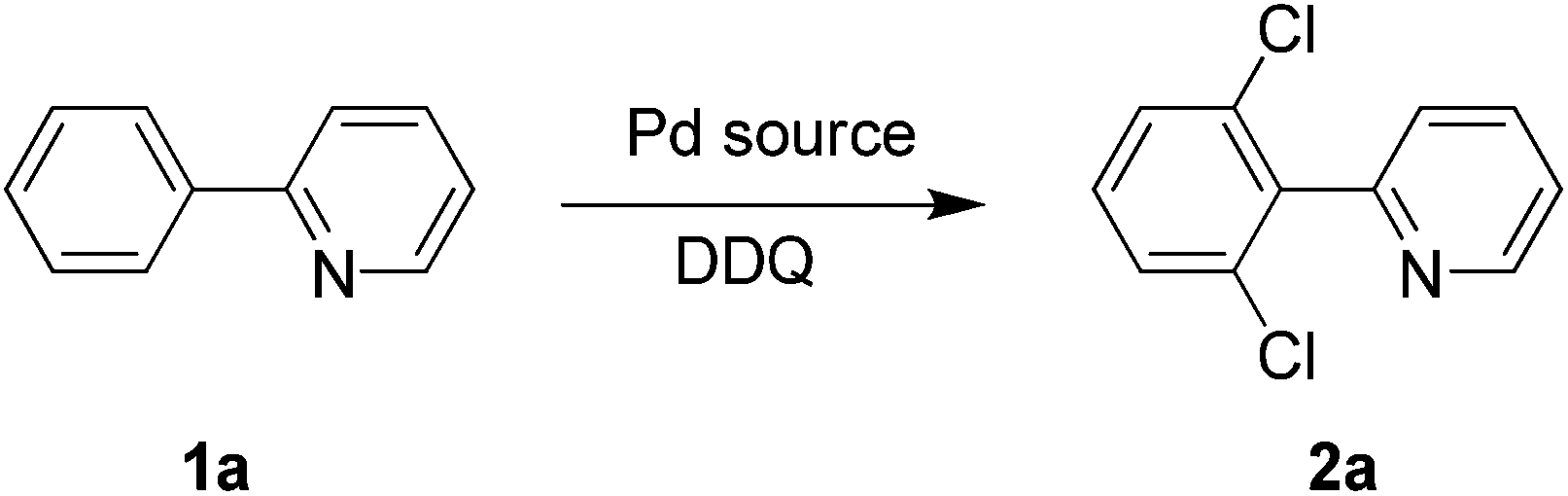
DDQ: the chlorinating reagent and oxidant for the ligand-directed ortho-chlorination of 2-arylpyridines†
Qian
Zhang
,
Fan
Yang
* and
Yangjie
Wu
*
The College of Chemistry and Molecular Engineering, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou 450052, People's Republic of China. E-mail: yangf@zzu.edu.cn; wyj@zzu.edu.cn
First published on 4th June 2014
Abstract
A new and simple protocol for palladium-catalyzed ligand-directed ortho-chlorination of 2-arylpyridines with DDQ was developed, generating the chlorinated products in good to excellent yields. Note that DDQ played dual roles as both the chlorinating reagent and oxidant in this reaction. Moreover, high regioselectivity was observed for 2-arylpyridines bearing a meta-substituent in the aryl ring moiety, and the chlorination could take place at the less sterically hindered ortho-C–H bond.
Aryl halides are one type of the prominent coupling partners in transition metal-catalyzed C–C, C–N, C–O, and C–S bond forming cross-coupling reactions, thereby becoming one of the most important organic intermediates and structural motifs in many natural products and synthetic drugs.1,2 In particular, aryl chlorides are often much cheaper than aryl bromides or iodides, and 85% pharmaceuticals are manufactured using chlorides.3 Nowadays, the most prevalent strategies for preparing aromatic chlorides are electrophilic aromatic substitution (EAS) or two-step directed ortho-lithiation/halogenation.4 However, both the routes suffered from some disadvantages, including low regioselectivity as well as tedious and sometimes dangerous procedures.
To address these limitations, transition metal-catalyzed ligand-directed ortho-halogenation has emerged as one of the most facile and efficient protocols in recent years.3c,5 Especially, in 2003, the first palladium-catalyzed direct ortho-chlorination of the C–H bond was introduced by Sanford and co-workers, who utilized NCS and air as the chlorinating reagent and oxidant, respectively.5a Since then, significant efforts have been made towards such ortho-chlorination by the groups of Shi, Yu, Glorius, and others, employing MClx (M = Cu, Ca), Cl2CHCHCl2, or NCS as the chlorinating reagent and Cu(OAc)2, Cu(OTFA)2, O2, or Na2S2O8 as the oxidant (Scheme 1).3c,5 Nevertheless, the reported procedures traditionally employed independent chlorinating and oxidative reagents, which would make the reaction conditions a little sophisticated and also did not conform to the viewpoint of atom-economy. Thus, simplifying the reaction conditions and developing a simpler and more facile protocol for the regioselective ortho-chlorination would be urgent and highly desirable.
