Fusarisetins: structure–function studies on a novel class of cell migration inhibitors†
Eduardo J. E.
Caro-Diaz
a,
Aereas
Aung
b,
Jing
Xu
a,
Shyni
Varghese
b and
Emmanuel A.
Theodorakis
*a
aDepartment of Chemistry & Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA. E-mail: etheodor@ucsd.edu
bDepartment of Bioengineering, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
First published on 7th February 2014
Abstract
Herein, we report the effects of fusarisetin A on cell morphology focusing in particular on actin and microtubules dynamics. We also report the synthesis and structure–function studies of a designed library of synthetic fusarisetins in cell-based assays.
While there have been tremendous advances in cancer research and treatment, the mortality caused by this disease represents still an enormous challenge.1 In fact, according to the American Cancer Society, more than 1.5 million new cancer cases are diagnosed every year and more than 0.5 million Americans will die annually from this disease.2 About 90% of these deaths are attributed to cancer metastasis, which is the ability of tumor cells to migrate from their tissue of origin and colonize elsewhere in the body.3 When cancer is detected at an early stage, before it has spread, it can often be treated successfully either by local therapy (surgery, radiation) or by systemic therapy (chemotherapy, targeted therapy, hormonal therapy).4 However, when cancer is detected after it has metastasized, such treatments are much less successful. Furthermore, for many patients for whom there is no evidence of metastasis at the time of their initial diagnosis, metastatic tumors could be detected at a later time.5 Thus, there is a clear need to develop novel and effective treatment strategies that target tumor progression by interfering with the metastatic process.
In principle, it is possible to halt (or retard) cancer metastasis at different stages with the help of cell migration inhibitors.6 A significant benefit of identifying such inhibitors stems from the observation that they do not need to be cytotoxic and thus they do not produce side effects associated with the use of current chemotherapeutic agents. This approach has led to drug discovery strategies that affect cancer metastasis by targeting critical regulators of extracellular matrix, cell motility and adhesion.7,8 These efforts demonstrate the great therapeutic potential of targeting cancer metastasis and the urgent need to identify potent inhibitors of this process. In the recent literature only few examples exist of natural product anti-migration agents that have been synthetically pursued leading to their evaluation in different biological models.9
Screening efforts to identify novel cell migration inhibitors led to the isolation of fusarisetins A (1) and B (2) (Fig. 1). Isolated from a Fusarium species, these fungal metabolites were found to inhibit acinar morphogenesis in MDA-MB-231 cells, a highly metastatic breast cancer cell line.10 Their peculiar biological activity, in combination with their novel pentacyclic motif, has attracted the attention of the synthetic community. To-date, several total syntheses of these natural products have been described in the literature.11–14 We have recently reported a scalable synthesis of fusarisetins,12,15 which yielded significant amounts of synthetic material in enantiopure form (greater than 95% ee) paving the way for a methodical structure–function study presented herein.
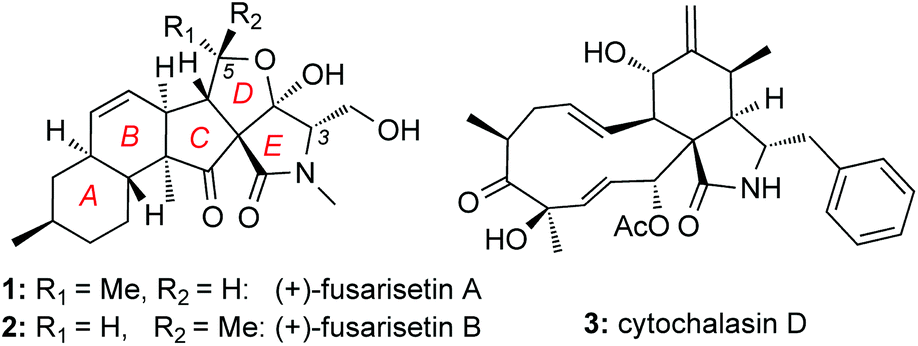 | ||
| Fig. 1 Selected natural products that inhibit cell migration. | ||
At the onset of this investigation, we evaluated the effects of 1 on the cell morphology and function. In general, motility inhibitors are known to interfere with microtubules,16 actin17 and/or cell adhesion processes.18 For instance cytochalasin D (3), a natural product that binds to actin filaments and induces actin depolymerization, is a well-known cell motility inhibitor.19 Intrigued by the observation that 3 is structurally and biogenetically related to 1,20 we sought to compare their effects in vitro. Incubation of cells with 3 induced the expected actin depolymerization as shown by lack of actin fibers (Fig. 2, column A). This effect was reversible, since removal of 3, by washing the cells with PBS and re-incubation with vehicle control, led to recovery of the actin network (Fig. 2, column B). Removal of 3 followed by re-incubation with growth media containing 1, led to reconstitution of actin filaments (Fig. 2, column C), indicating that fusarisetin A does not affect actin polymerization. Similar observations were made upon sole treatment of cells with 1 (Fig. 2, column D). Moreover, in all cases we observed healthy microtubules morphology (Fig. 2). Based on these findings we can conclude that fusarisetin A does not affect actin nor microtubules dynamics. These results, parallel previous findings10 and further support the notion that fusarisetin A acts via a novel mechanism of action that is distinctly different from those of known anti-migration agents.
