Concise synthesis of atorvastatin lactone under high-speed vibration milling conditions†
Verónica
Estévez
,
Mercedes
Villacampa
and
J. Carlos
Menéndez
*
Departamento de Química Orgánica y Farmacéutica, Facultad de Farmacia, Universidad Complutense, 28040 Madrid, Spain. E-mail: josecm@farm.ucm.es
First published on 16th April 2014
Abstract
The Hantzsch-type sequential three-component reaction under high-speed vibration milling conditions between 4-methyl-3-oxo-N-phenylpentanamide, tert-butyl 2-[(4R,6R)-6-(2-aminoethyl)-2,2-dimethyl-1,3-dioxan-4-yl]acetate and 1-(4-fluorophenyl)-2-iodo-2-phenylethanone in the presence of ytterbium triflate and silver nitrate, followed by hydrolytic deprotection/lactonization, afforded atorvastatin lactone in 38% overall yield, thus providing ready access to the top-selling antihyperlipidemic drug atorvastatin calcium.
Introduction
Mechanochemical synthesis is defined by the use of mechanical energy to induce chemical reactions.1,2 While milling has been long employed in the pharmaceutical, chemical and metallurgical industries, mechanochemistry is still in a very early stage of development. However, it is increasingly considered as one of the most exciting additions to the synthetic repertoire as an environmentally friendly alternative to solution-based reactions because of the possibility of carrying out solvent-free chemical processes. Recently, analytical techniques such as the diffraction of high-energy synchrotron X-rays3 and single-molecule force spectroscopy4 have allowed the in situ study of model mechanochemical reactions, leading to the conclusion that they proceed with reaction rates comparable to or greater than those in solution. Among the various mechanochemical techniques, high-speed vibration milling (HSVM) is particularly well suited for work on the laboratory scale and is based on the vibratory movement of an arm that is attached to a vessel containing a ball made of inert material that falls over the material up to 30 times per second. In spite of the current interest in the development of solvent-free synthetic methodologies prompted by environmental concerns, this technique has found relatively little application in synthesis, especially in terms of the generation of carbon–heteroatom bonds.5Multiple bond-forming transformations,6 and in particular, multicomponent reactions,7 have attracted much attention from the synthetic community in recent years because of the high efficiency due to the formation of several bonds in a single operation and also because the associated reduction in the number of isolation and purification steps is crucial for achieving a reduction of the use of organic solvents and chromatographic stationary phases, one of the main goals of green chemistry.8 Nevertheless, the coupling of multicomponent reactions with other methodologies aimed at the reduction of reaction waste has received little attention.
In this context, we present here the first example of the application of mechanochemical techniques to a target-oriented synthesis with a multicomponent reaction as the key step. Our chosen target was atorvastatin (Fig. 1),9,10 the best known representative of the statins, which are the main group of cholesterol-lowering drugs in therapeutic use.11 This compound was introduced in the market in 1997 as its calcium salt under the trade name Lipitor® and is the best-selling drug in history, having been at the top of the list of largest-selling branded pharmaceutical entities for almost a decade. The importance of atorvastatin has led to much interest in its synthesis, which has been the subject of several recent reviews and chapters.12,13 All these routes are aimed at the preparation of the so-called atorvastatin lactone, which can be easily transformed into the final drug molecule by hydrolysis and salt formation.