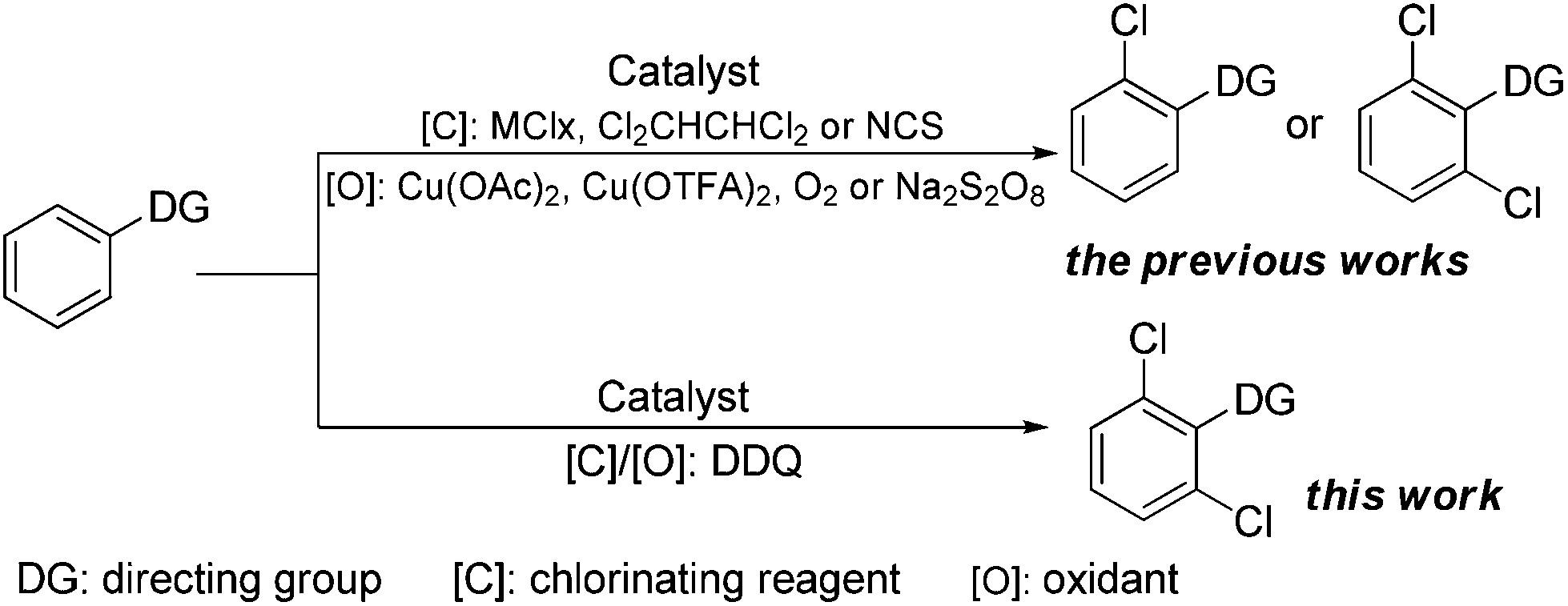 | ||
| Scheme 1 Transition metal-catalyzed direct ortho-acylation. | ||
DDQ (2,3-dichloro-5,6-dicyano-p-benzoquinone), as a one-electron oxidant giving a radical anion, has wide applications in the oxidation of steroid ketones, hydroaromatic compounds, alcohols, phenols, and heterocycles.6 However, DDQ contains two chlorine atoms and may act as a potential chlorinating source. Therefore, we reasoned that DDQ would act as both the chlorinating reagent and the oxidant, thus providing a new, simple and efficient way to obtain highly regioselective ortho-chlorinated arenes.