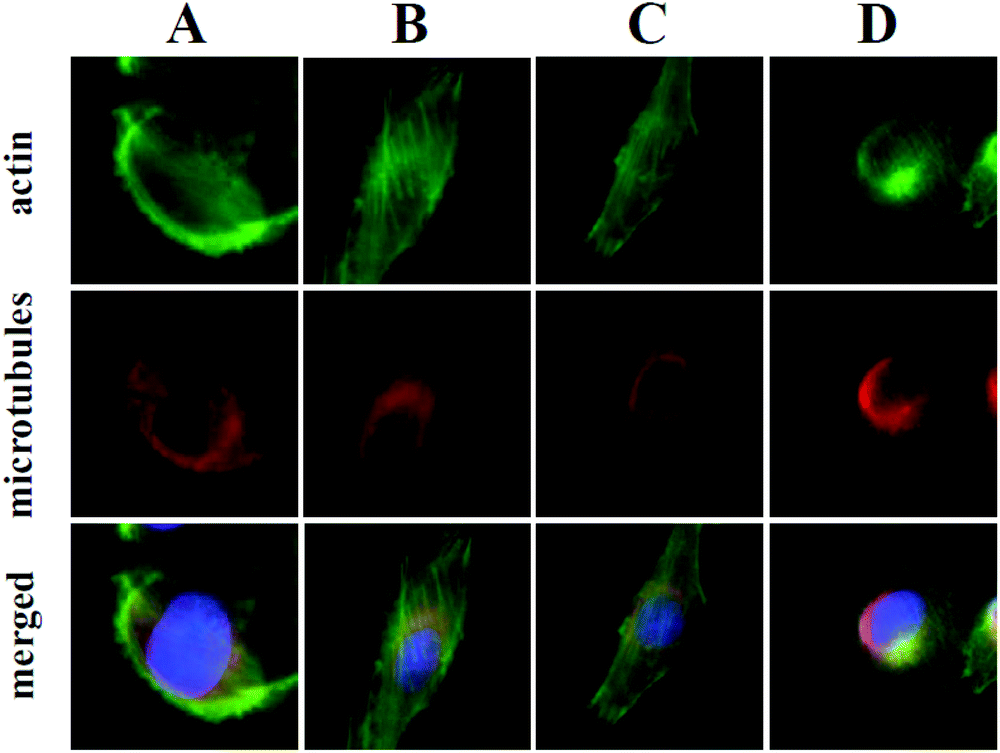 | ||
| Fig. 2 Effect of cytochalasin D (3) and fusarisetin A (1) on actin and microtubule dynamics. MDA-MB-231 cells stained for actin (phalloidin, green), microtubules (β-tubulin, red) and nuclei (DAPI, blue). Cells treated with 3 (4 h, 10 μg mL−1) (column A); were washed with PBS to remove 3 and then incubated with: vehicle control (column B); or 1 (12 h, 10 μg mL−1) (column C). Cells treated solely with 1 (12 h, 10 μg mL−1) (column D). | ||
At present, the key structural features of fusarisetins that account for their anti-motility properties are unknown. To address this issue, we sought to construct a library of analogs in which key reactive sites of 1 were systematically evaluated for their bioactivity. Interestingly, incorporation of various amino acids has been shown to occur in biosynthetically related natural products that derive from the same gene cluster of Fusarium species.20 We began this study by exploring the biological significance of the N-methyl serine moiety in fusarisetin A by varying the amino acid motif (C3 modification) (Scheme 1). Our synthetic strategy towards 1 allows coupling of N-methyl serine with β-keto acid 4, and conversion of the resulting amide to fusarisetin A via a key oxidative radical cyclization (ORC) reaction cascade. Using this strategy, β-keto acid 4 was coupled with the N-methyl amino methyl esters of phenyl alanine (5a), alanine (5b), isoleucine (5c) and glycine (5d) to produce compounds 6a–6d respectively. Treatment of these adducts with NaOMe led to tetramic acid analogs 7a to 7d (86–94% over 2 steps). It is worth noting that during this Dieckmann condensation we observed only minimal epimerization at the C3 center. On the other hand, significant racemization at the C3 center was observed when a serine analog was cyclized under the same conditions en route to the synthesis of 1.15 This difference is attributed to the methyl hydroxy group of serine that inductively increases the acidity of the C3 proton.21 Exposure to cerium ammonium nitrate (CAN) in acetic acid under oxygen atmosphere followed by reduction of the resulting endoperoxides,22 produced fusarisetin analogs 8a–8d (18–25% overall yield).