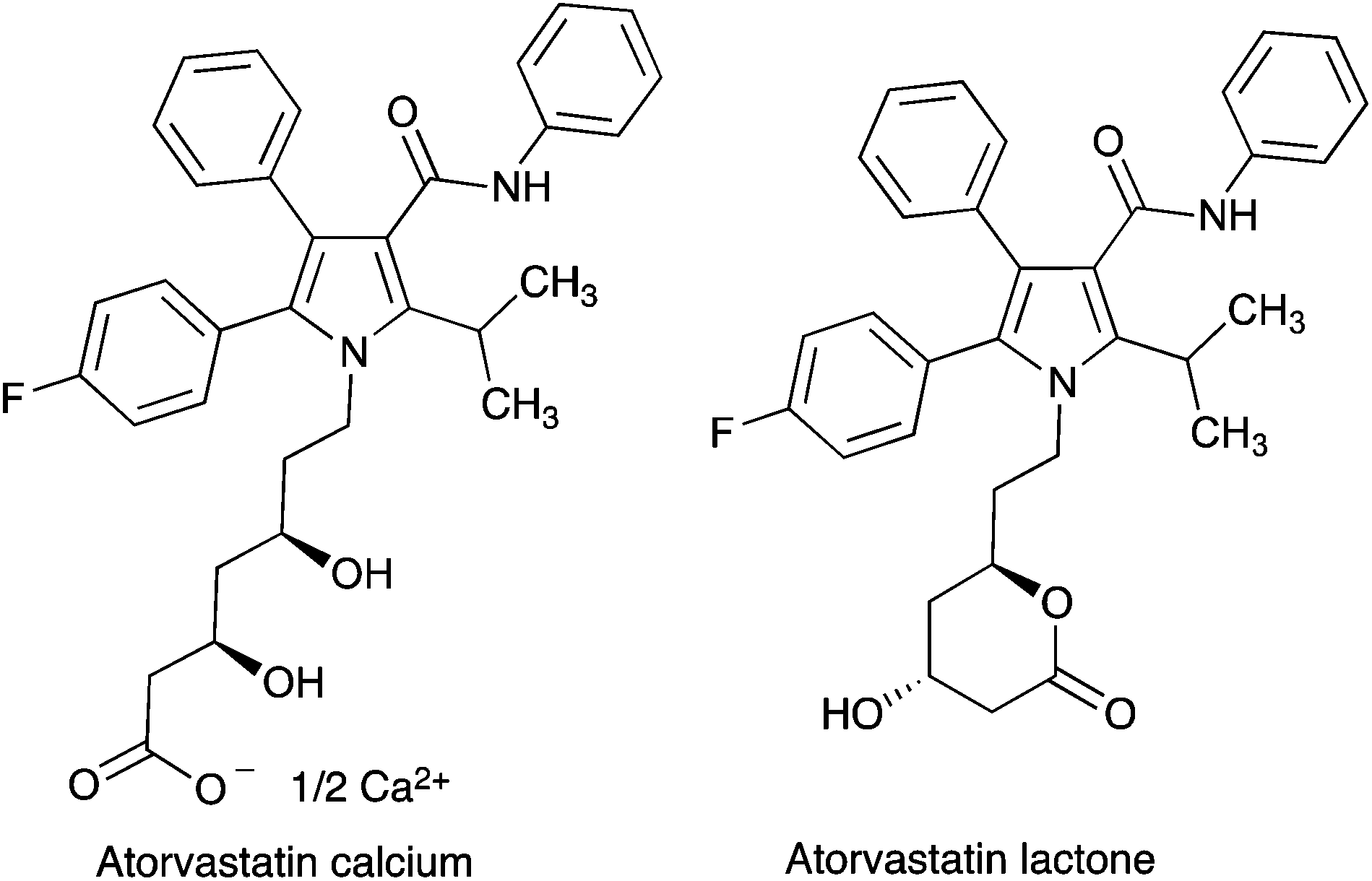 | ||
| Fig. 1 Structures of atorvastatin calcium and the atorvastatin lactone. | ||
Much effort has been directed towards the synthesis of the chiral side chain, and relatively little attention has been devoted to optimizing the construction of the atorvastatin pyrrole core. In published work, this has normally been achieved through linear sequences having as key steps the classical Paal–Knorr pyrrole synthesis or 1,3-dipolar cycloadditions.14 We realized that our recent development of a generalized version of the Hantzsch pyrrole synthesis under mechanochemical conditions in the presence of cerium(IV) ammonium nitrate and silver nitrate15 furnished an opportunity to develop a concise, convergent route to atorvastatin. Thus, the protected atorvastatin derivative 1 should be accessible along the lines summarized in Scheme 1 from α-iodoketone 2, β-ketoamide 3 and chiral amine 4. This disconnection is interesting in that, in spite of its name reaction status, the Hantzsch pyrrole synthesis has found little use in the literature because of its lack of generality, especially for the preparation of pentasubstituted pyrroles. The present paper describes the successful conclusion of this project.