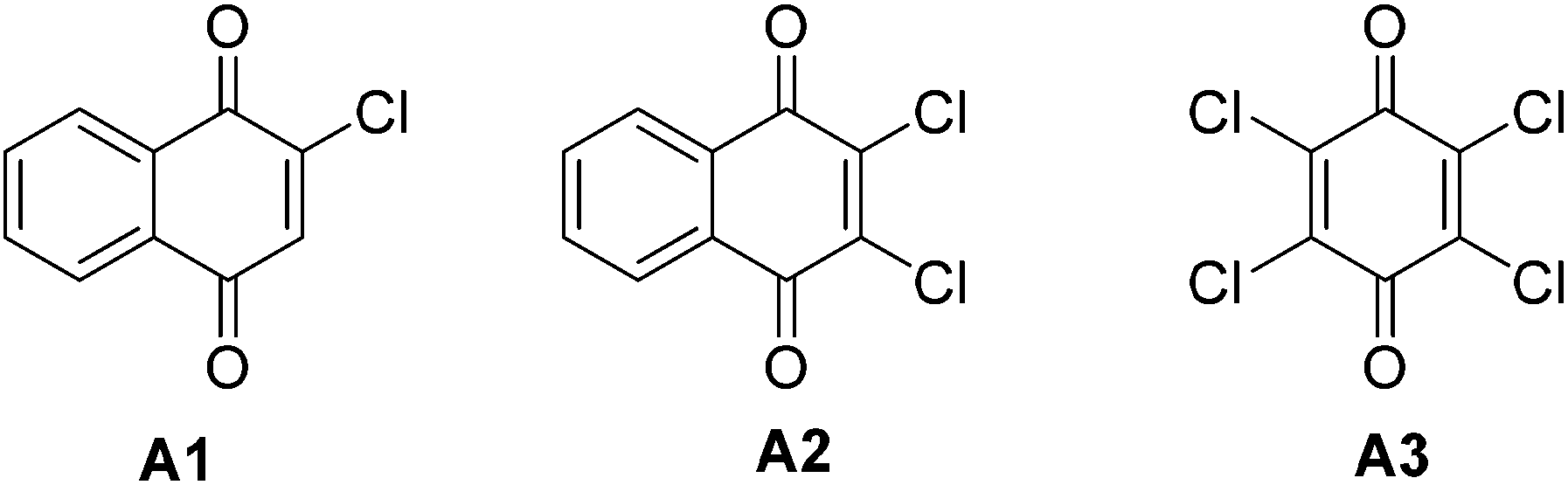
We started the optimization process by performing the chlorination of 2-phenylpyridine (1a) in DMSO under a nitrogen atmosphere at 120 °C for 3 h, and gratifyingly, the dichlorinated product was generated albeit with a low yield of 10% (Table 1, entry 1). The solvent effect (e.g., NMP, H2O, HOAc, toluene, chlorobenzene and DMF) was then screened, and to our delight, the DMF could deliver the dichlorinated product in up to 88% isolated yield (Table 1, entry 7). Some controlling experiments were also conducted such as adding water to the reaction, reducing the amount of DDQ to 2 equiv. or the reaction temperature to 100 °C, prolonging the reaction time to 5 h, and performing the reaction under air, but unfortunately, no better results were obtained (Table 1, entries 8–12). Other commercially available palladium source such as Pd(OAc)2 and Pd2dba3 were also checked, and both of them did not exhibit higher catalytic activity (Table 1, entries 13 and 14). Finally, three DDQ analogues (e.g., 2-chlorobenzoquinone, 2,3-dichloro-1,4-naphthoquinone and chloranil) were prepared and applied to the chlorination of 2-phenylpyridine (1a), but the reactions did not afford the product in better yields, which proves that DDQ played an irreplaceable role for the successful reaction (Table 1, entries 15–17).
|
|
||||
|---|---|---|---|---|
| Entry | Palladium source (mol%) | T (°C) | Solvent | Yieldb (%) |
 a Reaction conditions: 1a (0.3 mmol), palladium catalyst, and DDQ (3 equiv.) in 2 mL of solvent under a nitrogen atmosphere at 120 °C for 3 h.
b Isolated yield.
c DDQ (2 equiv.).
d For 5 h.
e Under air.
f 2-Chlorobenzoquinone (A1) (3 equiv.) was used as the chlorinating reagent.
g 2,3-Dichloro-1,4-naphthoquinone (A2) (3 equiv.) was used as the chlorinating reagent.
h Chloranil (A3) (3 equiv.) was used as the chlorinating reagent.
a Reaction conditions: 1a (0.3 mmol), palladium catalyst, and DDQ (3 equiv.) in 2 mL of solvent under a nitrogen atmosphere at 120 °C for 3 h.
b Isolated yield.
c DDQ (2 equiv.).
d For 5 h.
e Under air.
f 2-Chlorobenzoquinone (A1) (3 equiv.) was used as the chlorinating reagent.
g 2,3-Dichloro-1,4-naphthoquinone (A2) (3 equiv.) was used as the chlorinating reagent.
h Chloranil (A3) (3 equiv.) was used as the chlorinating reagent.
