieu et al. conditions | Yield% |
| g | DMTrCl (3 eq.) t = 18 h, T = 70 °C | 87 | DMTrCl (3 eq.) t = 4 h, T = 80 °C | 47 |
| Solv. dry Py/Et3N | Solv. dry Py | |||
| h | TBAF–THF 1 M (1.3 eq.) t = 1 h, T = r.t. | 89 | TBAF–THF 1 M (2 eq.) t = 4 h, T = r.t. | 59 |
| i | DIEA (3 eq.) | 85 | DIEA (1.9 eq.) | 80 |
| CEP-Cl (3 eq.) t = 30 min, T = r.t. | CEP-Cl (1.1 eq.) t = 30 min, T = r.t. | |||
| Solv. dry CH2Cl2 | Solv. dry CH2Cl2 | |||
Our routes to (5′S)- and (5′R)-5′,8-cyclo-2′-deoxypurine represent an important milestone in the subsequent investigations of biochemical, biological and biophysical implications. The phosphoramidite derivatives were incorporated, by automated synthesis following standard procedures,12 into specific sequences of oligodeoxynucleotides (ODNs), which represent the biomimetic models of DNA. In order to optimize both the yield at the step of coupling the phosphoramidite of the modified monomers 14, 17, 22 and 25, and also the overall yield of the oligonucleotide synthesis, the following changes were introduced, with respect to the standard automated synthesis protocol. The concentration of the 5′,8-cyclonucleoside phosphoramidite was increased from 0.061 M to 0.074 M; the coupling time was increased from 1.6 min to 13 min for each coupling throughout the oligonucleotide synthesis. The removal conditions of the 5′-DMTr-group from the 5′,8-cyclonucleotide as well as from the next nucleotide d(A, C, G, T) added in the oligonucleotide sequence did not require any change (see ESI‡).
We found that the total yields of the ODNs containing (5′S)-cdA or (5′S)-cdG were always higher than the total yields of the ODNs containing (5′R)-cdA or (5′R)-cdG. Moreover, the ratios between the two yield values (S/R) were comparable (Fig. 2).
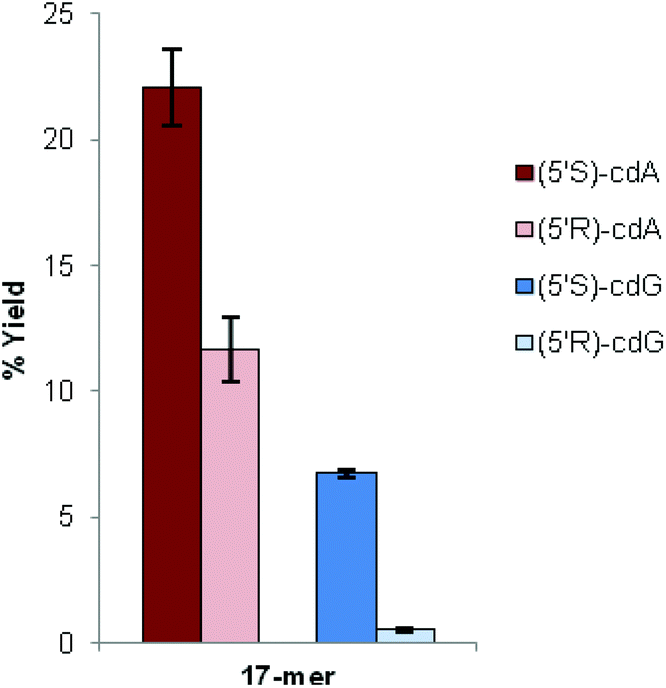 | ||
| Fig. 2 Comparison of the total yield of the oligonucleotide (17-mer) containing (5′S)-cdA or (5′R)-cdA and (5′S)-cdG or (5′R)-cdG. Each ODN was injected on HPLC three times, twice for each sample, and the concentration of the single final product was measured. | ||
In the case of the sequence containing cdG lesions, the coupling efficiency seems to be even more affected in cdG than in cdA, since in cdG the total yield of the ODNs containing the S diastereoisomer is significantly higher than that of the R one (Fig. 2). From the data obtained for oligonucleotide synthesis it can be highlighted that the stereochemistry of the cyclopurine lesions significantly influences the coupling yield and thus the total yield of the full-length oligonucleotide. According to these results, the improvement of the yields of both enantiomers (5′R)-cdG and (5′R)-cdA is a crucial achievement in order to provide sufficient quantities of oligonucleotides for further studies.