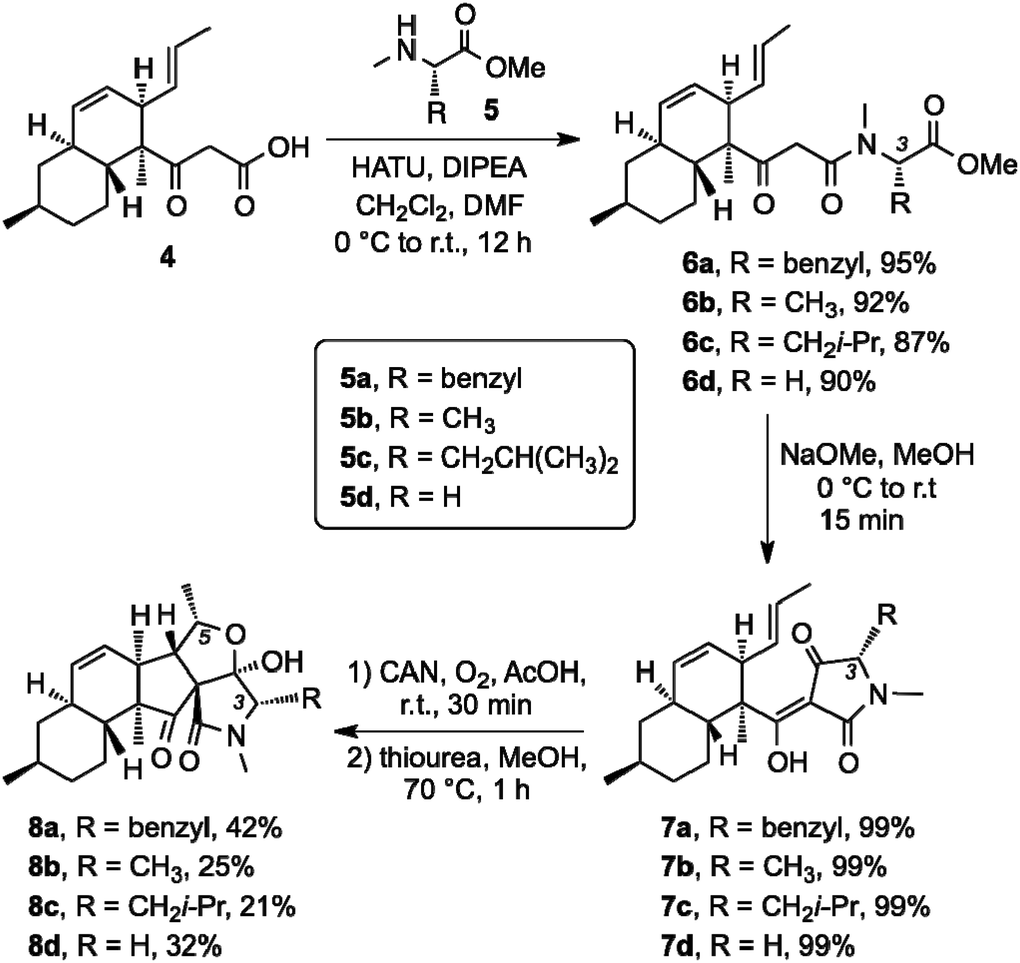 | ||
| Scheme 1 Synthesis of C3 analogs via a peptide coupling with diverse N-methyl amino ester fragments followed by ORC/reduction sequence. | ||
The AB decalin ring system of fusarisetins is also present in a wide array of natural products.23 It was hypothesized that the biological properties of 1 arise from the unique architectural motif of its CDE ring system. We also speculated that the stereochemistry at the C5 center is not critical to the fusarisetin bioactivity since both fusarisetin A (1) and B (2) are equipotent. With this in mind, we developed a synthesis of truncated analog 12 containing a C5 dimethylated center that, in turn, simplifies the stereoisomeric ratio obtained during the ORC reaction (Scheme 2). Compound 12 was synthesized via the following sequence: (a) kinetic alkylation of ethylacetoacetate (9) with prenyl bromide to form 10; (b) saponification of the ethyl ester followed by coupling of the resulting carboxylic acid with 5a to produce 11 (60% yield over 3 steps); (c) Dieckmann condensation (NaOMe/MeOH); and (d) ORC reaction (CAN, O2) followed by endoperoxide reduction (CuCl) to form 12 (34% yield over 2 steps).
