Palladium-catalyzed reductive cleavage of tosylated arenes using isopropanol as the mild reducing agent†
Wing Kin
Chow
,
Chau Ming
So
,
Chak Po
Lau
and
Fuk Yee
Kwong
*
State Key Laboratory of Chirosciences, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. E-mail: fuk-yee.kwong@polyu.edu.hk
First published on 17th April 2014
Abstract
The deoxygenation of tosylated arenes catalyzed by a palladium complex is described. This method represents one of the first general examples of reductive C–O bond cleavage of aryl tosylates via palladium catalysis. By simply employing isopropanol as a mild reducing agent, a variety of tosylated arenes can be smoothly reduced. Labelling experiments revealed that the H source is isopropanol.
The exploration of catalytic deoxygenation methods for alcohols is an important subject with versatile applications to modern chemical syntheses.1 In particular, the cleavage of C(sp2)–O bonds in phenolic derivatives remains a challenge due to the intrinsic inertness of the aromatic C–O bond.2 The robust nature of the C(sp2)–O bond can be weakened by installing an electron withdrawing group on to the oxygen atom. Indeed, one of the most reliable and frequently used protocols for the deoxygenation of phenols proceeds via the conversion of phenols into fluorinated sulfonates3 such as triflates and nonaflates, followed by a nickel- or palladium-catalysed reductive process.4,5 There have been a number of reported syntheses of known pharmaceutical intermediates employing this synthetic step (Scheme 1).6
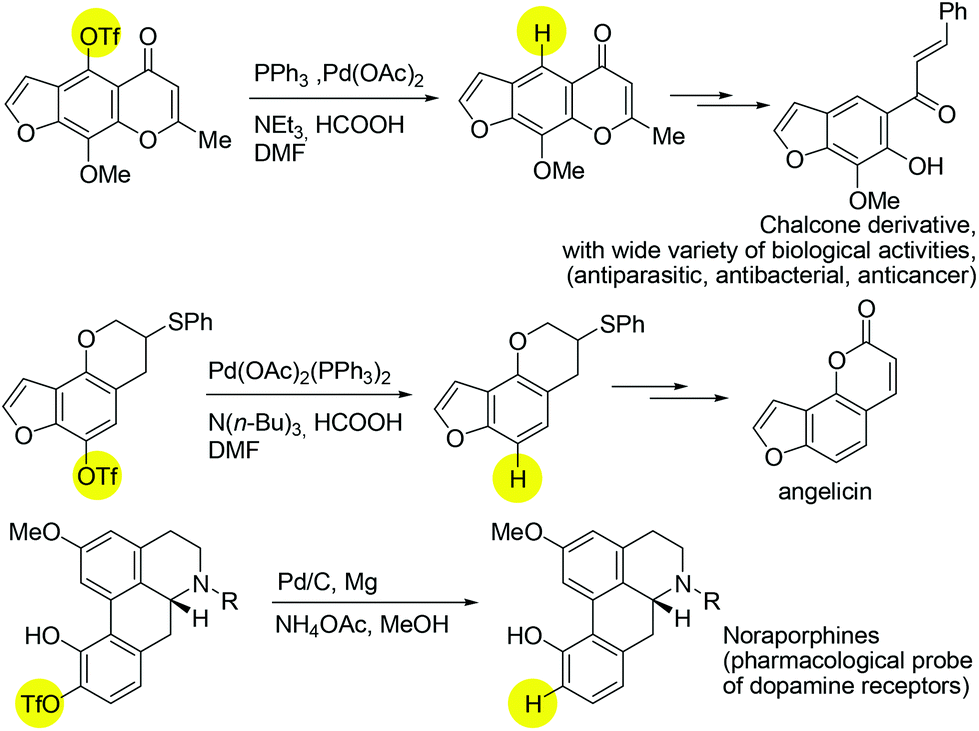 | ||
| Scheme 1 Selected pharmaceutical examples of using sulfonated precursors in deoxygenation/reduction reaction. | ||
New catalyst systems have emerged allowing for more challenging aryl sulfonates to be employed in this deoxygenation reaction.7,8 Aryl tosylates could be deoxygenated through the mediation of stoichiometric amounts of NiCl2–NaBH4 or RANEY®-Ni/NaOH/H2 (5 atm) systems.9 Sasaki achieved the reduction of aryl mesylate using NiBr2(Ph3P)2/dppp as the catalyst, zinc as the reductant and methanol as the hydrogen donor.10 Lipshutz disclosed that nickel-on-graphite-Ph3P (Ni/Cg/Ph3P) associated with Me2NH·BH3 facilitated the catalytic reduction of aryl tosylates and mesylates.11 Benzothiazole or –CN group-containing substrates were found not to be compatible with the established reaction conditions. Sajiki reported that 10 mol% of Pd/C with a stoichiometric amount of Mg and NH4OAc under MeOH promoted the single electron transfer (SET) reductive cleavage of aryl mesylates.12 Yet, in this system, aryl tosylate did not react. To the best of our knowledge, there has been no literature report describing the general palladium-catalysed reductive cleavage of aryl tosylates. Herein, we report a catalytic system, employing isopropanol as the H donor and featuring good functional compatibility, for the facile deoxygenation of tosylated arenes.