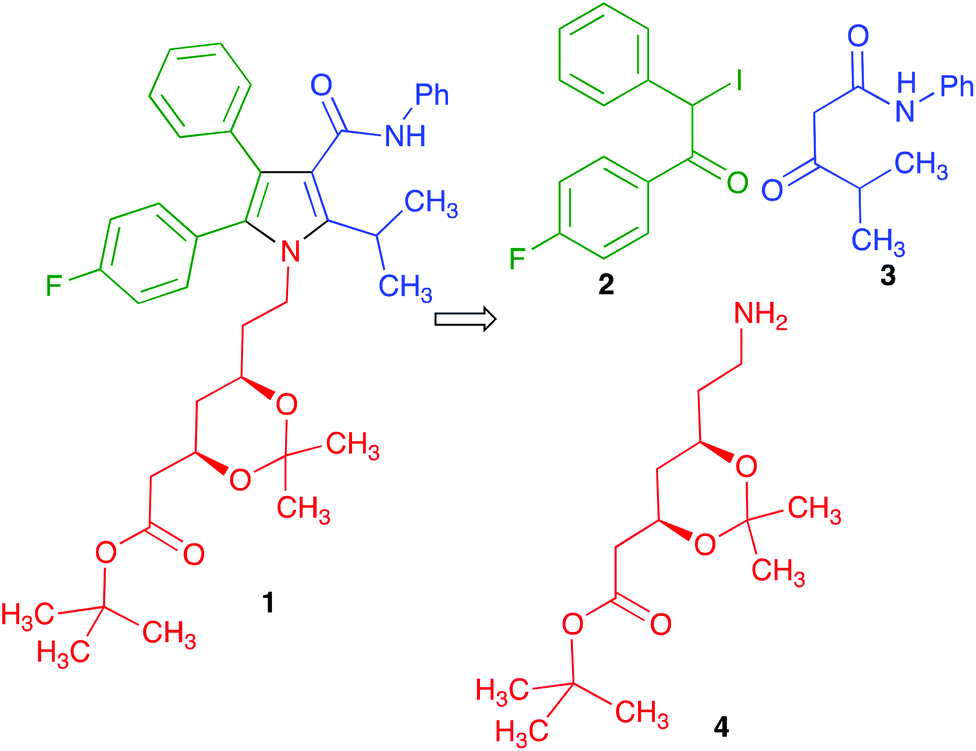 | ||
| Scheme 1 Our plan for the synthesis of a protected atorvastatin precursor (compound 1). | ||
Results and discussion
Regarding the synthesis of the required precursors, ketoamide 3 is known in the literature16 and amine 4 is commercially available. Compound 2 was prepared from the known acetophenone derivative 517 by α-bromination with hydrogen bromide in the presence of hydrogen peroxide,18 followed by halogen exchange with sodium iodide (Scheme 2).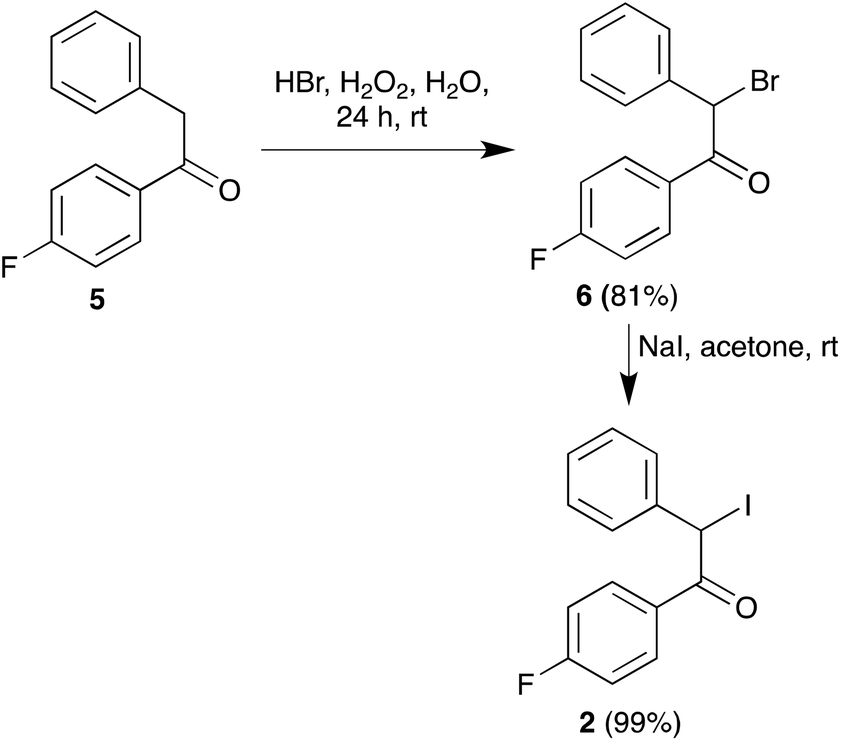 | ||
| Scheme 2 Synthesis of the α-iodoketone component (compound 2). | ||
We recognized that the structural features of atorvastatin took our Hantzsch-like reaction to its limits because it represents an unfavorable case for several of the substituents involved, including an amide function and a branched substituent in the β-dicarbonyl component, two aryl substituents in the α-haloketone and a complex primary amine component. For this reason, before committing the valuable chiral material 4, we decided to optimize the conditions for the pyrrole synthesis using hexylamine as a model primary amine. In doing so, we found that addition of a mixture of compound 3, CAN and hexylamine to iodoketone 2 and silver nitrate followed by high-speed vibration at 20 Hz for 1 h afforded the atorvastatin analog 7a in 65% yield. A similar experiment using benzylamine as a more hindered model primary amine furnished compound 7b in 40% yield (Scheme 3). Compound 7b was known in the literature,19 but it had been obtained as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with its 4-(p-fluorophenyl)-5-phenyl regioisomer via 1,3-dipolar cycloaddition chemistry.
