Triptycene-derived calix[6]arene analogues: synthesis, structure and complexation with paraquat derivatives†
Yu-Xiang
Xia
ab,
Tao
Xie
a,
Ying
Han
a and
Chuan-Feng
Chen
*a
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: cchen@iccas.ac.cn; Fax: +8610-62554449
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 30th January 2014
Abstract
A new kind of triptycene-derived calix[6]arene analogue was synthesized by two different routes. The triptycene-derived calix[6]arene analogues show highly symmetric structures and fixed conformations in solution, and can self-assemble into organic tubular structures with aromatic rings as the wall in the solid state. Moreover, the calix[6]arene analogue with two 1,8-dihydroxyltriptycene moieties in the cis position was also found to form stable complexes with paraquat derivatives with different terminal functional groups. Consequently, a [2]rotaxane assembly based on the calix[6]arene analogue was further constructed.
Introduction
As one of the most important macrocyclic hosts, calixarenes have been found wide applications in supramolecular chemistry owing to their functional characteristics and unique conformations.1 In particular, calix[6]arene and its derivatives have a large enough cavity to complex with ordinary organic molecules, so that they could find specific applications in molecular recognition and self-assembly.2–4 However, the complicated conformations restricted their applications to a great extent. For this reason, some methods to fix the conformations of calix[6]arenes have been developed. For example, the Atwood and Reinaud groups exploited strategies for conformational restriction by metal coordination.5 Appropriate functionalization has also been devoted to both the lower and upper rims of the calix[6]arene skeleton to fix the conformations of calix[6]arenes,6 which subsequently resulted in the synthesis of pseudorotaxanes and rotaxanes,7 and construction of biomimetic molecular receptors.8Triptycene has a unique Y-shaped rigid structure and rich reactive positions so that it could be used as a building block for the design and synthesis of novel hosts.9 Recently, we reported a class of novel triptycene-derived calix[6]arenes,10 and found that the triptycene building block with 3D rigid structure played a key role in expanding the cavity and fixing the conformation of the novel calixarenes.11 Moreover, the macrocycles could all encapsulate some solvent molecules inside their cavities.12 However, the complexation between these triptycene-derived calix[6]arenes and other organic guests except solvent molecules were still not reported so far.
In this paper, we report a new kind of triptycene-derived calix[6]arene analogue as shown in Fig. 1. It was found that the calix[6]arene analogues showed highly symmetric structures and fixed conformations in solution, and could self-assemble into organic tubular structures with aromatic rings as the wall and further 3D microporous architectures in the solid state. Moreover, we also found that the macrocycle with two 1,8-dihydroxyltriptycene moieties in the cis position could form stable complexes with different terminal functional paraquat derivatives. Furthermore, a [2]rotaxane assembly based on the calix[6]arene analogue was designed and constructed as well.
 | ||
| Fig. 1 Structures and proton designations of the macrocyclic hosts and the guests. | ||
Results and discussion
Synthesis of triptycene-derived calix[6]arene analogues
Synthesis of the triptycene-derived calix[6]arene analogues is depicted in Scheme 1. First, reaction of 1,8-dimethoxy-2,7-dihydroxymethyltriptycene 110b with excess of 2-substituted-1,3-dimethoxybenzene in dry CH2Cl2 with BF3·Et2O as the catalyst gave 5–7 in 68–76% yields. Then, the macrocyclization from reaction between 5 and 1 in dry CH2Cl2 in the presence of BF3·Et2O afforded the target products 8a and 8b in 29% and 23% yield, respectively.13,14 Similarly, 9 and 10 were obtained in total 39% and 35% yield, respectively. Compounds 8a and 8b were further demethylated by BBr3 in dry dichloromethane to give products 11a and 11b in 86% and 83% yield, respectively. | ||