We initially tested the feasibility of the reductive cleavage using our developed Pd/CM-Phos system (Table 1).13 Non-activated 4-tert-butylphenyl tosylate was chosen as the prototypical substrate for the deoxygenation reaction. A survey of commonly used inorganic bases revealed that K3PO4 and K2CO3 gave the best results (entries 1–2 and 5). Strong bases, e.g. NaOt-Bu, led to hydrolysis of the aryl tosylate and significant amounts of phenolic side products were observed (entry 3). Organic bases such as i-Pr2EtN and Et3N did not promote the reaction (entries 6–7). Solvent screening indicated that i-PrOH was the solvent of choice (entries 1 vs. 8–11). In fact, i-PrOH serves as both the solvent and the hydrogen donor in this reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Temp. (°C) | Yieldb |
| a Reaction conditions: ArOTs (1.0 mmol), base (3.0 mmol), Pd(OAc)2 (0.005 mmol, 0.5 mol%), CM-phos (0.02 mmol), solvent (3.0 mL) were stirred for 2 hours at the indicated temperature under nitrogen. b Calibrated GC yields were reported using dodecane as the internal standard. c Significant amount of phenolic product was observed. | ||||
| 1 | i-PrOH | K3PO4 | 90 | 88 |
| 2 | i-PrOH | K2CO3 | 90 | 78 |
| 3 | i-PrOH | NaOt-Bu | 90 | 7c |
| 4 | i-PrOH | Na3PO4 | 90 | 18 |
| 5 | i-PrOH | K3PO4·H2O | 90 | 85 |
| 6 | i-PrOH | i-Pr2EtN | 90 | Trace |
| 7 | i-PrOH | Et3N | 90 | Trace |
| 8 | MeOH | K3PO4 | 90 | 4 |
| 9 | EtOH | K3PO4 | 90 | 3 |
| 10 | t-BuOH | K3PO4 | 90 | Trace |
| 11 | t-AmOH | K3PO4 | 90 | 12 |
| 12 | i-PrOH | K3PO4 | 70 | 54 |
| 13 | i-PrOH | K3PO4 | 50 | Trace |
| 14 | i-PrOH | K3PO4 | r.t. | N.R. |
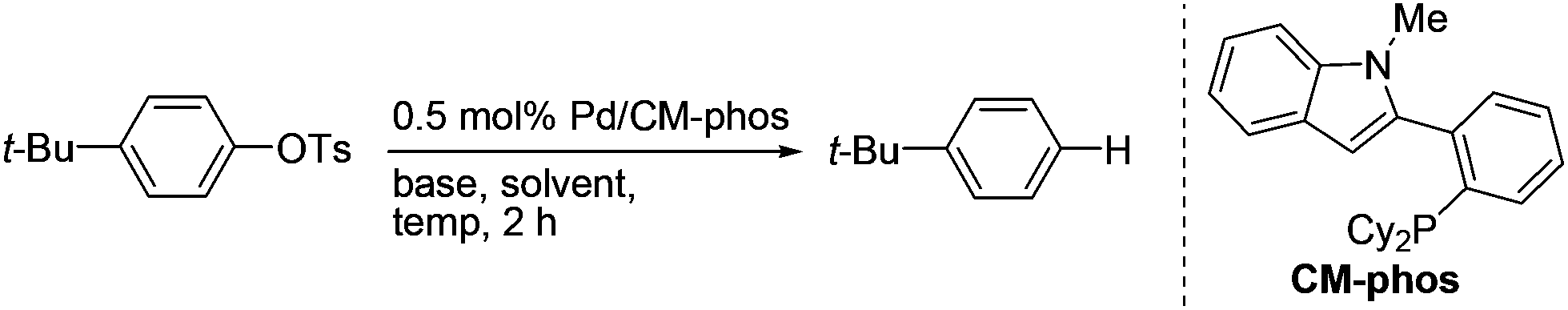
With the optimized reaction conditions in hand,14 we next investigated the scope of aryl tosylates in the deoxygenation reaction (Table 2). In general, this transformation proceeded well at 90 °C in 2 h. These mild reaction conditions tolerated keto, ester, nitrile and benzothiazolyl groups (entries 3–9), whereas other reducing agents were not fully compatible with them.15 To expand the substrate scope further, we tested the sterically hindered aryl tosylates (entries 8–10). To the best of our knowledge, there has been no successful example reported to date using 2,6-disubstituted aryl tosylates in a deoxygenation reaction. The electron-rich substrate screened (entry 12) was reduced smoothly.