:1 mixture with its 4-(p-fluorophenyl)-5-phenyl regioisomer via 1,3-dipolar cycloaddition chemistry.
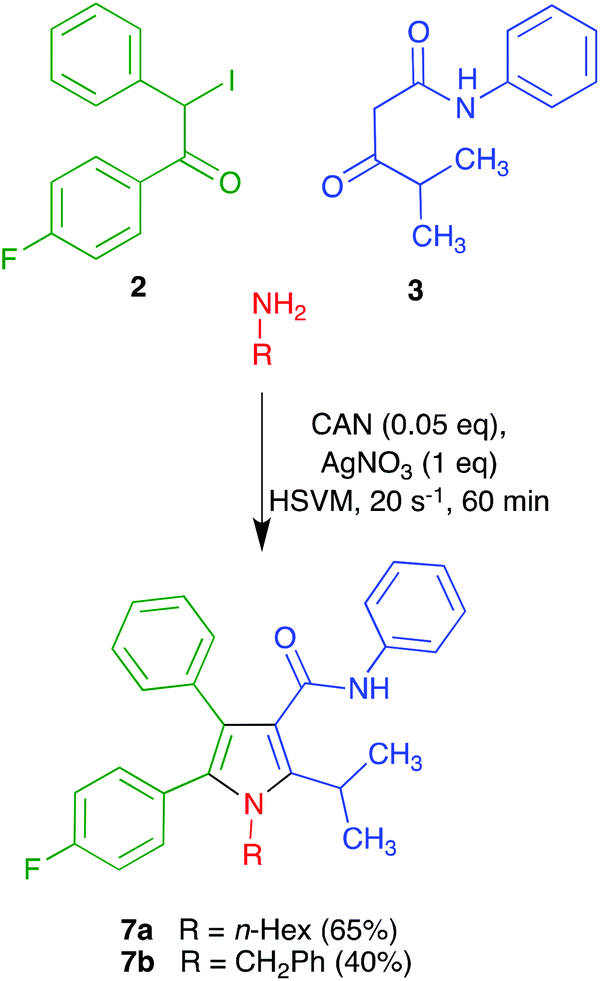 | ||
| Scheme 3 Model reactions leading to atorvastatin analogues 7. | ||
Unfortunately, when we applied the conditions established for the model compounds to the desired reaction between 2, 3 and 4, we obtained only complex mixtures instead of the target pyrrole derivative. We reasoned that this different behaviour of amine 4 with respect to the models could be associated to the fact that CAN is known to promote the hydrolysis of acetals20 and might therefore be affecting the acetonide protection. For this reason, a change in the Lewis acid was considered necessary and, after some experimentation, we discovered that ytterbium triflate was a suitable catalyst for the desired process. As a relatively minor downside, the generation of the intermediate β-enaminone involved in the mechanism of the Hantzsch reaction was slower than in the presence of CAN and required a mild heating of β-ketoamide 3 and amine 4 in the presence of 1% ytterbium triflate. The pure enaminone 8 thus obtained was mixed with compound 2, 1% ytterbium triflate and an equimolecular amount of silver nitrate, which was necessary to prevent reductive dehalogenation of 2 by the iodide anion liberated during the reaction.15 This mixture was submitted to high-speed vibration milling at 20 Hz for 1 h, leading to compound 1 in 40% overall yield. A final hydrolytic deprotection followed by acid-promoted spontaneous lactonization afforded atorvastatin lactone in 94% yield (Scheme 4). The transformation of compound 9 into atorvastatin calcium can be regarded as trivial.18,21
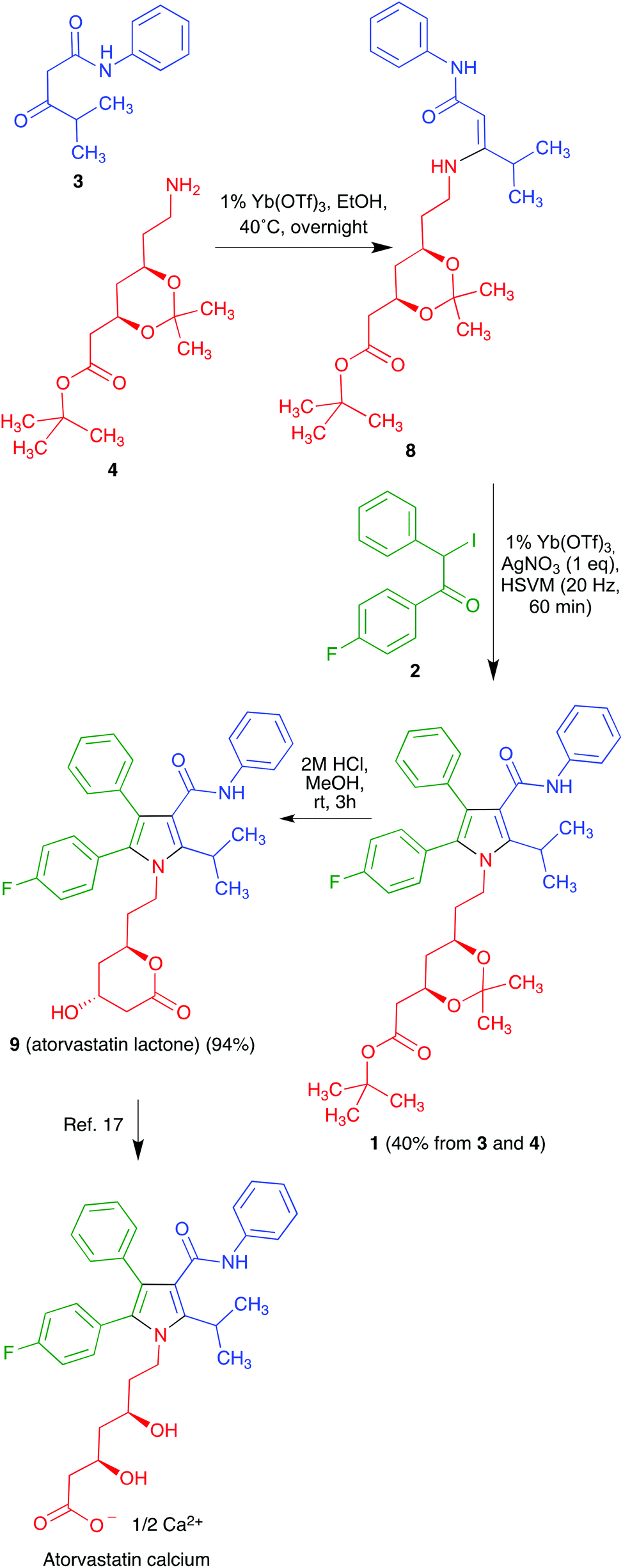 | ||
| Scheme 4 Synthesis of the atorvastatin lactone. | ||
Experimental
General experimental information