|
||||
| 1 | PdCl2 (5) | 120 | DMSO | 10 |
| 2 | PdCl2 (5) | 120 | NMP | 23 |
| 3 | PdCl2 (5) | Reflux | H2O | 0 |
| 4 | PdCl2 (5) | 120 | HOAc | 0 |
| 5 | PdCl2 (5) | Reflux | Toluene | 56 |
| 6 | PdCl2 (5) | 120 | Chlorobenzene | 70 |
| 7 | PdCl2 (5) | 120 | DMF | 88 |
| 8 | PdCl2 (5) | 120 | DMF–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
62 |
| 9c | PdCl2 (5) | 120 | DMF | 55 |
| 10 | PdCl2 (5) | 100 | DMF | 56 |
| 11d | PdCl2 (5) | 120 | DMF | 89 |
| 12e | PdCl2 (5) | 120 | DMF | 77 |
| 13 | Pd(OAc)2 (5) | 120 | DMF | 68 |
| 14 | Pd2dba3 (2.5) | 120 | DMF | 57 |
| 15f | PdCl2 (5) | 120 | DMF | 0 |
| 16g | PdCl2 (5) | 120 | DMF | 0 |
| 17h | PdCl2 (5) | 120 | DMF | 12 |
Under the optimized reaction conditions, the scope of 2-arylpyridines was explored and is summarized as Table 2. Generally, this chlorination could tolerate various functional groups (e.g., RO, F, Br and EtOOC), affording the desired products in moderate to excellent yields. Notably, the chlorination showed high regioselectivity for the substrates containing a meta-substituent in the benzene moiety, and the reaction could take place at less sterically hindered ortho-C–H bond, affording the monochlorinated products in