|
|
||||
|---|---|---|---|---|
| Entry | ArOTs | ArH | mol% Pd (time) | % Yieldb |
a Reaction conditions: ArOTs (1.0 mmol), K2CO3 (3.0 mmol), Pd(OAc)2 (0.005–0.04 mmol, 0.5–4.0 mol%), CM-phos (Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L = 1:4) and i-PrOH (3.0 mL) were stirred at 90 °C for the indicated time under nitrogen.
b Isolated yield.
c Reactions were performed at 110 °C.
d Only determined by GC-FID, yield calculated according to GC-FID using authentic sample calibration. :L = 1:4) and i-PrOH (3.0 mL) were stirred at 90 °C for the indicated time under nitrogen.
b Isolated yield.
c Reactions were performed at 110 °C.
d Only determined by GC-FID, yield calculated according to GC-FID using authentic sample calibration.
|
||||
| 1 |
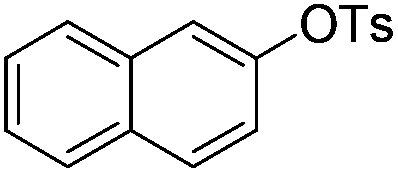
|
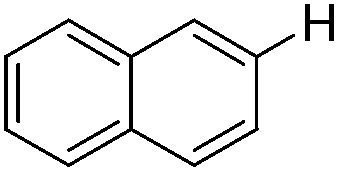
|
0.5 (2 h) | 82 |
| 2 |
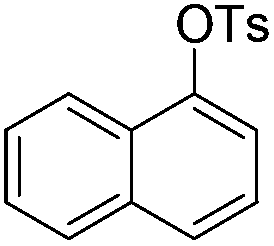
|
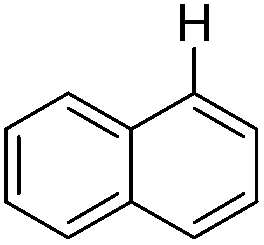
|
1.0 (2 h) | 81 |
| 3 |

|
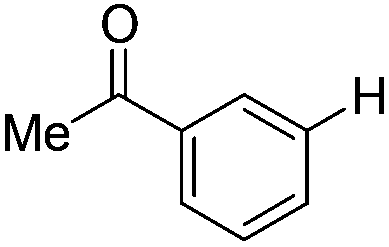
|
0.5 (2 h) | 81 |
| 4 |
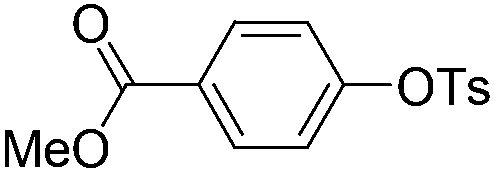
|

|
0.5 (2 h) | 82 |
| 5 |
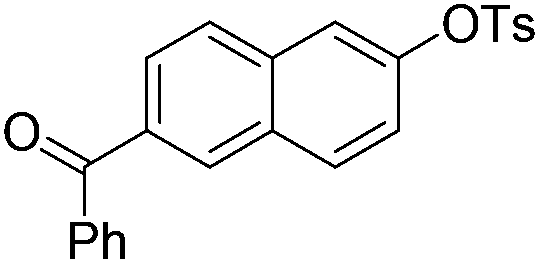
|
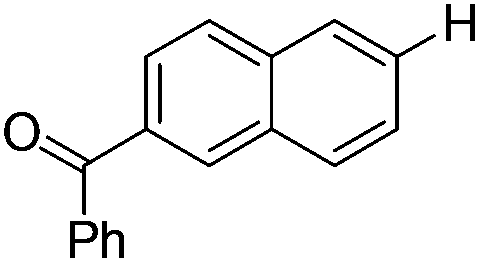
|
1.0 (2 h) | 86 |
