A green fullerene derivative as a fluoride ion sensor†
Liang
Xu
a,
Sisi
Liang
a and
Liangbing
Gan
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: gan@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Lu, Shanghai 200032, China
First published on 5th May 2014
Abstract
An open-cage fullerene derivative with an enamine moiety on the rim of the orifice is prepared through a lithium chloride induced dehydration process involving an intramolecular Friedel–Crafts reaction and ring expansion rearrangement steps. The enamine moiety reacts with fluoride ions selectively and acts as the active site for fluoride ion detection.
Introduction
Fluoride ion detection has received much attention due to its dual effects on human health. The intake of a small amount of fluoride is beneficial to dental health and the treatment of osteoporosis.1 But an overdose of fluoride ions can cause many problems such as collagen breakdown, bone disorder, thyroid activity and depression.2 Thus to determine the level of fluoride ions various detection methods have been reported including highly accurate yet expensive analytical instruments which use ion selective electrodes. On the other hand a colorimetric sensor does not need any spectroscopic instrument since the visible colour change can be detected by the naked eye. But such colorimetric sensors also have their drawbacks. The sensitivity of some of these sensors is limited because of their low absorption. The selectivity of fluoride ions is also a problem for some systems. As a result, development of alternative colorimetric sensors has been an active research area.3A number of colorimetric sensors have been prepared for fluoride ion detection.1–3 In most cases selective fluoride ion detection is based on H-bonding between fluoride and amino group(s) on the sensor molecule.4 Anion–π interaction,5 Lewis acid–base interaction6 and anion induced chemical reactions are employed in some sensors.7 