| Scheme 1 Synthesis of the macrocycles 8a–11a and 8b–11b. | ||
Triptycene-derived calix[6]arene analogues 11a–b could also be synthesized by a fragment-coupling method starting from triptycene derivative 1 and 2-methylresorcin 12. As shown in Scheme 2, the reaction of compound 1 with excess of 12 in dry CH3CN in the presence of p-toluenesulfonic acid gave compound 13 in 76% yield. Then, the reaction of 13 with 1 in o-dichlorobenzene (o-DCB) in the presence of p-toluenesulfonic acid gave 14a and 14b in 15% and 11% yield, respectively. Macrocycles 14a and 14b were further demethylated by BBr3 in dry dichloromethane to give calix[6]arene analogues 11a and 11b in 90% and 86% yield, respectively. The new compounds were all characterized by the 1H NMR, 13C NMR, MALDI-TOF mass spectra, and elemental analysis.15
 | ||
| Scheme 2 Synthesis of 14a–b and their demethylation. | ||
Structures and solid-state assemblies of the macrocycles
The structural characterization of the macrocycles was first studied by NMR experiments. The 1H NMR spectrum of 8a showed not only a singlet for the methyl protons, two singlets for the methoxy protons, and two singlets for the bridgehead protons of the triptycene moiety, but a pair of doublet signals for the methylene protons. Moreover, six signals for the aliphatic carbons were only observed in the 13C NMR spectrum of 8a. Similar results were found for 8b, and its variable-temperature 1H NMR experiments also showed no obvious changes of the methylene proton signals with the increase of the temperature even up to 380 K.15 These observations suggested that both 8a and 8b have highly symmetric structures and fixed conformations. Similarly, it was found that macrocycles 9a–11a and 9b–11b all have highly symmetric structures and fixed conformations in solution.15 The NMR and variable-temperature 1H NMR experiments also showed that macrocycles 14a–b have similar structural properties to those of macrocycles 8a–b.15Fortunately, we obtained single crystals of 8a and 8b suitable for X-ray analysis from slow evaporation of CH3OH into an acetone or chloroform solution, respectively.15 As shown in Fig. 2, the crystal structures clearly showed that compounds 8a and 8b are a pair of configurational isomers, in which 8a was a cis isomer with a boat-like conformation, while 8b was a trans isomer with a chair-like conformation. For cis-isomer 8a, the dihedral angle between the two dimethoxybenzene rings was 144.11° and the four methoxy groups point to the exterior of the cavity, while the dihedral angles between the face-to-face benzene rings in the triptycene moieties were 6.47° and 25.53°, respectively. While for trans-isomer 8b, it was found that the two dimethoxybenzene rings were opposite and in the same plane, and the four methoxy groups also pointed to the exterior of the cavity. Meanwhile, the face-to-face benzene rings in the triptycene moieties were all parallel to each other, and the centroid distances between the face-to-face benzene rings are 8.89 Å and 7.47 Å, respectively.
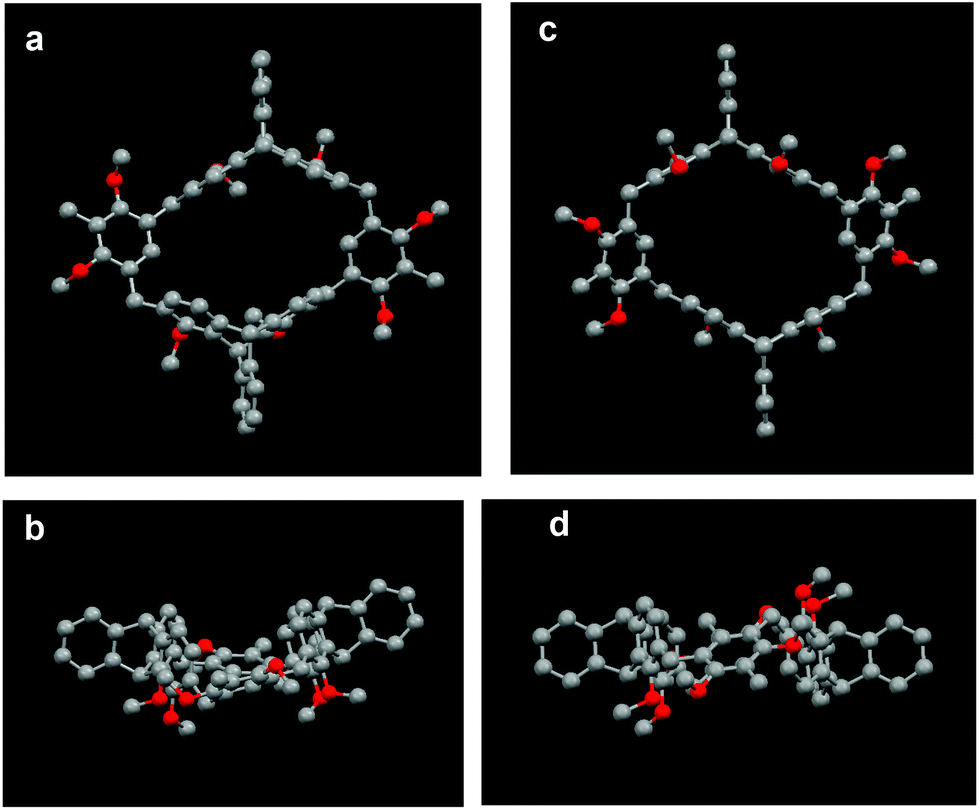 | ||
| Fig. 2 (a) Top view and (b) side view of the crystal structure of 8a. (c) Top view and (d) side view of the crystal structure of 8b. Solvent molecules and hydrogen atoms are omitted for clarity. | ||
The self-assembly of macrocycle 8b in the solid state was further studied. As shown in Fig. 3a, macrocyclic molecule 8b could form an organic tubular assembly with the aromatic rings as the wall by the bridges of adjacent molecules, and further 3D microporous architecture viewed along the c-axis15 by a pair of intermolecular complementary C–H⋯π interactions between the aromatic proton of the triptycene moiety of one molecule and the dimethoxybenzene ring of its adjacent molecule (dC–H⋯π = 2.78 Å), and another pair of C–H⋯π interactions between the methyl proton of the triptycene moiety of one molecule and the aromatic ring of the triptycene moiety of its adjacent molecule (dC–H⋯π = 2.89 Å).