| 6 |
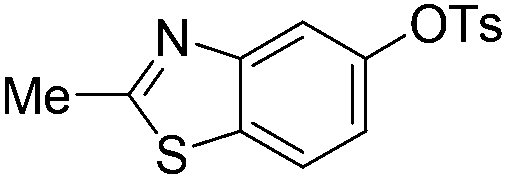
|
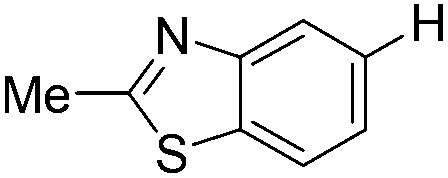
|
1.0 (2 h) | 86 |
| 7 |

|
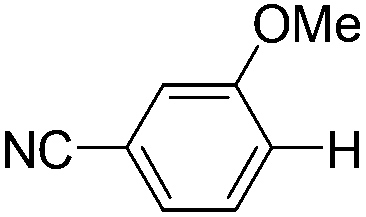
|
2.0 (2 h) | 86 |
| 8c |
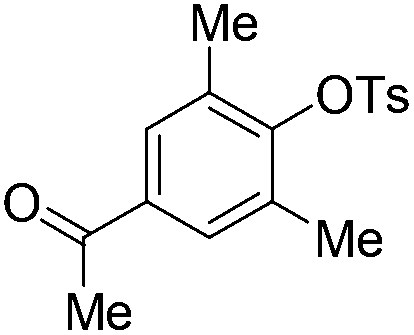
|
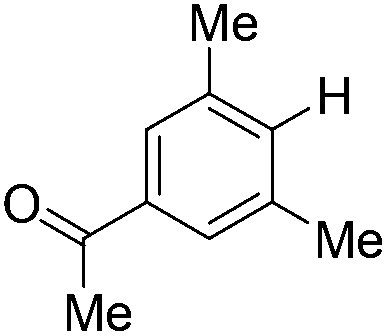
|
2.0 (24 h) | 82 |
| 9c |
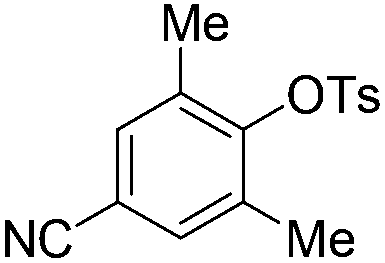
|
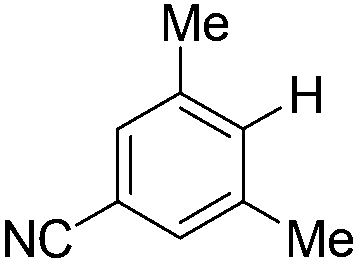
|
2.0 (24 h) | 82 |
| 10c |
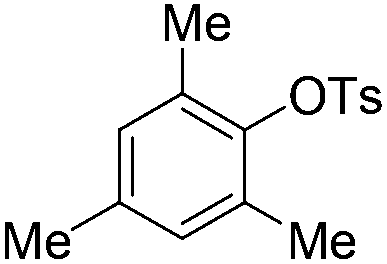
|
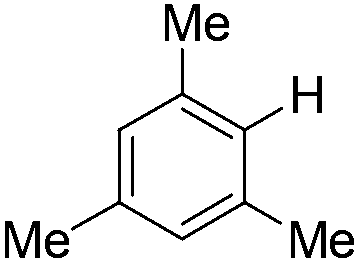
|
4.0 (24 h) | 72d |
| 11 |
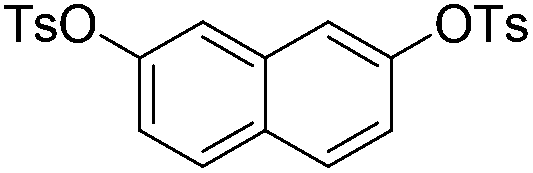
|
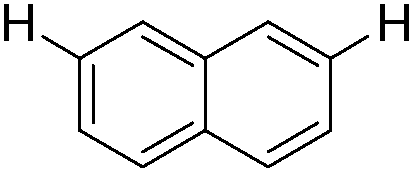
|
2.0 (2 h) | 77 |
| 12 |
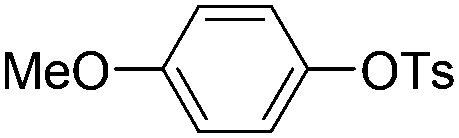
|
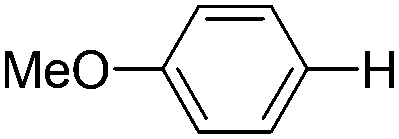
|
1.0 (2 h) | 70d |

In order to investigate the identity of the H source under these reaction conditions, we performed a labelling experiment (Scheme 2). Ethyl 4-tosylbenzoate was deoxygenated in deuterated isopropanol. The corresponding deuterated product was obtained in 76% yield (Scheme 2A). Moreover, we attempted to carry out the experiment under the i-PrOH–D2O conditions (Scheme 2B). These results unambiguously confirmed the H-source is not from the –OH group of isopropanol.