Fluoride induced deprotonation of highly acidic N–H protons has also been successfully applied in fluoride ion detection.8 All the reported colorimetric sensors have a planar aromatic system as the core of the chromophore. Fullerenes have a unique spherical π system and high absorption in the UV-Vis region. Selective opening of the fullerene cage and addition of functional groups can modify the π system of the fullerene cage significantly to form fullerene derivatives suitable for possible application.9 Fullerene modified electrodes have been investigated as possible sensors for certain biologically active molecules.10 Fullerenes immobilized in oxygen permeable polymer films are potential sensors for oxygen.11 So far there has been no report on fullerene-based fluoride ion sensors. We have been working on the preparation of open-cage fullerenes using a peroxide-mediated pathway.12 Some of our open-cage fullerene derivatives showed interesting properties such as the selective encapsulation of a single water molecule.13 Here we report the preparation of a green open-cage fullerene compound and its potential application as a colorimetric fluoride ion sensor.
Results and discussion
Compound 1 was prepared by heating a chlorobenzene solution of the known compound 214 in the presence of a large excess of lithium chloride hydrate and acetic acid (Scheme 1). The presence of some water is essential for the conversion. Under the same conditions anhydrous lithium hydrate could not induce the formation of 1. Due to the much increased size of the orifice, compound 1 encapsulated a water molecule under the reaction conditions. Separation of 1 and H2O@1 was not possible. This mixture was used for all the spectroscopic data measurements, X-ray diffraction analysis and fluoride ion detection in the following sections. The encapsulated water shows little effect on the spectroscopic data as shown before for similar compounds.13 Other Lewis acids were also tested for the conversion from 2 to 1. Only MgCl2 gave similar results but with a lower yield than that from LiCl. Lewis acids such as FeCl3 and ZnCl2 gave complicated products.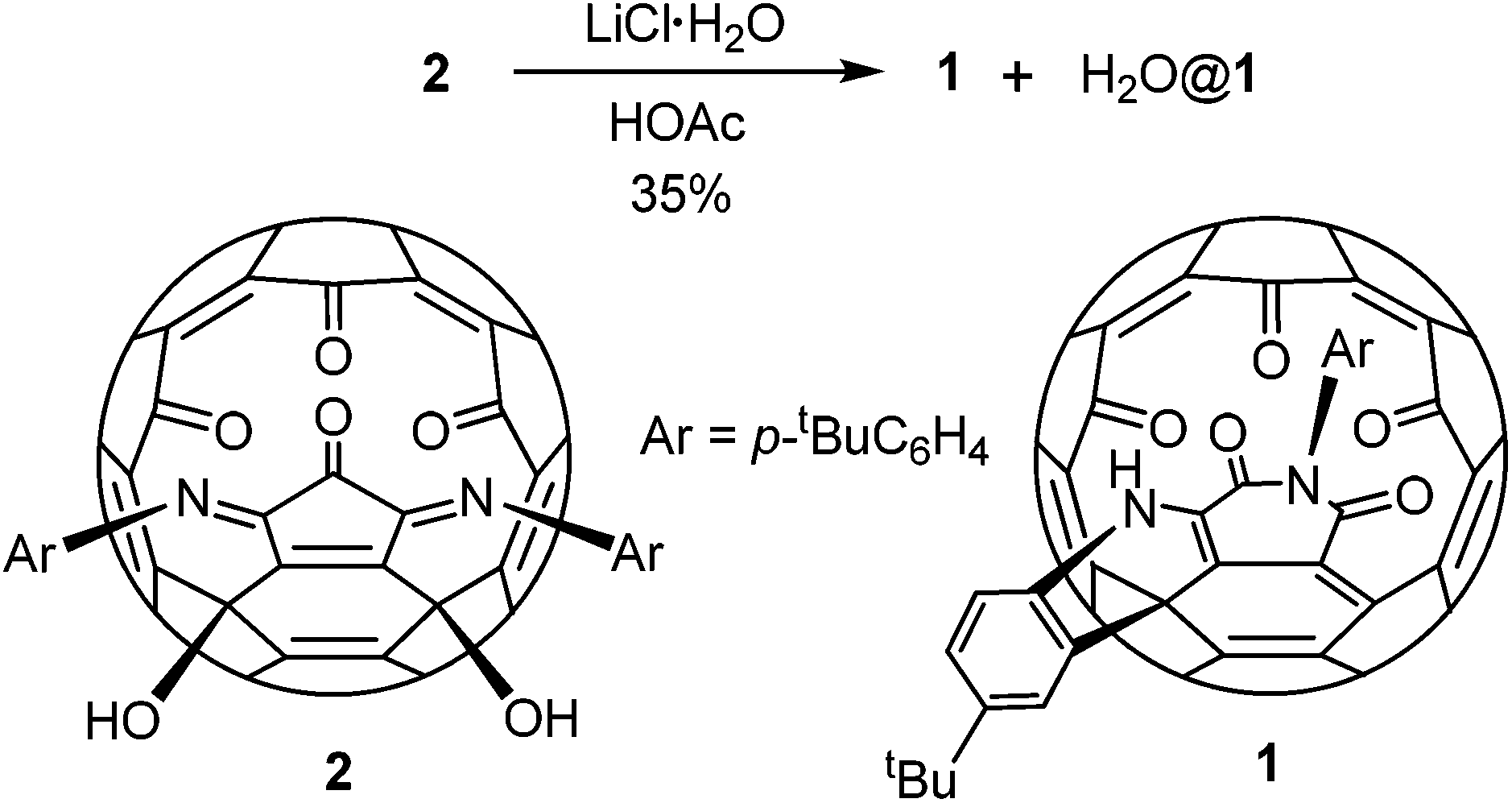 | ||