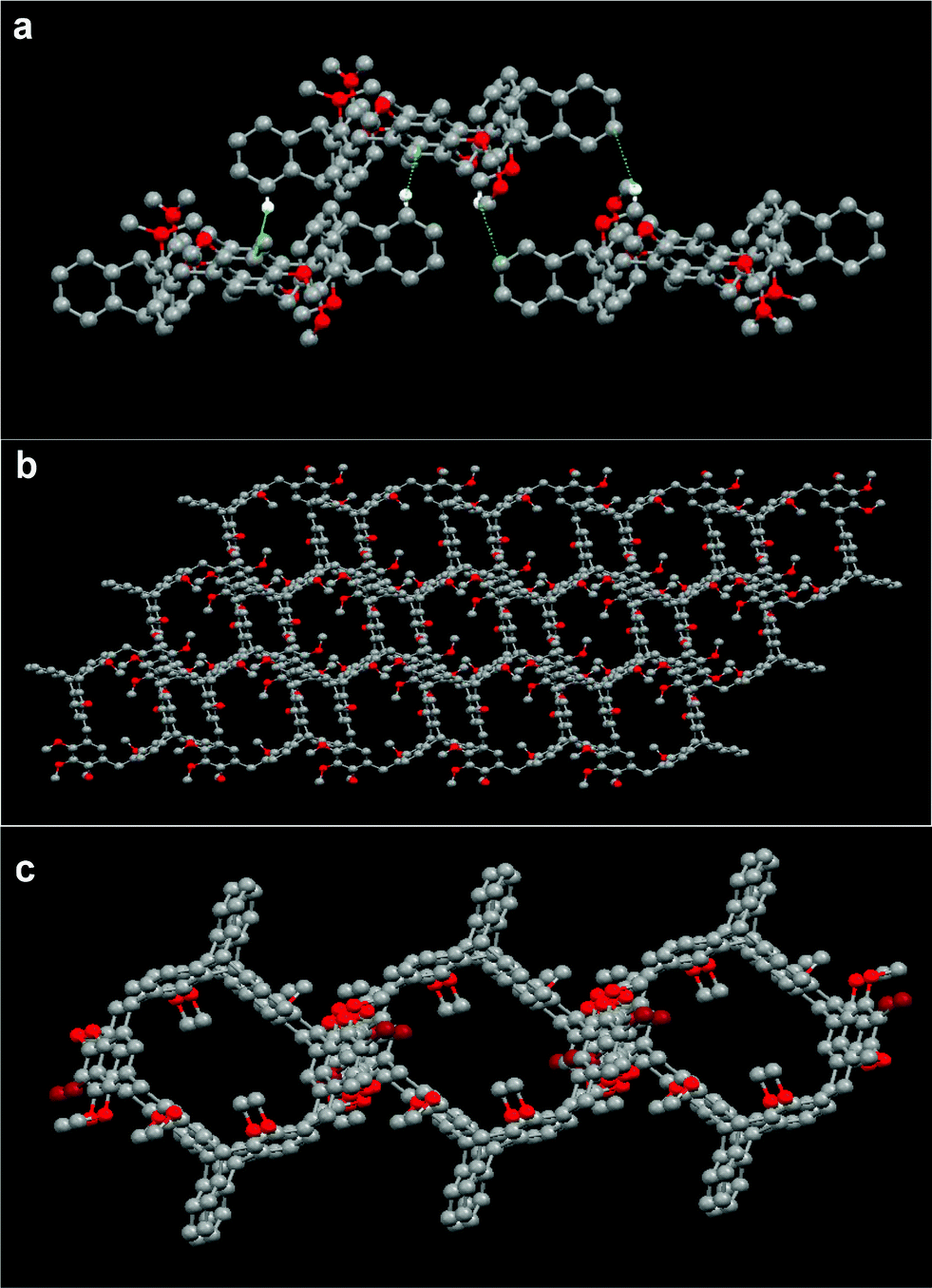 | ||
| Fig. 3 (a) Crystal structure of 8b, (b) and (c) tubular assembly of 9b and 10b. Dashed lines denote the non-covalent interactions of the adjacent molecules. Solvent molecules and hydrogen atoms not involved in the interactions are omitted for clarity. | ||
As shown in Fig. 3b and 3c, by the bridges of adjacent molecules, macrocycles 9b and 10b could form tubular assemblies with the aromatic rings as the wall and 3D microporous structures as well.15 Macrocycle 9b self-assembled in a tubular structure by a pair of C–H⋯O hydrogen bonds between the methyl proton of the triptycene moiety of one molecule and the methoxyl oxygen atom of the trimethoxybenzene of its adjacent molecule with the distance of 2.65 Å, and a pair of C–H⋯π interactions between the aromatic proton of the triptycene moiety of one molecule and the aromatic ring of the triptycene moiety of its adjacent molecule with the distance of 2.83 Å. For macrocycle 10b, a tubular assembly was also formed by a pair of C–H⋯O hydrogen bonds (dC–H⋯O = 2.36 Å) between the methyl proton of the triptycene moiety of one molecule and the methoxy oxygen atom of the dimethoxybenzene moiety of its adjacent molecule, and a pair of C–H⋯π interactions (dC–H⋯π = 2.62 Å) between the methyl proton of the dimethoxybenzene of one molecule and the aromatic ring of the triptycene moiety of its adjacent molecule. Furthermore, by the virtue of one pair of C–H⋯Cl (dC–H⋯Cl = 2.92 Å) interactions between the aromatic proton of the triptycene moiety of 10b and a chloride atom of the CH2Cl2 molecule, and six pairs of C–H⋯π (dC–H⋯π = 2.86 Å) interactions between protons of the CH2Cl2 molecules and the aromatic ring of the triptycene moiety of 10b, the tubular assembly can form a 2D superstructure, which further stacks into a 3D microporous architecture, and dichloromethane molecules were found to be situated in the channels.15
We also obtained a single crystal of 14a suitable for X-ray analysis from slow evaporation of i-PrOH into a chloroform solution. As shown in Fig. 4, the crystal structure clearly showed that macrocycle 14a was a cis isomer with a cone conformation, in which the four methoxy groups and four hydroxyl groups all point to the same side of the cavity. The dihedral angle between the two resorcin rings was 64.45°, and the dihedral angles between two face-to-face benzene rings in triptycene moieties are 36.13° and 37.66°, respectively. Moreover, it was found that macrocycle 14a can encapsulate one chloroform molecule inside the cavity by the C–Cl⋯π interaction (dC–Cl⋯π = 3.45 Å) between a chloride atom of the CHCl3 molecule and the resorcin ring of 14a.