Conclusions
In summary, we developed short and efficient syntheses of building blocks (5′R)-cdG (14), (5′S)-cdG (17), (5′R)-cdA (25) and (5′S)-cdA (22) which overcome the limitations present so far for the assessment of their biological and medical significance. Indeed, these lesions gain more and more attention for biological roles and disease implications, also because they can be used as reliable biomarkers of free radical stress in biological samples, with an advantage compared to the most known marker 8-oxo-2′-deoxyguanosine (8-oxodG), since it has been argued that oxidative artefacts may form during samples work-up.17 Having a library of the four diastereoisomers (2, 3, 20, 23) in our hands, it can be used for the production of analytical standards for individuation of lesions in biological samples of different origins, and as the starting material for the preparation of the corresponding phosphoramidites to access the automated synthesis of model ODNs containing such lesions. Further biochemical, biological and biophysical studies will clarify the role of these lesions, expanding the understanding of the DNA modifications as signalling or damage in the overall metabolic regulation of cell life.Acknowledgements
Financial support from the Ministero dell'Istruzione, dell'Università della Ricerca (PRIN 2009K3RH7N_002) and Marie Curie Intra-European Fellowship (CYCLOGUO-298555) are gratefully acknowledged. The sponsorship of the COST Action CM1201 “Biomimetic Radical Chemistry” is gratefully acknowledged in particular for the STSM grant to MT.References
- C. Chatgilialoglu, C. Ferreri and M. A. Terzidis, Chem. Soc. Rev., 2011, 40, 1368 RSC.
- B. Yuan, J. Wang, H. Cao, R. Sun and Y. Wang, Nucleic Acids Res., 2011, 39, 5945 CrossRef PubMed.
- C. You, X. Dai, B. Yuan, J. Wang, J. Wang, P. J. Brooks, L. J. Niedernhofer and Y. Wang, Nat. Chem. Biol., 2012, 8, 817 CrossRef PubMed.
- V. Pednekar, S. Weerasooriya, V. P. Jasti and A. K. Basu, Chem. Res. Toxicol., 2014, 27, 200 CrossRef PubMed.
- J. Wang, C. L. Clauson, P. D. Robbins, L. J. Niedernhofer and Y. Wang, Aging Cell, 2012, 11, 714 CrossRef PubMed.
- N. Belmadoui, F. Boussicault, M. Guerra, J. L. Ravanat, C. Chatgilialoglu and J. Cadet, Org. Biomol. Chem., 2010, 8, 3211 Search PubMed.
- F. Boussicault, P. Kaloudis, C. Caminal, Q. G. Mulazzani and C. Chatgilialoglu, J. Am Chem. Soc., 2008, 130, 8377 CrossRef PubMed.
- I. Kuraoka, C. Bender, A. Romieu, J. Cadet, R. D. Wood and T. Lindahl, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3832 CrossRef PubMed.
- K. Kropachev, S. Ding, M. A. Terzidis, A. Masi, Z. Liu, Y. Cai, M. Kolbanovskiy, C. Chatgilialoglu, S. Broyde, N. E. Geancitov and V. Shafirovich, Nucleic Acids Res., 2014, 42, 5020 CrossRef PubMed.
- M. L. Navacchia, C. Chatgilialoglu and P. C. Montevecchi, J. Org. Chem., 2006, 71, 4445 CrossRef PubMed.
- M. A. Terzidis and C. Chatgilialoglu, Aust. J. Chem., 2013, 66, 330 CrossRef.
- A. Romieu, D. Gasparutto and J. Cadet, Chem. Res. Toxicol., 1999, 12, 412 CrossRef PubMed.
- A. Manetto, D. Georganakis, L. Leondiadis, T. Gimisis, P. Mayer, T. Carell and C. Chatgilialoglu, J. Org. Chem., 2007, 72, 3659 CrossRef PubMed.
- C. Chatgilialoglu, R. Bazzanini, L. B. Jimenez and M. A. Miranda, Chem. Res. Toxicol., 2007, 20, 1820 CrossRef PubMed.
- M. H. Caruthers, Acc. Chem. Res., 1991, 24, 278 CrossRef.
- A. Romieu, D. Gasparutto, D. Molko and J. Cadet, J. Org. Chem., 1998, 63, 5245 CrossRef.
- J. Cadet, T. Douki and J. L. Ravanat, Mutat. Res., 2011, 711, 3 CrossRef PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria in recognition of his outstanding achievement, an authentic source of inspiration for friends and colleagues. |
| ‡ Electronic supplementary information (ESI) available: Materials including experimental procedures and detailed characterization data for all new compounds. See DOI: 10.1039/c4qo00133h |
| This journal is © the Partner Organisations 2014 |