| Scheme 1 Preparation of compound 1. | ||
A possible mechanism is proposed as shown in Scheme 2. The process initiates from an intramolecular Friedel–Crafts process through the cation A. The proximity of the phenyl group to the fullerene cage renders such an annulation process feasible. Selective hydration of the imino groups in B to form C is probably due to the rigidity of the unique local ring strain. The coordination of the lithium ion plays a key role in both the hydration of B and the ring expansion rearrangement from D to E. The ring expansion step is reminiscent of the Baeyer–Villiger reaction. Conjugation of the enamine with the cage π-system provides the driving force for the elimination of a water molecule from F to form 1.
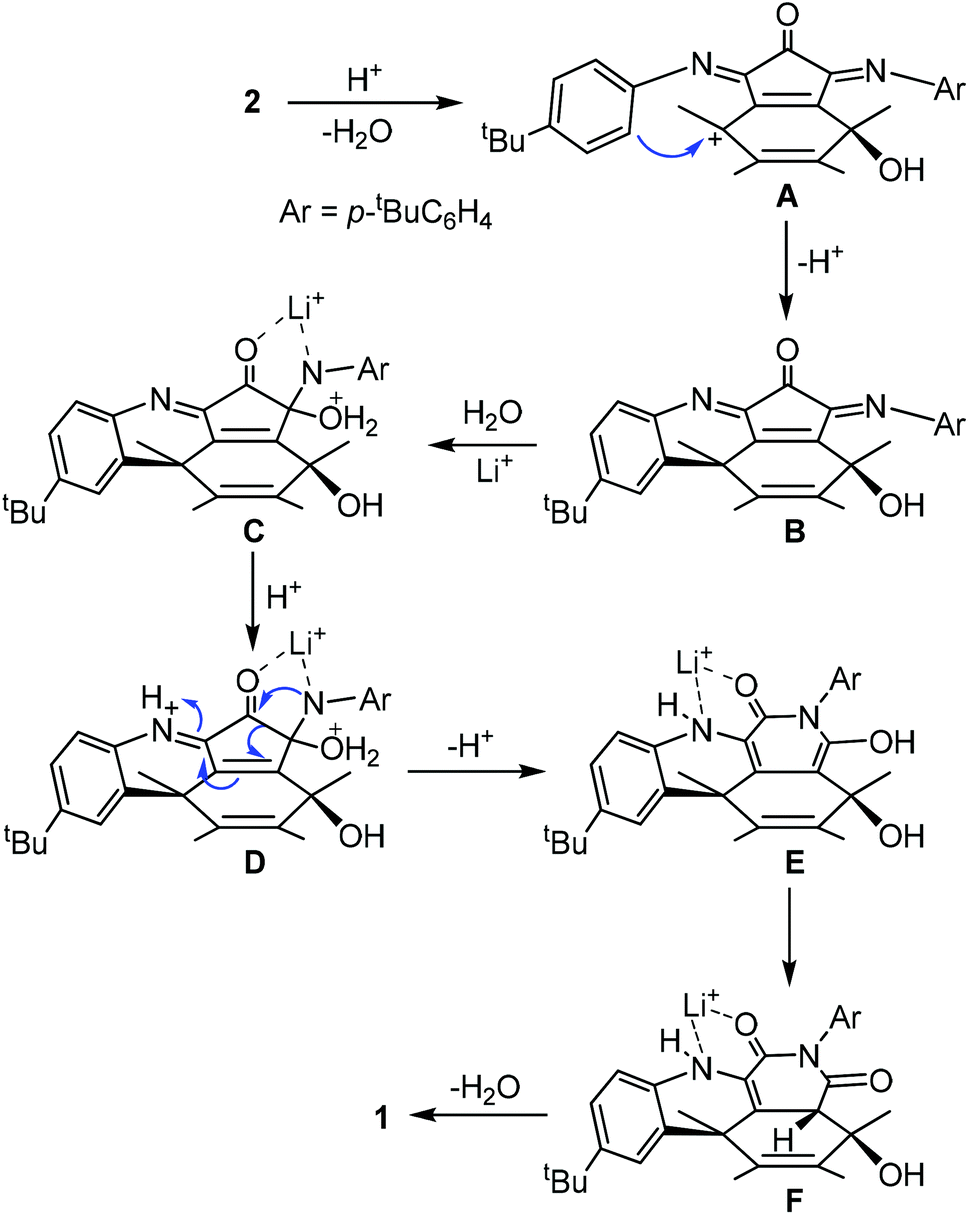 | ||
| Scheme 2 Possible mechanism for the formation of 1. | ||
Alternatively the conversion from 2 to 1 could start from hydration of an imino group, followed by ring expansion rearrangement and Friedel–Crafts processes. But this is less likely since electron poor aniline analogues of 2 such as the para bromo or nitro aniline derivatives failed to give any characterizable product under the same conditions. Compound 3 has been reported before in two steps from 2.15 It has an imino anhydride moiety instead of the imide moiety in 1. Both compounds 3 and 1 are very stable and their interconversion was not achieved (Scheme 3).
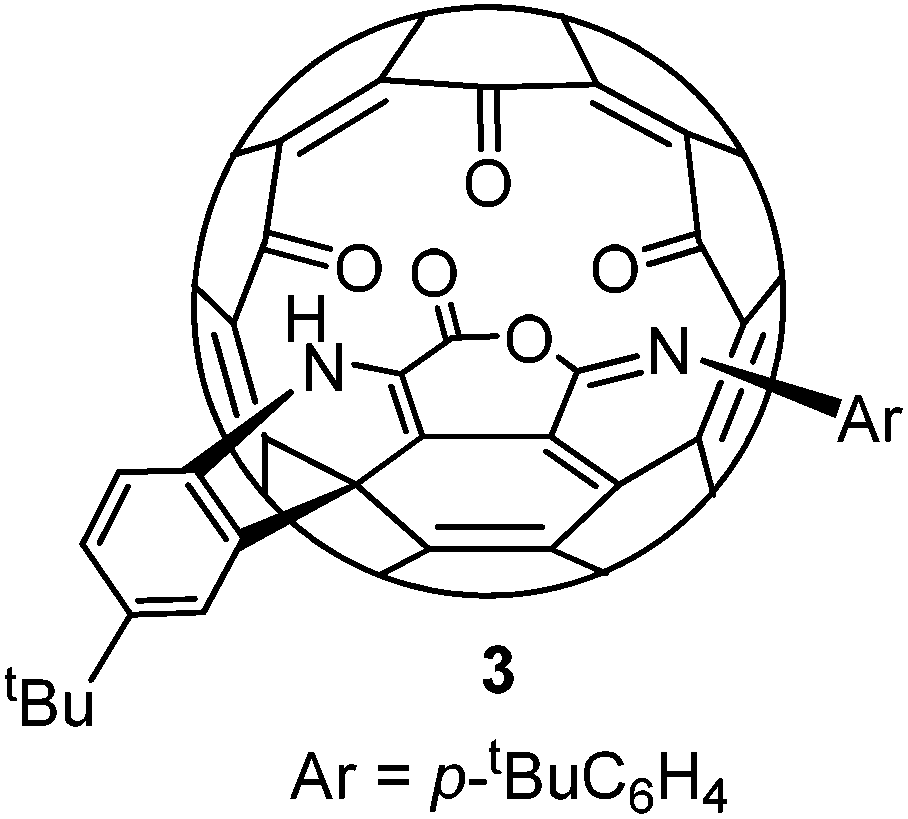 | ||
| Scheme 3 Structure of the known compound 3. | ||