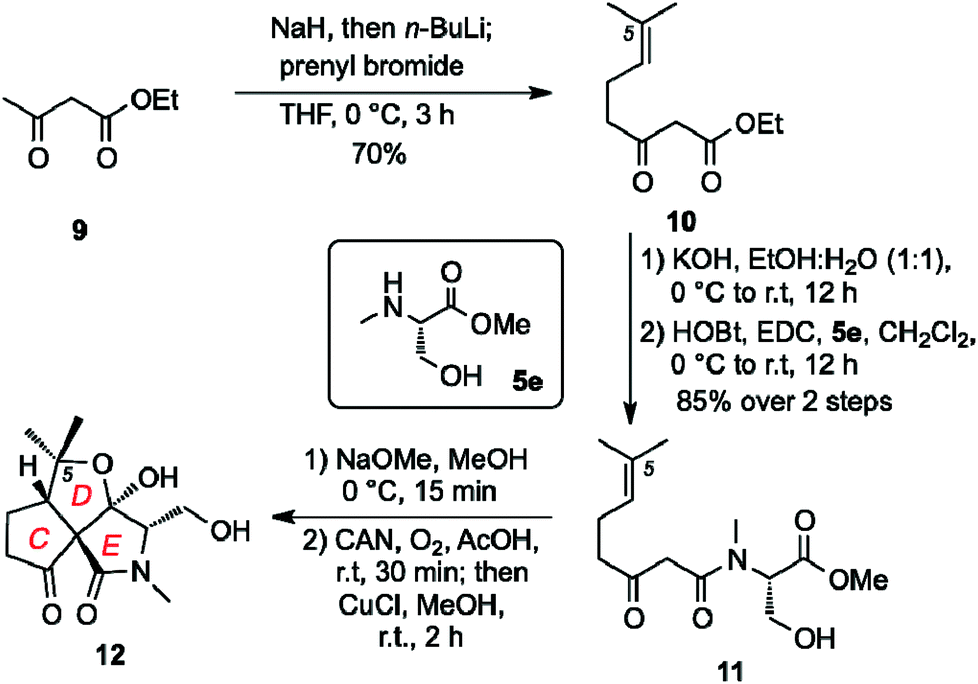 | ||
| Scheme 2 Synthesis of CDE core fusarisetin analog 12. | ||
We then explored the structure–function relationship of the decalin system of 1 by functionalizing the C8–C9 alkene (Scheme 3). OsO4-catalyzed dihydroxylation produced diol 13 (58% yield). The stereoselectivity of this reaction has been unambiguously confirmed by X-ray structure analysis.10 Similarly, treatment of 1 with mCPBA selectively afforded epoxide 14 (68% yield). Reduction of the C8–C9 alkene proceeded under Pd-catalyzed hydrogenation conditions to afford saturated analog 15 (95% yield). On the other hand, acetylation of the C18 hydroxyl group produced acetate 16 (66% yield).
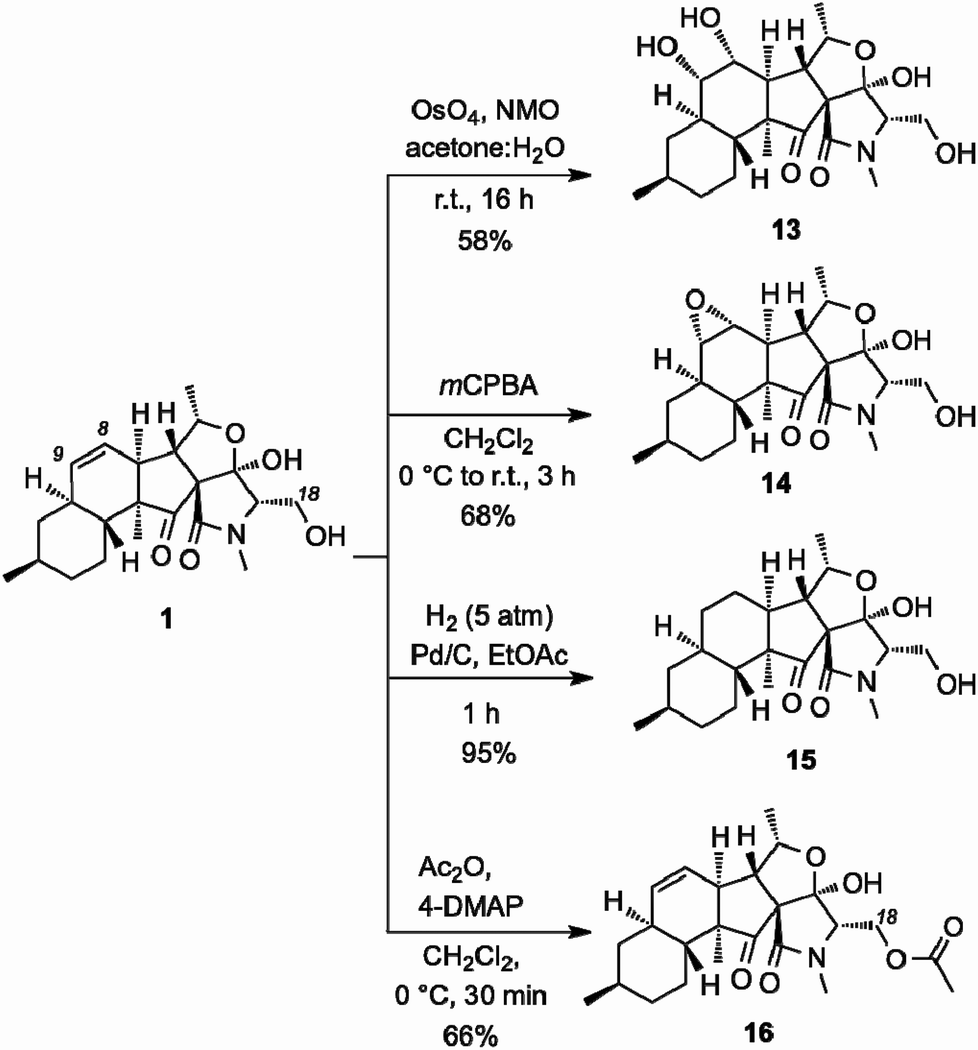 | ||
| Scheme 3 Synthesis of derivatives of fusarisetin A. | ||
Initial cell-based evaluation of all fusarisetins (synthetic material,15 1–100 μM) was performed using a well-described scratch wound assay (Fig. 3).24 MDA-MB-231 cells were grown as a confluent monolayer, scratched and treated with analogs over a 24 hour period. Compounds 8a–8d proved to be inactive up to 100 μM concentrations suggesting that the hydroxy-methyl group of serine plays a significant role in the biological activity of 1. Truncated fusarisetin 12, representing the CDE motif, was also inactive in the scratch wound assay even at high μM concentrations. Among the synthetic fusarisetin derivatives (compounds 13–16) neither epoxide 14 nor reduced analog 15 showed any activity in vitro. However, dihydroxylated derivative 13 and acetate 16 demonstrated inhibition of wound healing as shown in Fig. 3. These compounds were then subjected to a Boyden chamber Transwell assay to quantitatively determine their anti-motility activity (Fig. 4). The IC50 values of fusarisetin analogs are shown in Table 1.