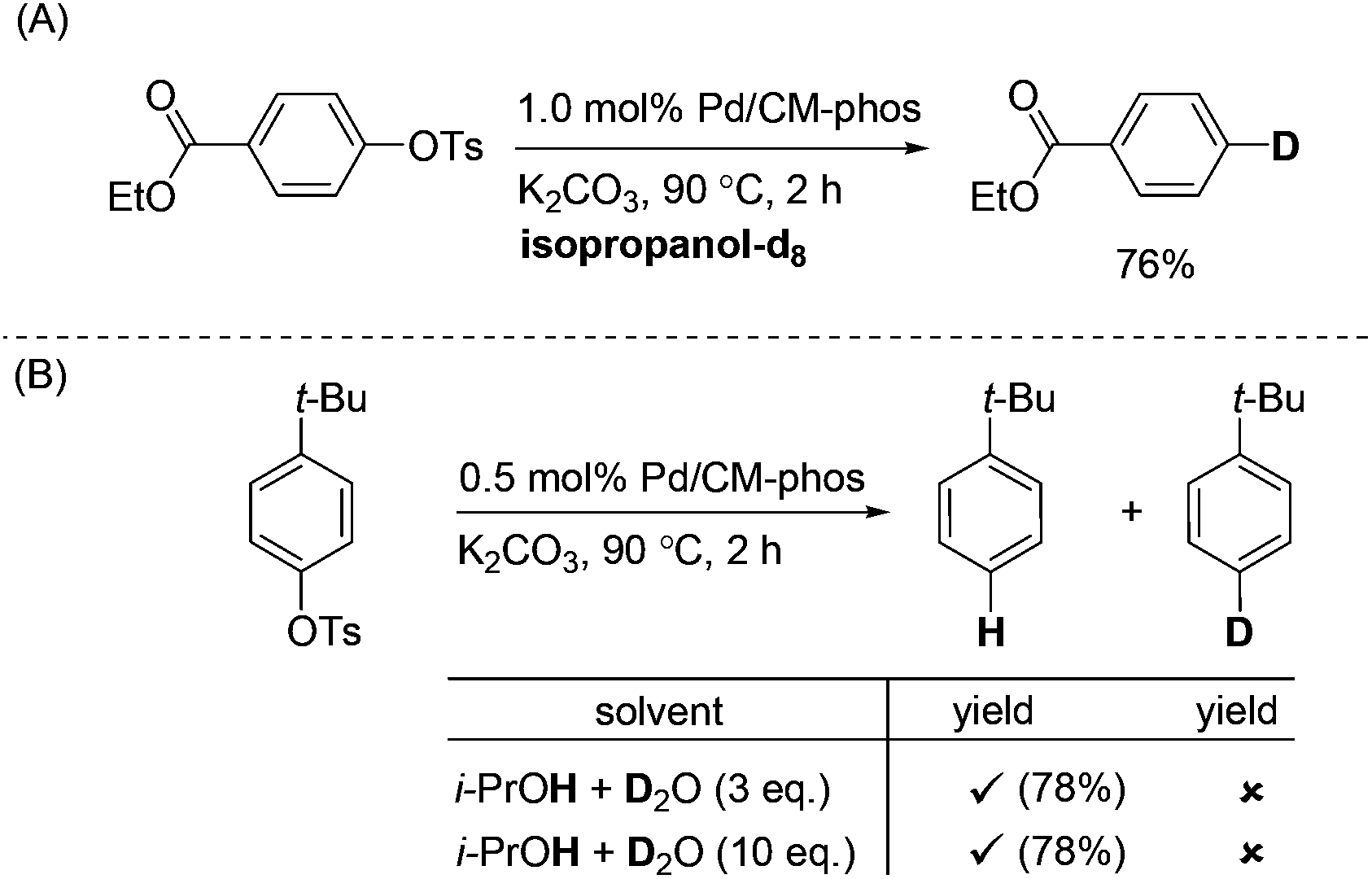 | ||
| Scheme 2 Labelling experiments of palladium-catalysed deoxygenation of aryl tosylates. | ||
The proposed mechanism is shown in Scheme 3. Aryl tosylate is oxidatively added to the palladium complex.16 Abstraction of TsOH by inorganic base generates an isopropoxy–palladium species. This intermediate undergoes β-hydride elimination to afford the corresponding aryl–palladium–hydride complex. Reductive elimination gives the deoxygenated product and regenerates the palladium catalyst.