The structure of 1 was established by X-ray diffraction analysis (Fig. 1). Single crystals of 1 were obtained from slow evaporation of its CH2Cl2–CHCl3 solution. The X-ray data showed strong intermolecular H-bonding between the NH group and one of the three carbonyl groups on the rim of the orifice in another molecule, thus forming a dimeric structure between two enantiomeric molecules. The trapped water molecule is in the center of the cavity. The distance from the water oxygen atom to the center of the pentagon at the bottom is 3.31 Å. Due to steric hindrance of the adjacent carbonyl oxygen on the rim of the orifice, the oxygen atom of the imide carbonyl group further away from the NH group is slightly pushed above the six-membered imide ring, thus preventing the conjugation between the imide nitrogen lone pair and this carbonyl π bond. As expected, the other imide carbonyl group is planar with the imide nitrogen and shows conjugation effects. The two imide N–C bonds are 1.381 and 1.484 Å respectively for the conjugated and non-conjugated cases.
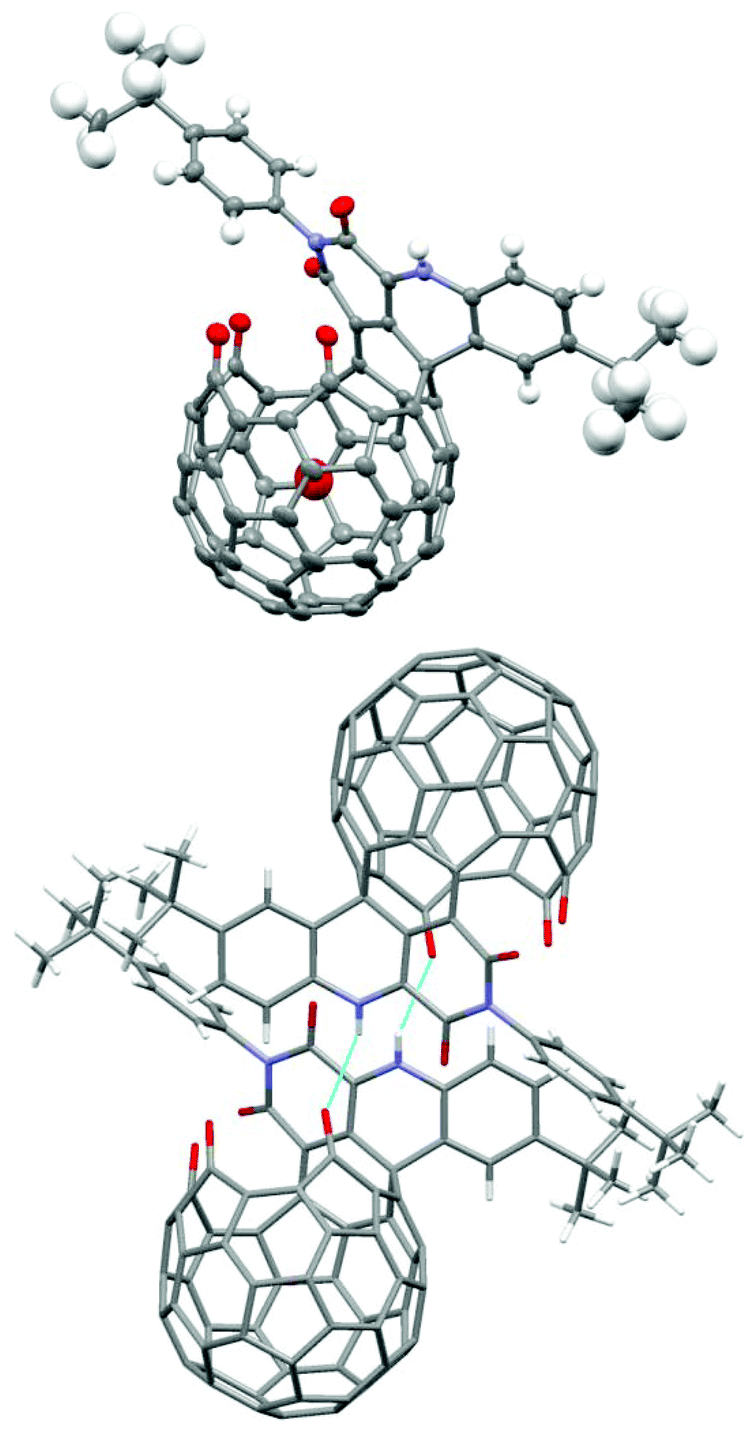 | ||
| Fig. 1 Single-crystal X-ray structure of H2O@1; colour scheme: grey = C, blue = N, red = O, white = H; light-blue lines indicate H-bonds in the dimeric structure. | ||
Spectroscopic data of 1 are in agreement with the X-ray structure. The 1H NMR spectrum showed the trapped water signal at −12.2 ppm. The encapsulation ratio was calculated from the integral ratio as 40%. There are two enamine proton signals at 9.31 and 9.36 ppm, which is probably due to the existence of equilibrium between the monomer and the dimer of 1. The t-butyl groups are not affected by the equilibrium and appear as two singlets at 1.28 and 1.43 pm. The 13C NMR spectrum also showed two sets of signals for some of the signals such as the carbonyl carbons and the sp3 carbon on the cage. But there is only one set of 13C signals for the t-butyl groups which are probably the same for both the monomer and the dimer as on the 1H NMR spectrum. So, unlike the above X-ray structure, which showed only the dimer, both the monomer and the dimer are present in solution.