All reagents and solvents were of commercial quality and were used as received. Reactions were monitored by thin layer chromatography on aluminium plates coated with silica gel and fluorescent indicator. Separations by flash chromatography were performed on silica gel. Mechanochemical reactions were carried out in a Retsch MM200 mixer ball mill at a frequency of 20 Hz using a 25 mL zirconium oxide grinding jar and a single zirconium oxide ball 20 mm in diameter. Melting points were measured with a Kofler-type heating platine microscope from Reichert, 723 Model, and are uncorrected. The optical rotation measurements were determined at 20 °C, on a 1 ml cell measuring 10 cm, using a Perkin Elmer 240 polarimeter operating at the emission wavelength of a sodium lamp (589 nm); the concentration c is given in g per 100 mL. Infrared spectra were recorded with a Perkin-Elmer FTIR Paragon-1000 spectrophotometer as thin films on NaCl disks; wavenumbers are given in cm−1. NMR spectroscopic data were recorded using spectrometers maintained by the CAI de Resonancia Magnética, UCM, operating at 250 or 500 MHz for 1H NMR, 235 MHz for 19F and 63 or 125 MHz for 13C NMR; chemical shifts are given in (δ) parts per million and coupling constants (J) in hertz. High-resolution mass spectra (HRMS) were recorded by the CAI de Espectrometría de Masas, UCM, on a Bruker FTMS APEX QIV mass spectrometer. Elemental analyses were determined by the CAI de Microanálisis, Universidad Complutense, using a Leco CHNS-932 combustion microanalyzer.:1), gave the desired bromide 6 as a yellow oil (2.365 g, 81%). IR (NaCl, neat) 1693, 991 cm−1; 1H NMR (CDCl3, 250 MHz) δ 8.07–8.02 (m, 2H), 7.54 (dd, J = 8.1, 1.9 Hz, 2H), 7.41–7.38 (m, 3H), 7.18–7.11 (m, 2H), 6.35 (s, 1H); 13C NMR (CDCl3, 63 MHz) δ 189.5 (C), 165.8 (d, J = 257.1 Hz, C), 135.6 (C), 131.8 (d, J = 9.5 Hz, C), 130.3 (d, J = 3.0 Hz, CH), 129.2 (CH), 129.0 (2 CH), 116.0 (d, J = 22.0 Hz, CH), 50.8 (CH); 19F NMR (CDCl3, 235 MHz) δ (−103.81) to (−103.93) (m). Anal. Calcd for C14H10BrFO: C, 57.36; H, 3.44. Found: C, 57.31; H, 3.39.