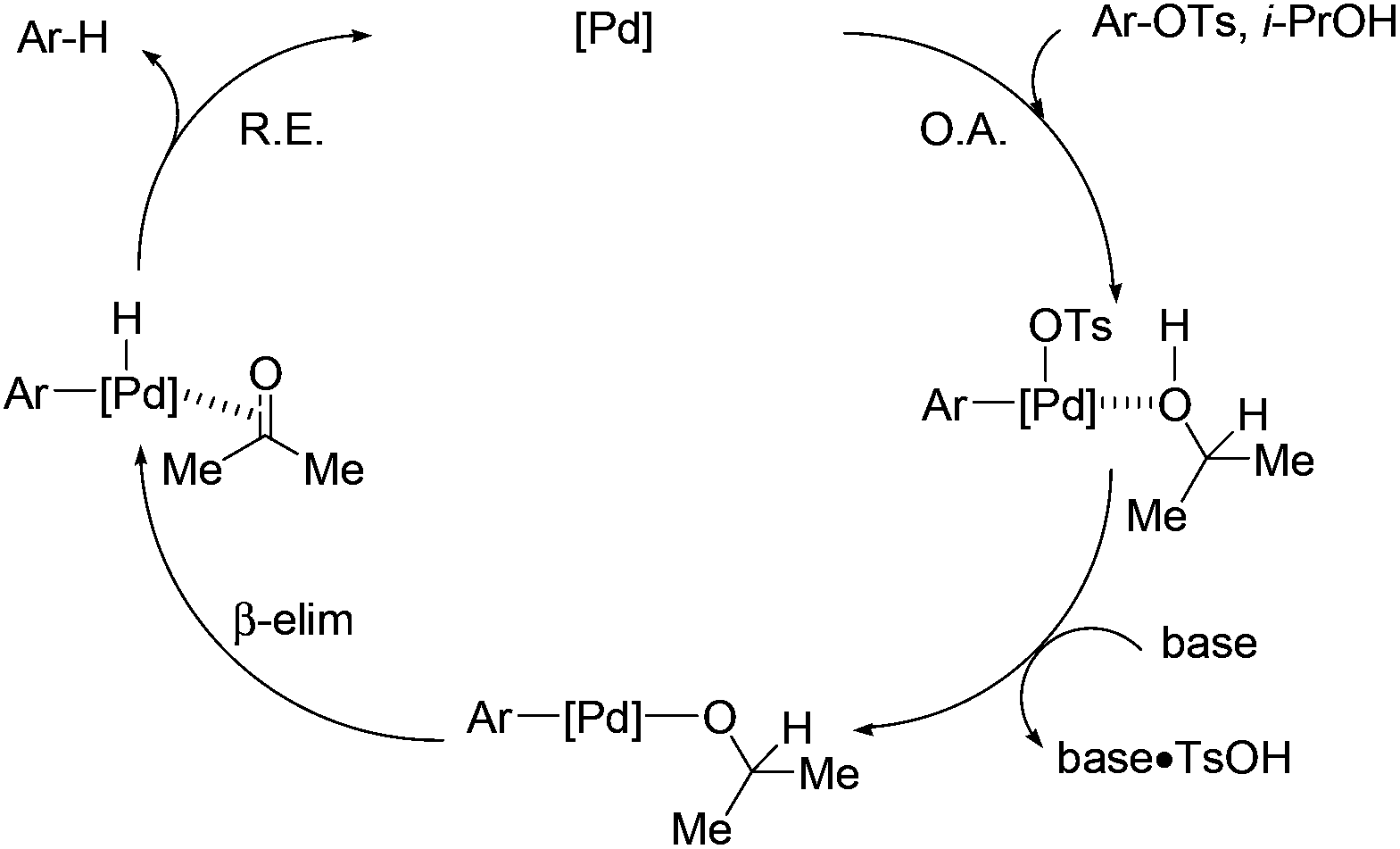 | ||
| Scheme 3 Proposed mechanism for the deoxygenation of ArOTs in isopropanol. | ||
General procedures
Pd(OAc)2 and CM-phos (Pd:L = 1:4, Pd loading as indicated) were loaded into a Schlenk tube (30 mL) equipped with a Teflon-coated magnetic stir bar (4 × 10 mm). The tube was evacuated and flushed with nitrogen for three times. Precomplexation was achieved by adding freshly distilled dichloromethane (∼1 mL) and Et3N (100 μL) into the tube. The solution was stirred and warmed using a hair drier for about 1 to 2 minutes until the solvent started boiling. The solvent was then evaporated under a high vacuum. Aryl tosylates (1.0 mmol) and K2CO3 (3.0 mmol) were loaded into the tube, and the system was further evacuated and flushed with nitrogen for three times. Isopropanol (3.0 mL) was then added. The tube was stirred at room temperature for several minutes and then placed into a preheated oil bath (90 °C) for the time period as indicated in the above tables. After completion of reaction as judged by GC analysis, the reaction tube was allowed to cool to room temperature and quenched with water and diluted with EtOAc. The organic layer was separated and the aqueous layer was washed with EtOAc. The filtrate was concentrated under reduced pressure. The crude products were purified by flash column chromatography on silica gel (230–400 mesh) to afford the desired product.
Conclusion
In summary, we have succeeded in showing the first general examples of deoxygenation of aryl tosylates under palladium catalysis. With the aid of the mild reaction conditions, a number of functional groups are tolerated, including keto, ester, nitrile and benzothiazolyl groups. Particularly noteworthy is that the Pd/CM-phos complex smoothly promotes the difficult deoxygenation of sterically hindered 2,6-disubstituted aryl tosylates. In fact, there has been no example reported so far for this type of substrate either with Ni or Pd catalysis. The deuterium-labelling experiments showed the possibility of incorporating a hydrogen atom into the aromatic ring via an aryl tosylate intermediate at a later stage of the synthetic sequence.17 Further study of this reaction towards the synthesis of pharmaceutically relevant intermediates is currently underway.Acknowledgements