Solutions of compound 1 in various solvents showed strong absorption in the near-IR region. A broad absorption band centered at 717 nm was observed with an absorption coefficient of 1.5 × 104 L mol−1 cm−1 for a 1.6 × 10−5 mol L−1 CHCl3 solution. The absorption coefficient showed hardly any change when the concentration was diluted 60 times. So, the intermolecular H-bonding shown in the X-ray crystal structure is probably too weak to form dimeric structures or aggregates in dilute solutions. In contrast, compound 3 showed much stronger intermolecular H-bonding in its X-ray crystal structure and a concentration dependent absorption coefficient.15 The near-IR absorption band of 1 shifted to 734 nm when the solvent was changed to benzene, indicating an intramolecular charge transfer nature.
Addition of fluoride ions into the solution of 1 resulted in a clear colour change observable by the naked eye as shown in Fig. 2. The band at 717 nm is red-shifted to 823 nm upon addition of excess TBAF. The shift could be reversed by adding excess acid. Addition of t-butylammonium hydroxide resulted in a red-shift similar to the addition of fluoride ions, but the process is not reversible because of complicated reactions of compound 1 under basic conditions. So, the origin of the fluoride induced red-shift should be the intermolecular proton transfer process between the enamine and the fluoride ion. The resulting deprotonated 1 shows stronger intramolecular charge transfer properties. Such a fluoride detection mechanism is similar to those fluoride sensors based on acid–base reactions between the acidic N–H group and fluoride ions, such as the diketopyrrolopyrrole reported by Tian et al.8a and benzoselenadiazole based diarylamine reported by Wang et al.8c
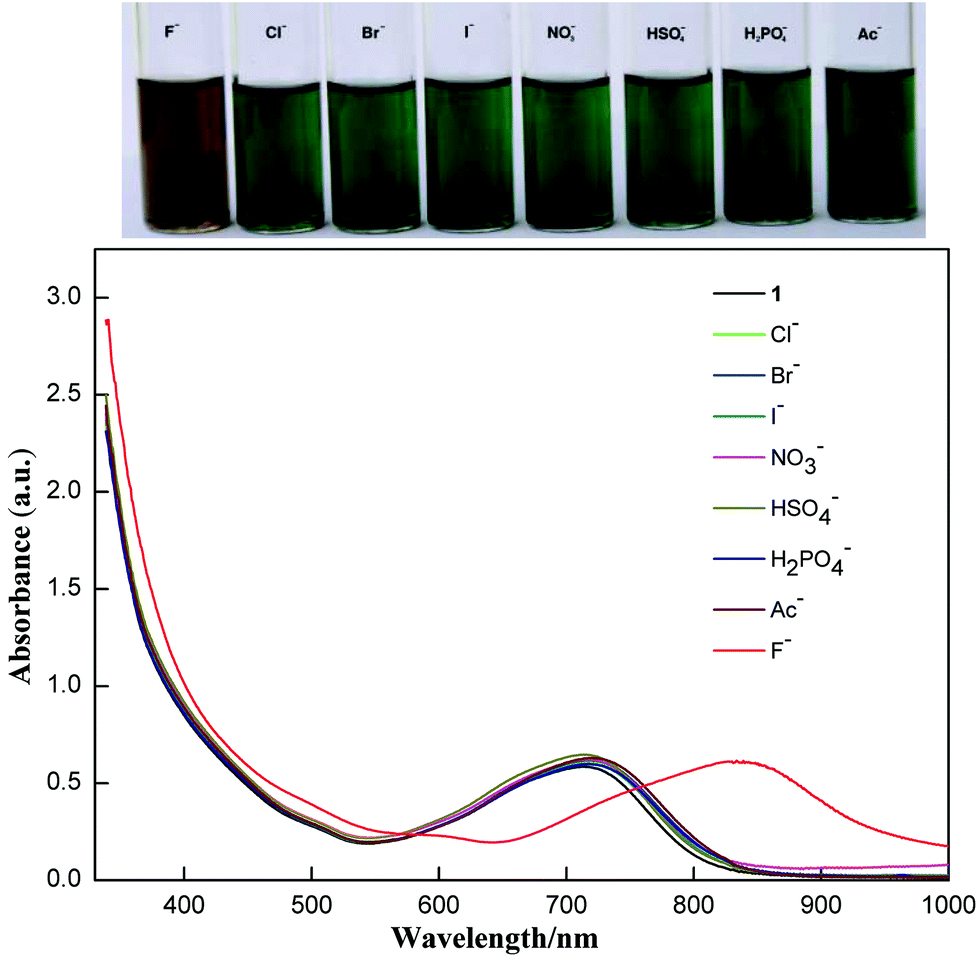 | ||
| Fig. 2 Visible color changes (above) and the corresponding absorption spectra (below) of 1 (10−5 M) in the presence of excess various anions (as their TBA salts) in CHCl3. | ||