good yields (Table 2, 2d–2h). On the other hand, electronic effect has influence on this reaction and the substrates bearing an electron-donating group in the benzene moiety could give the desired products in slightly higher yields than those bearing an electron-neutral and electron-withdrawing group in the benzene moiety. For example, the substrate bearing an electron-donating group (MeO) in the benzene moiety could be converted to the dichlorinated product in a yield of up to 99% (Table 2, 2j). Meanwhile, when the benzene moiety possessed a strong electron-withdrawing group (EtOCO), the desired products could be obtained in moderate yields (Table 2, 2t and 2u). The molecular structure of the dichlorinated product (2j) was unambiguously determined by the single crystal X-ray diffraction study (Fig. 1).7
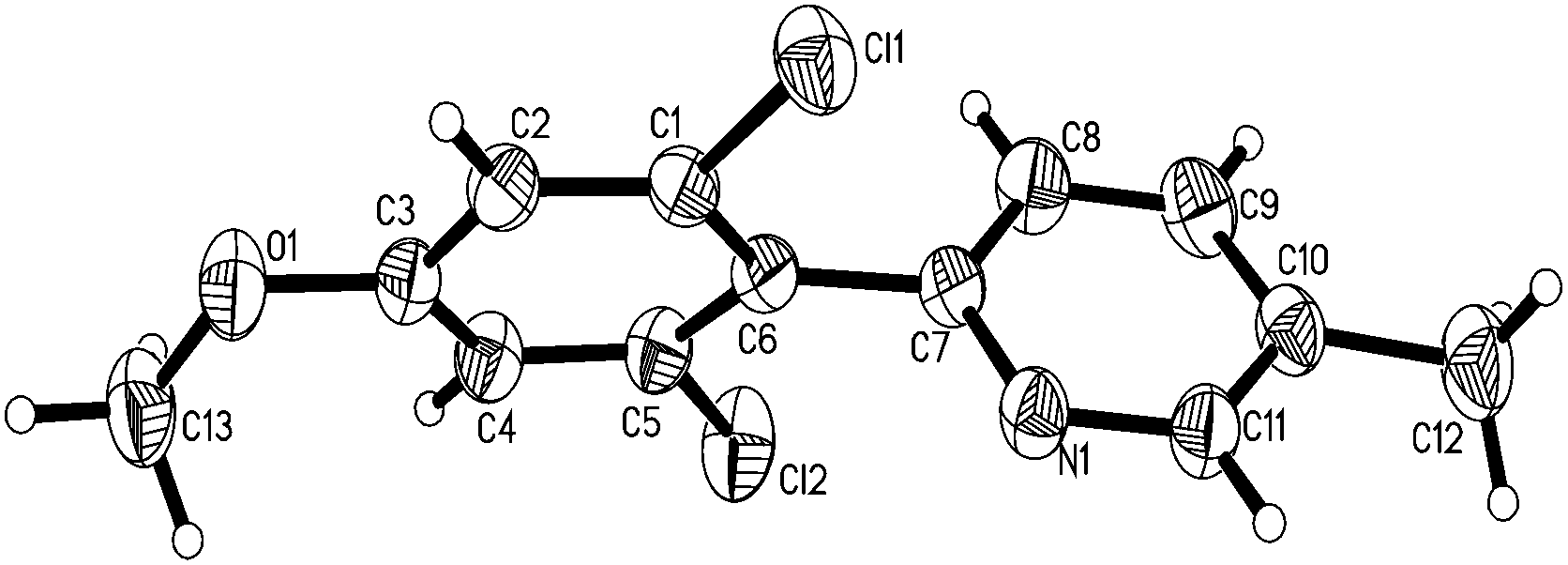 | ||
| Fig. 1 Molecular structure of 2j. | ||
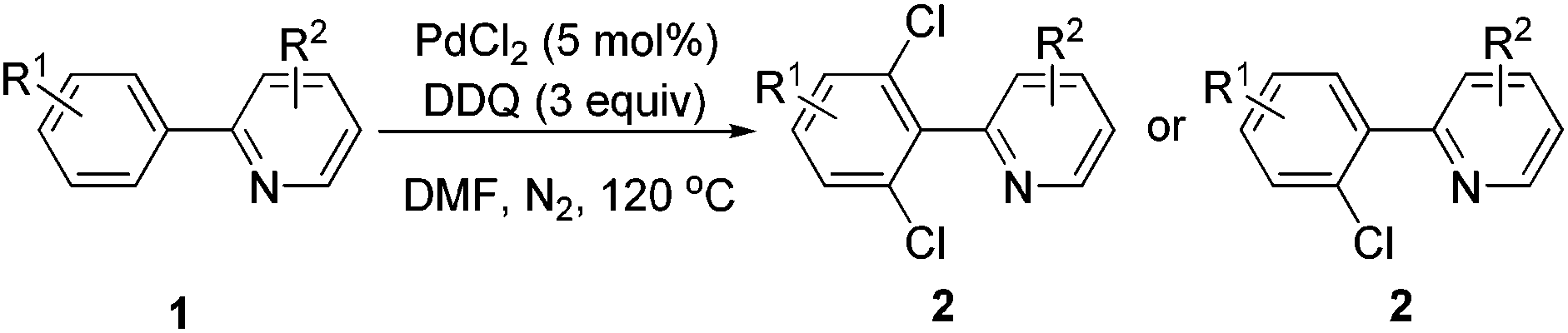
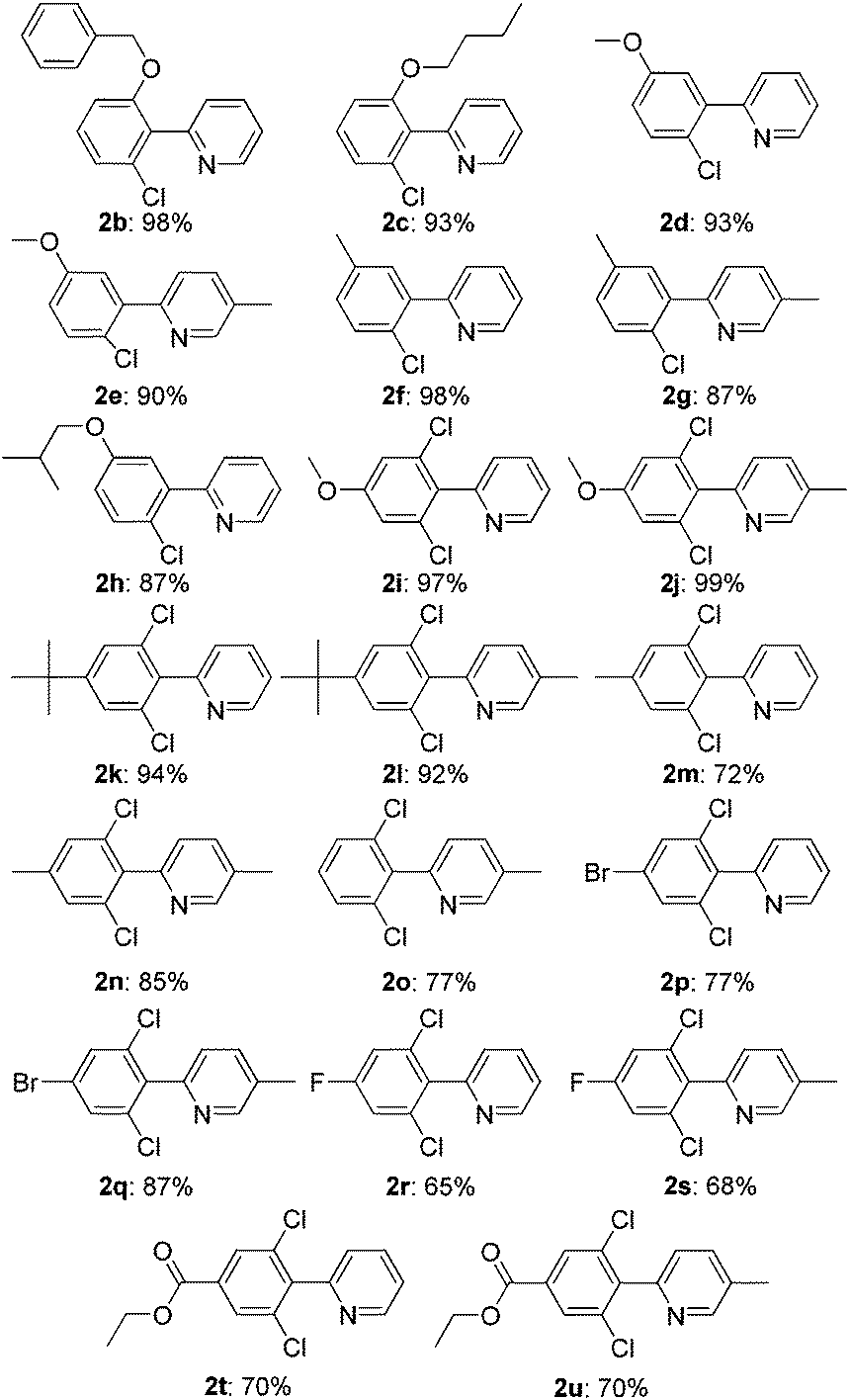
The scope of this ortho-chlorination could also be extended to heterocycles such as benzo[h]quinoline and 2-thienylpyridine derivatives, and all of them could provide the corresponding products in good to excellent yields (Scheme 3).