We thank the Research Grants Council of Hong Kong (CRF: PolyU5014/10P). F.Y.K. thanks the Croucher Foundation for the Croucher Senior Research Fellowship 2013. The authors are grateful to Ms Pui Ying Choy for her assistance in supplementary material preparation.Notes and references
- For a recent review, see: (a) J. M. Herrmann and B. Konig, Eur. J. Org. Chem., 2013, 7017 CrossRef CAS; for book chapters, see: (b) M. Beller and C. Bolm, Transition metals for organic synthesis: building blocks and fine chemicals, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1–2 Search PubMed; (c) Metal-catalyzed cross-coupling reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1–2 Search PubMed.
- (a) M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, NJ, 7th edn, 2013 Search PubMed. For recent reviews, see: (b) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728 CrossRef CAS PubMed; (c) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (d) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963 RSC.
- For reviews about aryl triflates as synthetic precursors in coupling reactions, see: (a) P. J. Stang, M. Hanack and L. R. Subramanian, Synthesis, 1982, 85 CrossRef CAS; (b) K. Ritter, Synthesis, 1993, 735 CrossRef CAS.
- For selected nickel catalyst systems, see: (a) K. Sasaki, M. Sakai, Y. Sakakibara and K. Takagi, Chem. Lett., 1991, 2017 CrossRef CAS; (b) L. R. Subramanian, A. G. Martinez, A. H. Fernandez and R. M. Alvarez, Synthesis, 1984, 481 CrossRef CAS.
- For selected palladium catalyst systems, see: (a) S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 5541 CrossRef CAS; (b) J. M. Saá, M. Dopico, G. Martorell and A. Garcia-Raso, J. Org. Chem., 1990, 55, 991 CrossRef; (c) B. H. Lipshutz, D. J. Buzard and R. W. Vivian, Tetrahedron Lett., 1999, 40, 6871 CrossRef CAS; (d) Y. Pan and C. P. Holmes, Org. Lett., 2001, 3, 2769 CrossRef CAS PubMed; (e) J. D. Revell and A. Ganesan, Chem. Commun., 2004, 1916 RSC; (f) A. N. Cammidge and Z. Ngaini, Chem. Commun., 2004, 1914 RSC.