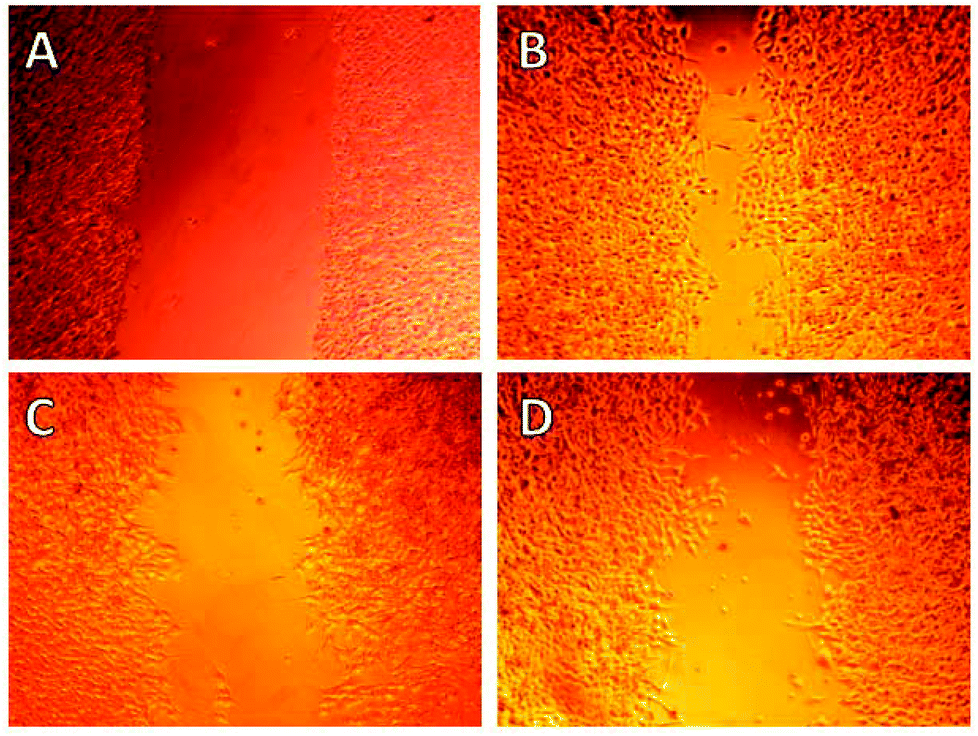 | ||
| Fig. 3 Evaluation of synthetic fusarisetins in a scratch wound assay. MDA-MB-231 cells, grown as a confluent monolayer, were scratched to generate a cell-free zone and photographed at time 0 (A). Cells were incubated for 24 h with DMSO as control (B) and in the presence of compounds 13 (100 μM) (C) and 16 (100 μM) (D). | ||
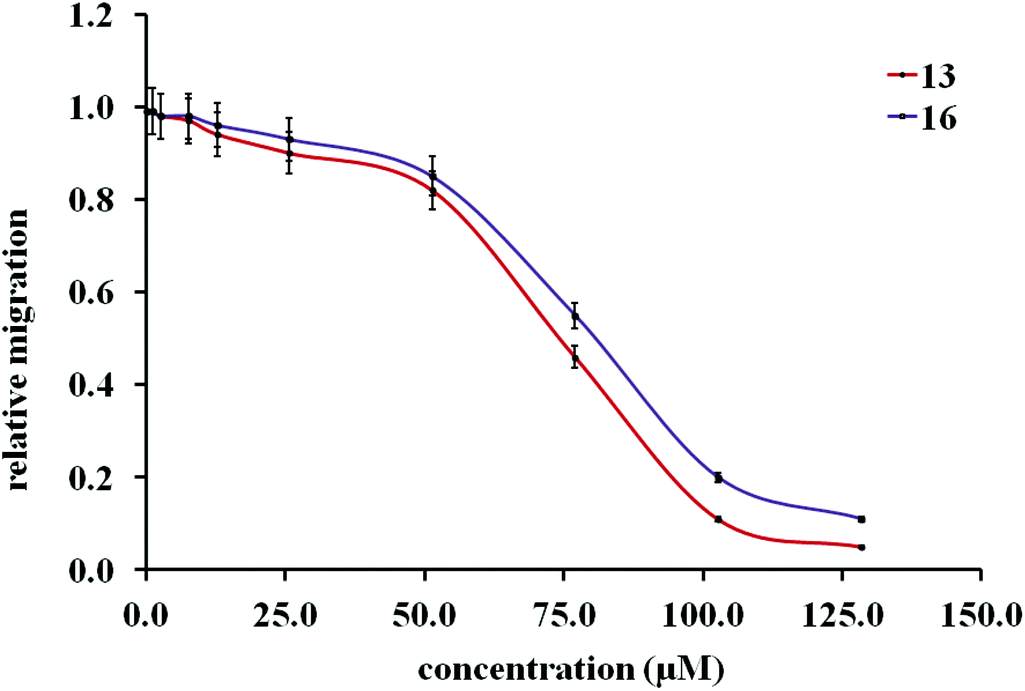 | ||
| Fig. 4 Dose-dependent inhibition of cell migration by 13 and 16 in a Transwell assay. Cells were plated in a Boyden chamber with 13, 16 or vehicle control (DMSO) for 24 h at the indicated concentrations. Amount of migrated cells is presented relative to the corresponding DMSO control. | ||
| Compound | IC50 (μM) |
|---|---|
| a Compounds with no significant activity in the scratch wound assay (up to 100 μM) were not submitted to the Transwell assay. IC50 values of compounds 13 and 16 were determined by the Transwell assay. b Value obtained from ref. 10 and 15. c Compound 12 was tested as the racemate. | |
| 1 | 7.715 |
| 2 | 7.7b |
| 8a | >100 |
| 8b | >100 |
| 8c | >100 |
| 8d | >100 |
| 12 | >100c |
| 13 | 74.5 ± 3.2 |
| 14 | >100 |
| 15 | >100 |
| 16 | 85.3 ± 3.8 |
Fig. 5 summarizes the observed structure–function relationship of fusarisetins. Comparison of the bioactivity data indicates that: (a) both the AB decalin motif and the serine amino acid are critical to the biological profile of fusarisetins; (b) the C5 stereochemistry is insignificant to the activity; and (c) although the B ring alkene is significant to bioactivity, its dihydroxylation can lead to analogs that maintain the bioactivity albeit at a higher concentration. Similarly, acetylation of the C18 oxygen produces a compound that inhibits cell migration at high μMolar concentration (Fig. 5).
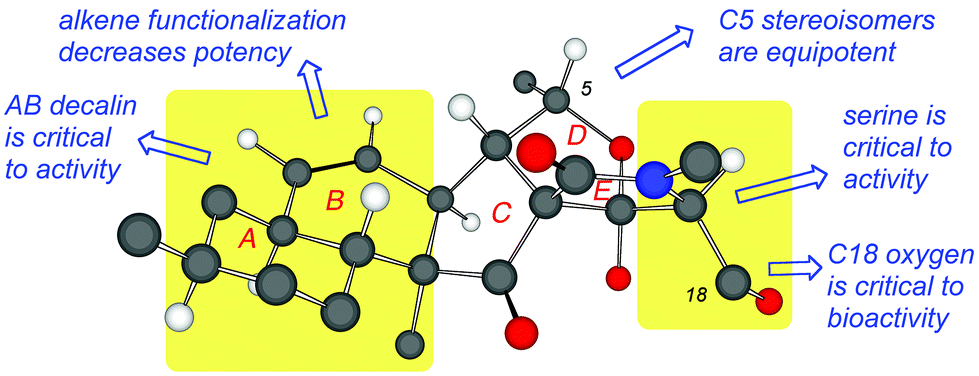 | ||
| Fig. 5 Structure–function relationship map of fusarisetins. | ||