In addition, arylboronates are valuable and robust organic intermediates, since they can be utilized as the coupling partners in many catalytic reactions.8 An alternative and elegant synthesis of arylboronates could be fulfilled via a two-step reaction of ligand-directed ortho-chlorination/borylation to afford the ortho-borylated product in a moderate yield of 67% (Scheme 4).
A tentative mechanism for the palladium-catalyzed ligand-directed ortho-C–H chlorination is depicted in Scheme 2. The reaction would be initiated by ortho-cyclometalation of the substrate with PdCl2 to form the palladacycle I. Then, the reaction of DDQ with the intermediate I (the chlorinating step) took place, affording the oxidative addition product as the intermediate II. Finally, the reductive elimination of the intermediate II would lead to the desired product and the reductive product of DDQ was determined by GC-MS (see ESI†), regenerating the active palladium species to fulfil the catalytic cycle.
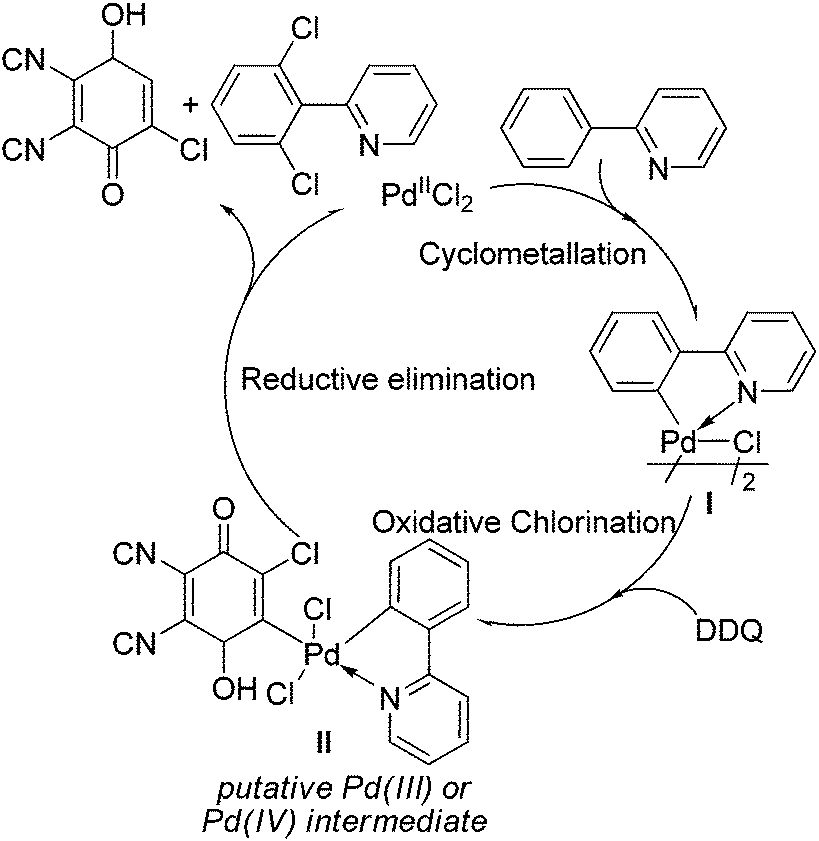 | ||
| Scheme 2 Proposed mechanism for catalytic ortho-C–H chlorination. | ||
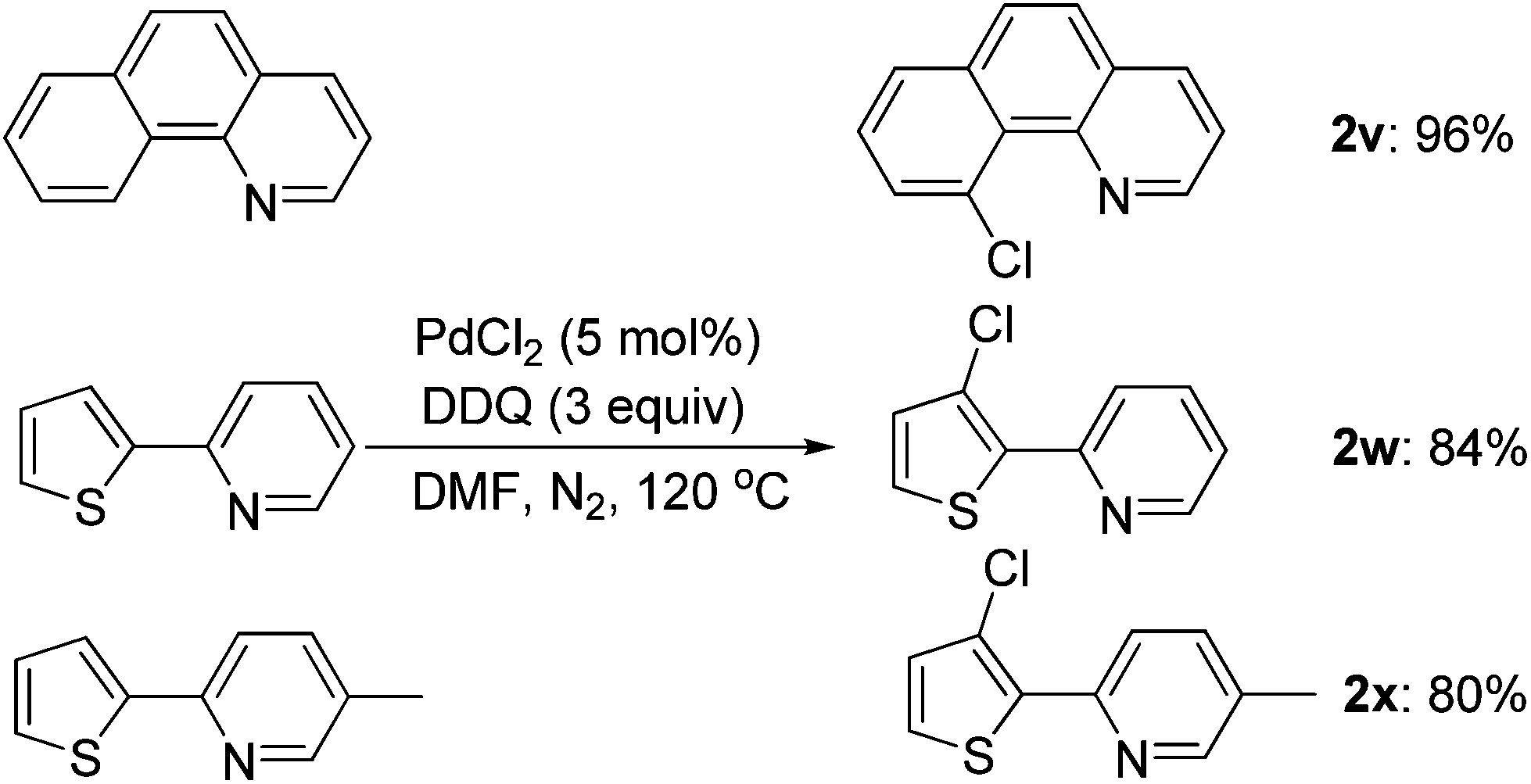 | ||
| Scheme 3 The palladium-catalyzed ortho-chlorination of heterocycles. | ||
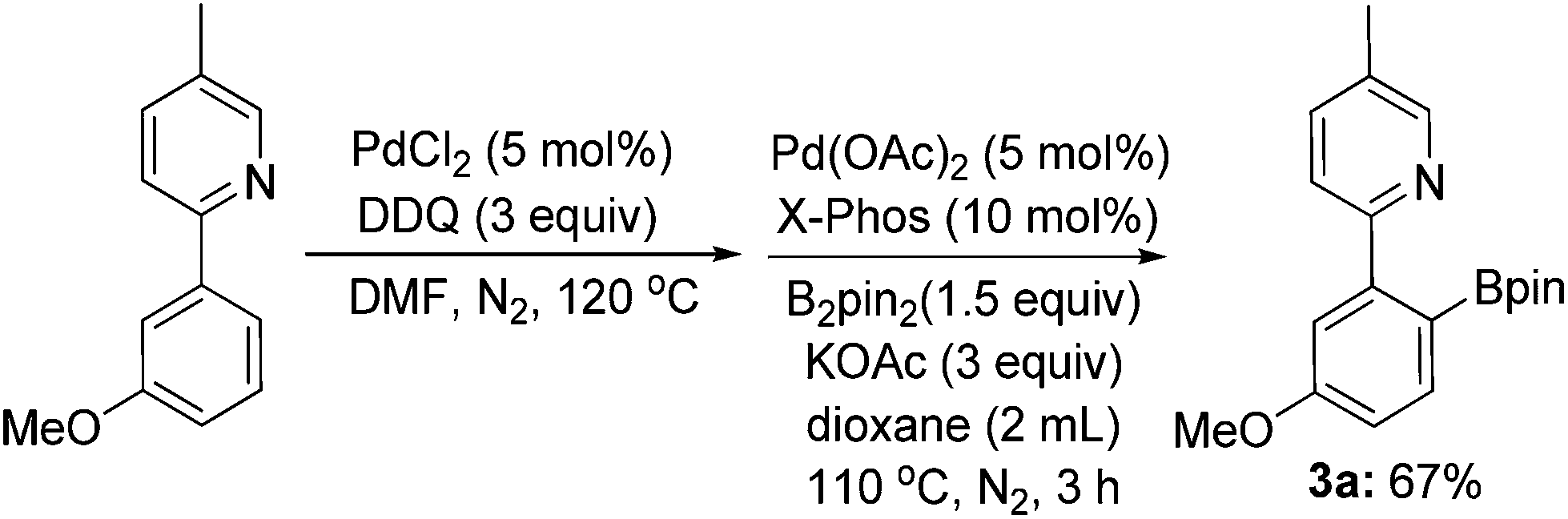 | ||
| Scheme 4 The new synthetic strategy for ligand-directed ortho-borylation. | ||
In summary, we have developed a new and simple protocol for palladium-catalyzed ortho-C–H chlorination of 2-arylpyridines and some heterocycles. Notably, DDQ played a dual role as the chlorinating reagent and oxidant for the successful reaction. Moreover, this reaction showed high regioselectivity for the substrates bearing a meta-substituent in the benzene moiety. Further application of this synthetic methodology is currently underway in our laboratory.