- For selected examples, see: (a) Y.-G. Si, M. P. Gardner, F. I. Tarazi, R. J. Baldessarini and J. L. Neumeyer, J. Med. Chem., 2008, 51, 983 CrossRef CAS PubMed; (b) J. Cianci, J. B. Baell, B. L. Flynn, R. W. Gable, J. A. Mould, D. Paul and A. J. Harvey, Bioorg. Med. Chem. Lett., 2008, 18, 2055 CrossRef CAS PubMed; (c) G. A. Peterson, F.-A. Kunng, J. S. McCallum and W. D. Wulff, Tetrahedron Lett., 1987, 28, 1381 CrossRef CAS; (d) S. Megati, S. E. Ealick, F. N. M. Naguib, M. H. el Kouni, R. S. Klein and B. A. Otter, Nucleosides Nucleotides, 1994, 13, 2151 CrossRef CAS.
- Besides sulfonated phenols, Ar–OR (R = alkyl) substrates were recently found to be deoxygenated by nickel catalyst systems, using HSiMe(OMe)2 or H2 as the reducing agents; see: (a) M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946 RSC; (b) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed; (c) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed.
- Aryl tosylates are far more inexpensive when compared with the corresponding aryl triflates, interms of attaining the precursors from phenols. The suggested prices of the sulfonating agents are: Tf2O, approx. USD$1.3 g−1; TsCl, approx. USD$0.042 g−1 from commercial suppliers.
- (a) F. Wang, K. Chiba and M. Tada, J. Chem. Soc., Perkin Trans. 1, 1992, 1897 RSC; (b) P. Sebök, T. Tímár, T. Eszenyi and T. Patonay, J. Org. Chem., 1994, 59, 6318 CrossRef.
- K. Sasaki, T. Kubo, M. Sakai and Y. Kuroda, Chem. Lett., 1997, 617 CrossRef CAS.
- B. H. Lipshutz, B. A. Frieman, T. Butler and V. Kogan, Angew. Chem., Int. Ed., 2006, 45, 800 CrossRef CAS PubMed.
- (a) H. Sajiki, A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa and K. Hirota, Org. Lett., 2006, 8, 987 CrossRef CAS PubMed; (b) A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2007, 13, 1432 CrossRef CAS PubMed.
- (a) C. M. So, C. P. Lau, Z. Zhou and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402 CrossRef CAS PubMed; (b) C. M. So, C. P. Lau, A. S. C. Chan and F. Y. Kwong, J. Org. Chem., 2008, 73, 7731 CrossRef CAS PubMed; (c) C. M. So, C. P. Lau and F. Y. Kwong, Chem. – Eur. J., 2011, 17, 761 CrossRef CAS PubMed; (d) P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Org. Lett., 2011, 13, 648 CrossRef CAS PubMed; (e) W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2010, 75, 5109 CrossRef CAS PubMed; (f) C. M. So, W. K. Chow, P. Y. Choy, C. P. Lau and F. Y. Kwong, Chem. – Eur. J., 2010, 16, 7996 CrossRef CAS PubMed; (g) P. Y. Choy, W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Chem. – Eur. J., 2010, 16, 9982 CrossRef CAS PubMed; (h) K. H. Chung, C. M. So, S. M. Wong, C. H. Luk, Z. Zhou, C. P. Lau and F. Y. Kwong, Chem. Commun., 2012, 48, 1967 RSC; (i) W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Chem. – Eur. J., 2011, 17, 6913 CrossRef CAS PubMed; (j) P. Y. Wong, W. K. Chow, K. H. Chung, C. M. So, C. P. Lau and F. Y. Kwong, Chem. Commun., 2011, 47, 8328 RSC.
- We used K2CO3 for the substrate scope study as K3PO4 resulted in the formation of more undesirable products from the alkaline hydrolysis of electron-deficient ArOTs in our system. Thus, K2CO3 is a more general base in this reaction.
- For example, the Ni/Cg/Ph3P/Me2NH·BH3 system is not compatible with benzothiazolyl and –CN containing substrates, see ref. 11.
- For a study of oxidative addition of ArOTs, see: A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704 CrossRef CAS PubMed.
- Ar-OTs groups are difficult to activate. The –OTs group could serve as the protecting group in the early synthetic sequence.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectral data. See DOI: 10.1039/c4qo00103f |
| This journal is © the Partner Organisations 2014 |