Conclusions
In summary, we present our studies toward deciphering the pharmacophoric motif of fusarisetins, a new family of potent cell migration inhibitors. Fusarisetin A, the archetype of this family, inhibits cell motility without directly targeting actin or microtubule networks. Empowered by our robust synthetic strategy,12,15 we have produced several analogs that were used to interrogate the biological significance of the fusarisetin framework. We found that structural modifications of this rigid scaffold, such as deletion of the AB ring system or replacement of the E-ring serine with other amino acids, result in acute loss of potency. However, subtle changes at the periphery of the fusarisetin motif, such as shuffling of the stereochemistry at the C5 center, result in retention of activity. Moreover, acetylation of the C18 hydroxyl group or dihydroxylation of the C8–C9 alkene produces compounds that maintain biological function albeit at higher concentration, suggesting that these functionalities could be used as tethering sites for further functionalization. Our observations strongly support the notion that fusarisetins operate via an unexplored mechanism of action associated with cell motility. Importantly, these findings attest to the uncharted and highly promising potential of fusarisetins as novel leads for the development of cancer metastasis inhibitors.Acknowledgements
We gratefully acknowledge the National Institutes of Health (NIH) for financial support of this work through grant number R01 CA133002. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr Yongxuan Su (UCSD MS Facility), Dr Anthony Mrse (UCSD NMR Facility) as well as Michelle H. Lacoske and Chao-I. Hung (UCSD, Chemistry Department) for technical help.Notes and references
- A. Jemal, F. Bray, M. Center, J. Ferlay, E. Ward and D. Forman, CA-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2013 .