The selectivity of compound 1 was tested by using various anions as the tetrabutylammonium salt. Only the fluoride ion showed a clear colour change, and the other ions Cl−, Br−, I−, NO3−, HSO4−, H2PO4−, and CH3COO− showed no noticeable colour change as shown in Fig. 2. Progressive addition of fluoride ions resulted in a clear isosbestic point at around 745 nm (Fig. 3). The appearance of the isosbestic point indicates that there are only two species, the neutral compound 1 and the deprotonated 1. A near-linear correlation between intensity ratios of absorbance at 823 nm to those at 717 nm (A823/A717) vs. fluoride ion concentration in CHCl3 was obtained for compound 1 (Fig. 4). These results indicate that compound 1 could sense fluoride ions selectively by colorimetric and ratiometric methods. The fluoride ion detection limit was found to be at the micromolar level with a 10−5 M solution of 1.
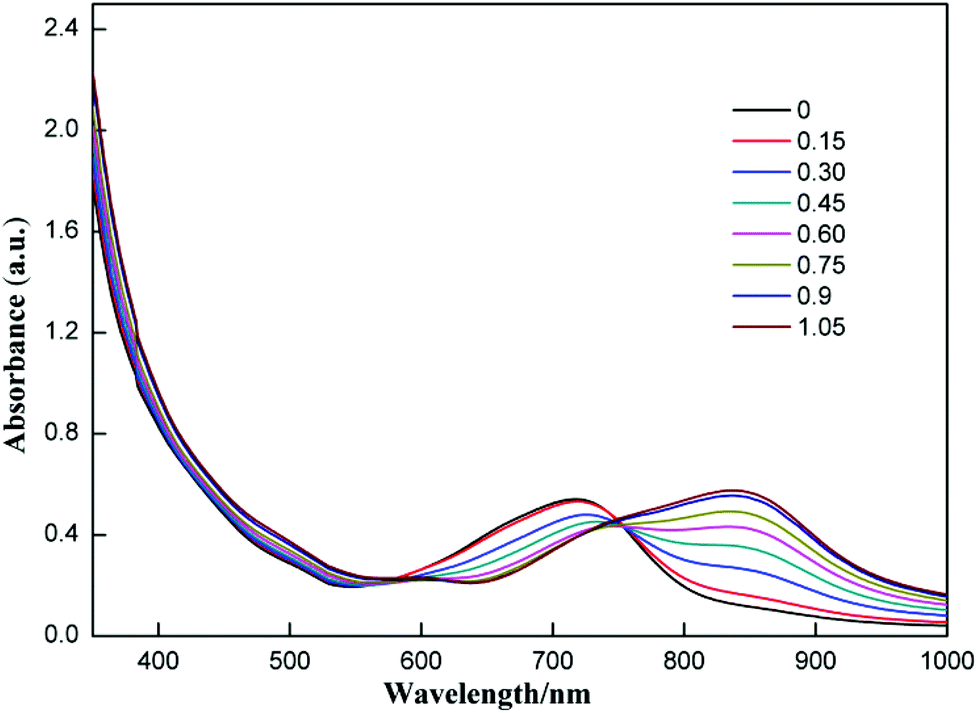 | ||
| Fig. 3 UV-Vis absorption spectra of 1 (3.5 × 10−5 M) in CHCl3 upon addition of TBAF; the numbers in the inset are equivalents of fluoride ions added. | ||
 | ||
| Fig. 4 Plot of the absorbance ratio of 1 between 823 nm and 717 nm (A823/A717) vs. concentration of F− in CHCl3. | ||
Experimental
Compound 1
30 ml acetic acid and 2.1 g LiCl·H2O were added to a solution of compound 2 (110.4 mg, 0.099 mmol) in 30 ml chlorobenzene. The resulting solution was stirred at 110 °C for 4 h. The lithium salt was removed by a flash chromatograph eluting with toluene–ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Another chromatograph eluting with toluene–ethyl acetate (50:1) was employed to purify the mixture. A green band was collected and evaporated to give compound 1 (37.6 mg, 0.034 mmol, 35%) as a green solid. 1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 0.5 H), 9.31 (s, 0.5 H), 8.00 (m, 3H), 7.75 (m, 2H), 7.44 (m, 1H), 7.34 (m, 1H) 1.43 (s, 9H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ: 184.40, 184.30, 184.27, 184.08, 182.41, 182.23, 170.34, 170.20, 162.61, 159.98, 152.74, 151.70, 151.45, 150.59, 149.97, 149.82, 149.73, 149.55, 149.48, 149.28, 149.22, 149.14, 148.84, 148.74, 148.54, 148.30, 148.07, 147.94, 147.68, 147.39, 147.30, 147.11, 146.89, 146.42, 146.26, 146.04, 145.99, 144.99, 144.46, 144.34, 144.27, 143.79, 142.72, 142.26, 141.88, 141.64, 140.90, 140.69, 140.65, 140.27, 137.57, 137.42, 136.74, 136.56, 136.47, 135.50, 135.34, 134.42, 133.29, 133.21, 132.71, 132.57, 132.27, 132.17, 130.51, 129.04, 128.92, 128.23, 126.55, 126.49, 125.30, 124.87, 123.02, 117.30, 117.22, 110.58, 61.80, 61.76, 54.44, 54.41, 34.92, 34.87, 31.51, 31.41; the signals are due to a mixture of monomers and dimers. ESI-HRMS: C79H27N2O4 (M − CO + H+) calculated 1067.1965, found 1067.1962.