We are grateful to the National Natural Science Foundation of China (no. 21172200, 21102134) for financial support.
Notes and references
- (a) D. Prim, J. M. Campagne, D. Joseph and B. Andrioletti, Tetrahedron, 2002, 58, 2041 CrossRef CAS; (b) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS PubMed; (c) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125 CrossRef CAS; (d) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (e) G. Bringmann, R. Walter and R. Weirich, Angew. Chem., Int. Ed. Engl., 1990, 29, 977 CrossRef; (f) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125 CrossRef CAS.
- (a) D. A. Evans, J. L. Katz, G. S. Peterson and T. Hintermann, J. Am. Chem. Soc., 2001, 123, 12411 CrossRef CAS; (b) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937 CrossRef CAS.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed; (c) X. Y. Sun, G. Shan, Y. H. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed.
- (a) R. Taylor, Electrophilic Aromatic Substitution, John Wiley, New York, 1990 Search PubMed; (b) H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS; (c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS; (d) D. W. Young and D. L. Comins, Org. Lett., 2005, 7, 5661 CrossRef CAS PubMed.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) X. B. Wan, Z. X. Ma, B. J. Li, K. Y. Zhang, S. K. Cao, S. W. Zhang and Z. J. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS PubMed; (c) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523 CrossRef CAS PubMed; (d) X. Chen, X. S. Hao, C. E. Goodhue and J. Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; (e) R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chem., Int. Ed., 2011, 50, 5524 CrossRef CAS PubMed; (f) B. R. Song, X. J. Zheng, J. Mo and B. Xu, Adv. Synth. Catal., 2010, 352, 329 CrossRef CAS; (g) N. Schröder, J. Wencel-Delord and F. Glorius, J. Am. Chem. Soc., 2012, 134, 8298 CrossRef PubMed; (h) J. M. Murphy, X. B. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed.
- (a) Y. H. Zhang and C. J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949 CrossRef CAS PubMed; (b) W. Y. Tu and P. E. Floreancig, Angew. Chem., Int. Ed., 2009, 48, 4567 CrossRef CAS PubMed; (c) L. Liu and P. E. Floreancig, Angew. Chem., Int. Ed., 2010, 49, 3069 CrossRef CAS PubMed; (d) C. Guo, J. Song, S. W. Luo and L. Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 5558 CrossRef CAS PubMed; (e) L. Liu and P. E. Floreancig, Angew. Chem., Int. Ed., 2010, 49, 5894 CrossRef CAS PubMed; (f) Y. Hayashi, T. Itoh and H. Ishikawa, Angew. Chem., Int. Ed., 2011, 50, 3920 CrossRef CAS PubMed; (g) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153 CrossRef CAS; (h) L. Minuti, A. Taticchi, A. Marrocchi, D. Lanari, E. Gacs-Baitz and A. Gomory, Tetrahedron Lett., 2005, 46, 949 CrossRef CAS PubMed.
- CCDC 917218 contains the supplementary crystallographic data for 2j. Crystal data for compound 2j: C13H11Cl2NO, M = 268.13, triclinic, a = 6.2427(4) Å, α = 74.492(6)°, b = 8.5337(6) Å, β = 77.526(6)°, c = 12.7695(10) Å, γ = 81.552(5)°, V = 637.16(8) Å3, T = 291.15 K, space group = P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, number of reflections = 4499, independent reflections = 2279, [R(int) = 0.0212], final R indices [I > 2σ(I)] R1 = 0.0389, wR2 = 0.1078, R indices (all data) R1 = 0.0443, wR2 = 0.1131.
, Z = 2, number of reflections = 4499, independent reflections = 2279, [R(int) = 0.0212], final R indices [I > 2σ(I)] R1 = 0.0389, wR2 = 0.1078, R indices (all data) R1 = 0.0443, wR2 = 0.1131. - (a) J. Yan, H. Fang and B. H. Wang, Med. Res. Rev., 2005, 25, 490 CrossRef CAS PubMed; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS; (c) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271 CrossRef CAS PubMed; (d) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 917218. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00076e |
| This journal is © the Partner Organisations 2014 |