- For selected recent reviews see: (a) C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed; (b) J. E. Talmadge and I. J. Fidler, Cancer Res., 2010, 70, 5649–5669 CrossRef CAS PubMed; (c) A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed; (d) M. Bacac and I. Stamenkovic, Annu. Rev. Pathol.: Mech. Dis., 2008, 3, 221–247 CrossRef CAS PubMed; (e) A. C. Chiang and J. Massague, N. Engl. J. Med., 2008, 359, 2814–2823 CrossRef CAS PubMed; (f) D. X. Nguyen, P. D. Bos and J. Massague, Nat. Rev. Cancer, 2009, 9, 274–284 CrossRef CAS PubMed; (g) D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS.
- (a) C. Coghlin and G. I. Murray, J. Pathol., 2010, 222, 1–15 CrossRef CAS PubMed; (b) D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- A. Giubellino, Y. Gao, S. M. Lee, M. J. Lee, J. R. Vasselli, S. Medepalli, J. B. Trepel, T. R. Burke and D. P. Bottaro, Cancer Res., 2007, 67, 6012–6016 CrossRef CAS PubMed.
- (a) D. Shan, L. Chen, J. T. Njardarson, C. Gaul, X. Ma, S. J. Danishefsky and X.-Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3772–3776 CrossRef CAS PubMed; (b) S. Braig, C. A. Kressirer, J. Liebl, F. Bischoff, S. Zahler, L. Meijer and A. M. Vollmar, Cancer Res., 2013, 73, 6004–6012 CrossRef CAS PubMed; (c) W. Wu, C. Bi, K. Credille, J. R. Manro, V. L. Peek, G. P. Donoho, L. Yan, J. A. Wijsman, S.-C. B. Yan and R. A. Walgren, Clin. Cancer Res., 2013, 19, 5699–5710 CrossRef CAS PubMed.
- T. K. Sawyer, Expert Opin. Invest. Drugs, 2004, 13, 1–19 CrossRef CAS PubMed.
- P. Elvin and A. P. Garner, Curr. Opin. Pharmacol., 2005, 5, 374–381 CrossRef CAS PubMed.
- (a) C. Gaul, J. T. Njardarson, D. Shan, D. C. Dorn, K. D. Wu, W. P. Tong, X. Y. Huang, M. A. Moore and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 11326–11337 CrossRef CAS PubMed; (b) A. Cipres, D. P. O'Malley, K. Li, D. Finlay, P. S. Baran and K. Vuori, ACS Chem. Biol., 2010, 5, 195–202 CrossRef CAS PubMed; (c) K. Micoine and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 14064–14066 CrossRef CAS PubMed; (d) M. Sawada, S. Kubo, K. Matsumura, Y. Takemoto, H. Kobayashi, E. Tashiro, T. Kitahara, H. Watanabe and M. Imoto, Bioorg. Med. Chem. Lett., 2011, 21, 1385–1389 CrossRef CAS PubMed.
- (a) J. H. Jang, Y. Asami, J.-P. Jang, S. O. Kim, D. O. Moon, K. S. Shin, D. Hashizume, M. Muroi, T. Saito, H. Oh, B. Y. Kim, H. Osada and J. S. Ahn, J. Am. Chem. Soc., 2011, 133, 6865–6867 CrossRef CAS PubMed; (b) J. S. Ahn, J.-H. Jang, B. Y. Kim, J. Jang, Y. Asami and H. Oh, Novel fusarisetin compounds and use thereof, US2013/0116297 A1, publication date 05/09/2013 Search PubMed.
- J. Deng, B. Zhu, Z. Y. Lu, H. X. Yu and A. Li, J. Am. Chem. Soc., 2012, 134, 920–923 CrossRef CAS PubMed.
- J. Xu, E. J. E. Caro-Diaz, L. Trzoss and E. A. Theodorakis, J. Am. Chem. Soc., 2012, 134, 5072–5075 CrossRef CAS PubMed.
- (a) J. Yin, C. Wang, L. Kong, S. Cai and S. Gao, Angew. Chem., Int. Ed., 2012, 51, 7786–7789 CrossRef CAS PubMed; (b) J. Yin, L. Kong, C. Wanf, Y. Shi, S. Cai and S. Gao, Chem.–Eur. J., 2013, 19, 13040–13046 CrossRef CAS PubMed; (c) J. Yin, L.-L. Kong and S. Gao, Chin. J. Org. Chem., 2013, 33, 259–266 CrossRef CAS; (d) J. Yin and S. Gao, Synlett, 2014, 1–7 Search PubMed.