General procedure for the CAN-catalyzed synthesis of pyrroles
To a ball mill vessel was added α-iodoketone 2 (340 mg, 1 mmol), along with a zirconium oxide ball. Then, a mixture of the suitable amine (1.95 mmol), β-ketoamide 3 (308 mg, 1.5 mmol) and cerium ammonium nitrate (CAN, 27 mg, 0.05 eq.), previously stirred together at room temperature over 30 min, and silver nitrate (169 mg, 1 mmol) were added. The vessel was fitted to one of the horizontal vibratory arms of the ball mill, while the other arm was occupied with an empty vessel. The ball mill was set to vibrate at a frequency of 20 Hz for 60 min at room temperature. The reaction vessel was cleansed with ethyl acetate (5 mL) and the suspension thus obtained was filtered to remove silver iodide. The organic layer was washed with water (2 mL) and dried over anhydrous sodium sulphate, and the solvent was evaporated under reduced pressure. Purification by column chromatography on silica gel eluting with a petroleum ether–ethyl acetate mixture (9:1) afforded the desired pyrroles.
Compound 2 (102 mg, 0.3 mmol) was added to a ball mill vessel, followed by ytterbium triflate (2 mg, 0.003 mmol) and silver nitrate (85 mg, 0.5 mmol). Then, a solution of the intermediate β-enaminone 8 (230 mg, 0.5 mmol) in ethanol (0.5 mL) was also added to the reaction vessel and the solvent was evaporated under a stream of argon. The vessel was fitted to one of the horizontal vibratory arms of the ball mill, while the other arm was occupied with an empty vessel. The ball mill was set to vibrate at a frequency of 20 Hz for 60 min. After extracting the reaction mixture from the ball mill vessel with ethyl acetate (5 mL), the suspension obtained was filtered and the organic layer was washed with water (2 mL) and dried over anhydrous sodium sulphate. Finally, the solvent was evaporated under reduced pressure. Purification was achieved through silica gel column chromatography, using a mixture of petroleum ether–ethyl acetate (8:2) as eluent to yield compound 1 as a yellow solid (79 mg, 40%).
Data for 8: [α]20D +12.8 (c 1.2, CHCl3); IR (NaCl, neat) 3331, 1726, 1677, 1596, 1254 cm−1; 1H-NMR (250 MHz, CDCl3) δ 9.37 (br s, 1H), 7.46 (d, J = 7.4 Hz, 2H), 7.29 (t, J = 7.4 Hz, 2H), 7.02 (t, J = 7.4 Hz, 1H), 6.72 (br s, 1H), 4.47 (s, 1H), 4.33–4.22 (m, 1H), 4.07–3.96 (m, 1H), 3.74 (q, J = 7.0 Hz, 2H), 3.42–3.29 (m, 2H), 2.75–2.69 (m, 1H), 2.38 (qd, J = 15.0, 7.0 Hz, 2H), 1.72 (q, J = 6.0 Hz, 2H), 1.39 (s, 9H), 1.27 (s, 6H), 1.16 (d, J = 6.7 Hz, 6H); 13C NMR (63 MHz, CDCl3) δ 170.3 (C), 170.1 (C), 169.6 (C), 139.3 (C), 128.7 (CH), 122.5 (CH), 119.5 (CH), 98.7 (C), 80.6 (C), 79.8 (CH), 66.1 (CH), 65.7 (CH), 42.6 (CH2), 37.7 (CH2), 37.2 (CH2), 36.4 (CH2), 28.4 (CH), 28.0 (CH3), 21.5 (CH3), 21.4 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C26H40N2O5Na 483.2835; found 483.2125.