:1). Another chromatograph eluting with toluene–ethyl acetate (50:1) was employed to purify the mixture. A green band was collected and evaporated to give compound 1 (37.6 mg, 0.034 mmol, 35%) as a green solid. 1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 0.5 H), 9.31 (s, 0.5 H), 8.00 (m, 3H), 7.75 (m, 2H), 7.44 (m, 1H), 7.34 (m, 1H) 1.43 (s, 9H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ: 184.40, 184.30, 184.27, 184.08, 182.41, 182.23, 170.34, 170.20, 162.61, 159.98, 152.74, 151.70, 151.45, 150.59, 149.97, 149.82, 149.73, 149.55, 149.48, 149.28, 149.22, 149.14, 148.84, 148.74, 148.54, 148.30, 148.07, 147.94, 147.68, 147.39, 147.30, 147.11, 146.89, 146.42, 146.26, 146.04, 145.99, 144.99, 144.46, 144.34, 144.27, 143.79, 142.72, 142.26, 141.88, 141.64, 140.90, 140.69, 140.65, 140.27, 137.57, 137.42, 136.74, 136.56, 136.47, 135.50, 135.34, 134.42, 133.29, 133.21, 132.71, 132.57, 132.27, 132.17, 130.51, 129.04, 128.92, 128.23, 126.55, 126.49, 125.30, 124.87, 123.02, 117.30, 117.22, 110.58, 61.80, 61.76, 54.44, 54.41, 34.92, 34.87, 31.51, 31.41; the signals are due to a mixture of monomers and dimers. ESI-HRMS: C79H27N2O4 (M − CO + H+) calculated 1067.1965, found 1067.1962.
Single crystal X-ray diffraction data of 1. Formula: C83H34N2O6Cl6, T = 180.00(10) K, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , unit cell dimensions: a = 10.2605(6) Å, b = 16.8131(9) Å, c = 17.3175(10) Å, α = 101.471(5)°, β = 103.442(5)°, γ = 99.530(5)°. V = 2776.4(3)(5) Å3. Z = 2, ρcalcd = 1.636 Mg m−3. Reflections collected/unique 21545/10481 [R(int) = 0.0430]. Final R indices [I > 2σ(I)] R1 = 0.0992, wR2 = 0.2565. CCDC 995293 contains the supplementary crystallographic data for this paper.
, unit cell dimensions: a = 10.2605(6) Å, b = 16.8131(9) Å, c = 17.3175(10) Å, α = 101.471(5)°, β = 103.442(5)°, γ = 99.530(5)°. V = 2776.4(3)(5) Å3. Z = 2, ρcalcd = 1.636 Mg m−3. Reflections collected/unique 21545/10481 [R(int) = 0.0430]. Final R indices [I > 2σ(I)] R1 = 0.0992, wR2 = 0.2565. CCDC 995293 contains the supplementary crystallographic data for this paper.