- J. Huang, L. Fang, R. Long, L.-L. Shi, H.-J. Shen, C.-C. Li and Z. Yang, Org. Lett., 2013, 15, 4018–4021 CrossRef CAS PubMed.
- J. Xu, E. J. E. Caro-Diaz, M. H. Lacoske, C.-I. Hung, C. Jamora and E. A. Theodorakis, Chem. Sci., 2012, 3, 3378–3386 RSC.
- For selected references on this topic see: (a) D. M. Bollag, P. A. McQueney and J. Zhu, Cancer Res., 1995, 55, 2325–2333 CAS; (b) Y. A. Elnakady, F. Sasse, H. Lunsdorf and H. Reichenbach, Biochem. Pharmacol., 2004, 67, 927–935 CrossRef CAS PubMed For reviews on this class of compounds see: (c) K.-H. Altmann, Curr. Opin. Chem. Biol., 2001, 5, 424–431 CrossRef CAS; (d) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- For selected references on this topic see: (a) J. F. Casella, M. D. Flanagan and S. Lin, Nature, 1981, 293, 302–305 CrossRef CAS; (b) S. Saito, S. Watabe, H. Ozaki, N. Fusetani and H. Karaki, J. Biol. Chem., 1994, 269, 29710–29714 CAS; (c) S. Saito, S. Watabe, H. Ozaki, H. Kigoshi, K. Yamada, N. Fusetani and H. Karaki, J. Biochem., 1996, 120, 552–555 CrossRef CAS; (d) L. Haviv, D. Gillo, F. Backouche and A. Bernheim-Groswasser, J. Mol. Biol., 2008, 327, 325–330 CrossRef PubMed.
- For selected references on this topic see: (a) T. Yamada, K. Minoura, R. Tanaka and A. Numata, J. Antibiot., 2007, 60, 370–375 CrossRef CAS PubMed; (b) Z. Wang, S. Castellano, S. S. Kinderman, C. E. Argueta, A. B. Beshir, G. Fenteany and O. Kwon, Chem.–Eur. J., 2011, 17, 649–654 CrossRef CAS PubMed; (c) A. W. Kahsai, S. Zhu, D. J. Wardrop, W. S. Lane and G. Fenteany, Chem. Biol., 2006, 13, 973–983 CrossRef CAS PubMed; (d) G. Fenteany and S. Zhy, Curr. Top. Med. Chem., 2003, 3, 593–616 CrossRef CAS.
- For selected references on this topic see: (a) M. Binder and C. Tamm, Angew. Chem., Int. Ed. Engl., 1973, 12, 370–380 CrossRef CAS PubMed; (b) G. S. Pendse and A. M. Mujumdar, Recent Advances in Cytochalasans, Chapmann & Hall, London, 1987 Search PubMed; (c) D. W. Goddette and C. Frieden, J. Biol. Chem., 1987, 261, 15974–15980 Search PubMed; (d) J. A. Cooper, J. Cell Biol., 1987, 105, 1473–1478 CrossRef CAS; (e) M. Schliwa, J. Cell Biol., 1982, 92, 79–91 CrossRef CAS; (f) K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC.
- (a) J. W. Sims, J. P. Fillmore, D. D. Warner and E. W. Schmidt, Chem. Commun., 2005, 186–188 RSC; (b) J. Schumann and C. Hertweck, J. Am. Chem. Soc., 2007, 129, 9564–9565 CrossRef PubMed.
- (a) K. Yuki, M. Shindo and K. Shishido, Tetrahedron Lett., 2001, 42, 2517–2519 CrossRef CAS; (b) M. H. Wheeler, R. D. Stipanovic and L. S. Puckhaber, Mycol. Res., 1999, 103, 967–973 CrossRef CAS; (c) S. V. Ley, S. C. Smith and P. R. Woodward, Tetrahedron, 1992, 48, 1145–1174 CrossRef CAS.
- In this case the resulting endoperoxides (C5 epimeric structures) have not been isolated. Their formation is proposed based on the previously reported syntheses of fusarisetin A in which the corresponding endoperoxides have been isolated and spectroscopically/analytically characterized (see ref. 13, 15).
- For selected examples see: (a) H. R. Burmeister, G. A. Bennet, R. F. Vesonder and W. J. C. Hesseltine, Antimicrob. Agents Chemother., 1974, 5, 634–639 CrossRef CAS; (b) Y. Sugie, H. Hirai, H. Kachi-Tonai, Y. J. Kim, Y. Kojima, Y. Shiomi, A. Sugiura, A. Sugiura, Y. Suzuki, N. Yoshikawa, L. Brennan, J. Duignan, L. H. Huang, J. Sutcliffe and N. Kojima, J. Antibiot., 2001, 54, 917–925 CrossRef CAS; (c) Y. Igarashi, H. Ogura, K. Furihata, N. Oku, C. Indananda and A. Thamchaipenet, J. Nat. Prod., 2011, 74, 670–674 CrossRef CAS PubMed.
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protocols, 2007, 2, 329–333 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00067b |
| This journal is © the Partner Organisations 2014 |