Data for 1: mp 72–74 °C; [α]20D +5.5 (c 1.5, CHCl3); IR (NaCl, neat) 3315, 1724, 1667, 1599, 1231, 1158 cm−1; 1H-NMR (CDCl3, 250 MHz) δ 7.24–6.94 (m, 14H), 6.88 (br s, 1H), 4.24–4.04 (m, 2H), 3.93–3.81 (m, 1H), 3.77–3.69 (m, 1H), 3.60–3.54 (m, 1H), 2.42 (dd, J = 15.3, 6.2 Hz, 1H), 2.26 (dd, J = 15.3, 6.2 Hz, 1H), 1.73–1.65 (m, 2H), 1.55 (d, J = 7.1 Hz, 6H), 1.46 (s, 9H), 1.39 (s, 3H), 1.32 (s, 3H), 0.86 (m, 2H); 13C-NMR (CDCl3, 125 MHz) δ 170.2 (C), 164.8 (C), 162.2 (d, J = 246.2 Hz, C), 141.5 (C), 138.3 (C), 134.6 (C), 133.1 (d, J = 8.1 Hz, CH), 130.5 (CH), 128.7 (C), 128.6 (CH), 128.3 (CH), 128.2 (d, J = 3.4 Hz, C), 126.5 (CH), 123.5 (CH), 121.7 (C), 119.5 (CH), 115.3 (d, J = 21.3 Hz, CH), 115.2 (C), 98.6 (C), 80.7 (C), 66.4 (CH), 65.8 (CH), 42.4 (CH2), 40.8 (CH2), 38.0 (CH2), 35.9 (CH2), 28.0 (CH3), 26.0 (CH), 21.7 (CH3), 21.5 (CH3), 19.6 (CH3); 19F NMR (CDCl3, 235 MHz) δ (−114.08) to (−114.18) (m). Anal. Calcd for C40H47FN2O5 C, 73.37; H, 7.23; N, 4.28. Found: C, 73.31; H, 7.19; N, 4.24.
:1, 6 mL). Thereafter, 2 M aqueous HCl was added to the aqueous layer until its pH was neutral and it was then extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to give atorvastatin lactone 9 as a white solid (63 mg, 94%); mp 150–153 °C; [α]20D +24.4 (c 1.0, CHCl3) [lit.22 +25.5 (c 0.2, CHCl3); lit.23 +26.05 (c 1.0, CHCl3)]; IR (NaCl, neat) 3406, 1715, 1651, 1596, 1248, 1157 cm−1; 1H-NMR (CDCl3, 250 MHz) δ 7.24–6.96 (m, 14H), 6.91 (br s, 1H), 4.20–3.90 (m, 1H), 4.25–4.09 (m, 3H), 3.61–3.55 (m, 1H), 2.67–2.62 (m, 2H), 2.35 (br s, 1H), 1.78–1.54 (m, 4H), 1.67–1.57 (m, 6H); 13C-NMR (CDCl3, 63 MHz) δ 169.4 (C), 163.1 (C), 162.5 (d, J = 246.6 Hz, C), 141.3 (C), 138.2 (C), 134.3 (C), 133.1 (d, J = 8.1 Hz, CH), 133.0 (C), 130.4 (CH), 128.7 (CH and C), 128.4 (CH), 128.3 (d, J = 3.9 Hz, C), 126.6 (CH), 123.7 (CH), 119.6 (CH and C), 115.7 (d, J = 21.7 Hz, CH), 115.6 (C), 73.0 (CH), 65.5 (CH), 42.4 (CH2), 40.7 (CH2), 38.5 (CH2), 37.1 (CH2), 35.6 (CH2), 26.1 (CH), 22.0 (CH3), 21.7 (CH3). HRMS (ESI) m/z [M + Na]+ calcd for C33H33FN2O4Na 563.2322, found 563.2318. Anal. Calcd for C33H33FN2O4: C, 73.31; H, 6.15; N, 5.18. Found: C, 73.28; H, 6.12; N, 5.22.
Conclusions
In conclusion, we have developed a very short, convergent synthesis of atorvastatin lactone based on a mechanochemical Hantzsch-like pyrrole synthesis in the presence of ytterbium triflate and silver nitrate. Our method provides the target compound in 38% overall yield from precursors that are either commercially available or can be prepared in very high yield from commercial materials. This synthesis illustrates the power of the mechanochemical approach by providing the first example of a target-oriented synthesis using as the key step a reaction employing high-speed vibration milling as the only source of energy.Acknowledgements
Financial support from Ministerio de Economía y Competitividad, MINECO (grant CTQ2012-33272-BQU) is gratefully acknowledged. VE also acknowledges a predoctoral contract from Comunidad Autónoma de Madrid.Notes and references
- For reviews, see: (a) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC; (b) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (c) G. A. Bowmaker, Chem. Commun., 2013, 49, 334 RSC . See also ref. 3.
- See also the following Themed Issues: (a) Chemical Communications: S. James and T. Friščić (ed.) Mechanochemistry: fundamentals and applications in synthesis (http://pubs.rsc.org/en/journals/articlecollectionlanding?sercode=cc&themeid=6608cfd9-e113-499d-8069-3691fd2efb77&journalname=chemical%20communications); (b) Chemical Society Reviews: S. James and T. Friščić (ed.) Mechanochemistry. http://pubs.rsc.org/en/journals/journalissues/cs#!issueid=cs042018&type=current&issnprint=0306-0012.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66 CrossRef PubMed.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, Nat. Chem., 2013, 5, 110 CrossRef CAS PubMed.