Conclusions
A lithium chloride induced dehydration process is found to produce a green open-cage fullerene 1 with an enamine moiety on the rim of the orifice. The process involves an intramolecular Friedel–Crafts reaction and ring expansion rearrangement as the key steps. Conjugation of the active enamine moiety with the spherical cage system makes compound 1 a sensitive fluoride ion sensor. To develop more practical fullerene based fluoride sensors, water soluble fullerenes need to be prepared.Acknowledgements
This work was supported by NSFC (21272013 and 21132007) and the Major State Basic Research Development Program (2011CB808401).Notes and references
- (a) K. L. Kirk, in Biochemistry of the Elemental Halogens and Inorganic Halides, Plenum, New York, 1991, pp. 19–68 Search PubMed; (b) H. S. Horowitz, J. Public Health Dent., 2003, 63, 3 CrossRef.
- (a) B. L. Riggs, Bone Miner. Res., 1984, 366 CAS; (b) M. Laisalmi, H. Kokki, A. Soikkeli, H. Markkanen, A. Yli-Hankala, P. Rosenberg and L. Lindgren, Acta Anaesthesiol. Scand., 2006, 50, 982 CrossRef CAS PubMed.
- (a) M. Cametti and K. Rissanen, Chem. Soc. Rev., 2013, 42, 2016 RSC; (b) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (c) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaı, Chem. Rev., 2010, 110, 3958 CrossRef CAS PubMed.
- For example, I. Basaran, M. E. Khansari, A. Pramanik, B. M. Wong and Md. A. Hossain, Tetrahedron Lett., 2014, 55, 1467 CrossRef CAS PubMed.
- For example, S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679 CrossRef CAS PubMed.
- For example, Z. Liu, M. Shi, F. Li, Q. Fang, Z. Chen, T. Yi and C. Huang, Org. Lett., 2005, 7, 5481 CrossRef CAS PubMed.
- (a) J. F. Zhang, C. S. Lim, S. Bhuniya, B. R. Cho and J. S. Kim, Org. Lett., 2011, 13, 1190 CrossRef CAS PubMed; (b) K. M. Mahoney, P. P. Goswami and A. H. Winter, J. Org. Chem., 2013, 78, 702 CrossRef CAS PubMed; (c) R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915 CrossRef CAS PubMed.
- (a) Y. Qu, J. Hua and H. Tian, Org. Lett., 2010, 12, 3320 CrossRef CAS PubMed; (b) C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172 RSC; (c) C. Saravanan, S. Easwaramoorthi, C.-Y. Hsiow, K. Wang, M. Hayashi and L. Wang, Org. Lett., 2014, 16, 354 CrossRef CAS PubMed.
- (a) Z. Xiao, G. Ye, Y. Liu, S. Chen, Q. Peng, Q. Zuo and L. Ding, Angew. Chem., Int. Ed., 2012, 51, 9038 CrossRef CAS PubMed; (b) Y. Yu, L. Xu, X. Huang and L. Gan, J. Org. Chem., 2014, 79, 2156 CrossRef CAS PubMed.
- B. S. Sherigara, W. Kutner and F. D'Souza, Electroanalysis, 2003, 15, 753 CrossRef CAS.
- Y. Amao, Microchim. Acta, 2003, 143, 1 CrossRef CAS.
- For recent examples, see: (a) L. Xu, Q. Y. Zhang, G. Zhang, S. Liang, Y. Yu and L. Gan, Eur. J. Org. Chem., 2013, 7272 CrossRef CAS; (b) N. Xin, X. Yang, Z. Zhou, J. Zhang, S. Zhang and L. Gan, J. Org. Chem., 2013, 78, 1157 CrossRef CAS PubMed; (c) L. Shi, D. Yang, F. Colombo, Y. Yu, W.-X. Zhang and L. Gan, Chem. – Eur. J., 2013, 19, 16545 CrossRef CAS PubMed.
- L. Shi and L. Gan, J. Phys. Org. Chem., 2013, 26, 766 CrossRef CAS.
- Q. Zhang, Z. Jia, S. Liu, G. Zhang, Z. Xiao, D. Yang, L. Gan, Z. Wang and Y. Li, Org. Lett., 2009, 11, 2772 CrossRef CAS PubMed.
- S. Liu, C. Zhang, X. Xie, Y. Yu, Z. Dai, Y. Shao, L. Gan and Y. Li, Chem. Commun., 2012, 48, 2531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for compound 1. CCDC 995293. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00111g |
| This journal is © the Partner Organisations 2014 |