- B. Rodríguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213 CrossRef.
- For a recent conceptual review on multiple bond forming transformations, see: D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Chem. – Eur. J., 2013, 19, 2218–2231 CrossRef CAS PubMed.
- For selected reviews on multicomponent reactions, see: (a) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (b) M. M. Sánchez-Duque, C. Allais, N. Isambert, T. Constantieux and J. Rodriguez, Top. Heterocycl. Chem., 2010, 23, 227 Search PubMed; (c) B. Jiang, T. Rajale, W. Wever, S.-J. Tu and G. Li, Chem. – Asian J., 2010, 5, 2318 CrossRef CAS PubMed; (d) H. Eckert, Molecules, 2012, 17, 1074 CrossRef CAS PubMed; (e) M. J. Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16 CAS; (f) C. de Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969 RSC; (g) G. van der Heijden, E. Ruijter and R. V. A. Orru, Synlett, 2013, 666–685 CAS.
- For a recent review of the importance of multicomponent reactions in green chemistry, see: R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16 10.1039/c4gc00013g.
- For a review of the discovery and development of atorvastatin, see: B. D. Roth, Prog. Med. Chem., 2002, 40, 1 CrossRef CAS.
- For a review of the pharmacology of atorvastatin, see: A. P. Lea and D. McTavish, Drugs, 1997, 53, 828 CrossRef CAS PubMed.
- For general information about the statins family, see: (a) J. A. Tobert, Nat. Rev. Drug Discovery, 2003, 2, 517 CrossRef CAS PubMed; (b) E. S. Istvan and J. Deisenhofer, Science, 2001, 292, 1160 CrossRef CAS PubMed.
- For reviews of atorvastatin synthesis, see: (a) L. Jie-Jack, S. J. Douglas, R. S. Drago and D. R. Bruce, Contemporary Drug Synthesis. Wiley-Interscience, 2004, ch. 9 Search PubMed; (b) Z. Casar, Curr. Org. Chem., 2010, 14, 816 CrossRef CAS.
- For a review of process chemistry related to the preparation of atorvastatin, see: P. J. Harrington, Pharmaceutical process chemistry for synthesis: Rethinking the routes to scale-up, Wiley, 2011, ch. 9 Search PubMed.
- For reviews of the synthesis of pyrroles, see: (a) V. F. Ferreira, M. C. B. V. de Souza, A. C. Cunha, L. O. R. Pereira and M. L. G. Ferreira, Org. Prep. Proced. Int., 2001, 33, 411 CrossRef CAS; (b) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (c) S. D. Joshi, U. A. More, V. H. Kulkarni and T. M. Aminabhavi, Curr. Org. Chem., 2013, 17, 2279 CrossRef CAS; (d) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed.
- V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Commun., 2013, 49, 591 RSC.
- For the synthesis of β-ketoamide 3, see: H. Jendralla, E. Baader, W. Bartmann, G. Beck, A. Bergmann, E. Granzer, B. V. Kerekjarto, K. Kesseler, R. Krause, W. Schubert and G. Wess, J. Med. Chem., 1990, 33, 61 CrossRef CAS.
- For the synthesis of 5, see: R. Ramajayan, R. Giridhar, M. R. Yadav, R. Balaraman, H. Djaballah, D. Shum and C. Radu, Eur. J. Med. Chem., 2008, 43, 2004 CrossRef PubMed.
- For the bromination method, see: A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212 RSC.
- P. S. Pandey and T. S. Rao, Bioorg. Med. Chem. Lett., 2004, 14, 129 CrossRef CAS PubMed.
- For precedent for the cerium(IV)-catalyzed hydrolysis of acetals and ketals, see: I. E. Markó, A. Ates, A. Gautier, B. Leroy, J. M. Plancher, Y. Quesnel and J. C. Vanherck, Angew. Chem., Int. Ed., 1999, 38, 3207 CrossRef.
- H. W. Lee, Y. M. Kim, C. L. Yoo, S. K. Kang and S. K. Ahn, Biomol. Ther., 2008, 16, 28 CrossRef CAS.
- P. Sawant and M. E. Maier, Tetrahedron, 2010, 66, 9738 CrossRef CAS PubMed.
- K. L. Baumann, D. E. Butler, C. F. Deering, K. E. Mennen, A. Millar, T. N. Nanninga, C. W. Palmer and B. D. Roth, Tetrahedron Lett., 1992, 33, 2283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR (including DEPT-135) and 19F-NMR spectra. See DOI: 10.1039/c4qo00052h |
| This journal is © the Partner Organisations 2014 |