 | ||
| Fig. 4 (a) Top view and (b) side view of crystal structure of CHCl3–14a. The dashed line denotes the non-covalent interaction between the adjacent molecules. Hydrogen atoms are omitted for clarity. | ||
Complexation between 11a and paraquat derivatives
We also investigated the complex ability of 11a toward paraquat derivatives. Thus, when mixing 11a and guest G1 (2.0 mM each) in acetone, we found that the color of the solution changed from colorless to yellow, possibly due to the charge transfer interaction between the electron-rich aromatic rings of 11a and the electron-poor bipyridinium rings of G1. As shown in Fig. 5, the 1H NMR spectrum of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 11a and G1 in acetone-d6 showed a significant difference from those for 11a and G1. The spectrum exhibited only one set of different signals from those of the separated host and guest, which suggested that a new complex was formed, and the complexation between the host and the guest was a fast exchange process. The signals of aromatic protons H1 and H2 and methyl proton H3 of guest G1 shifted significantly upfield, which might be due to the strong shielding effect of the aromatic rings of 11a. Similarly, the signals of bridgehead protons He and Hf of 11a also shifted upfield, which was attributed to their positions in the shielding region of the bipyridinium rings of G1. Moreover, the aromatic protons Hi and Hc and the methylene protons Hg and Hh of 11a shifted downfield, which might be due to the charge transfer interaction between 11a and G1. These observations suggested that a stable complex between host 11a and guest G1 was formed. Furthermore, 1H NMR spectroscopic titrations afforded a quantitative estimate between 11a and G1 by monitoring of the changes of the chemical shift of the proton He in 11a. The results showed that a 1:1 complex between 11a and G1 was formed by a mole ratio plot14d,15 and the apparent association constant Ka,exp was calculated to be 505(±34.8) M−1.16 Similarly, guests G2–G4 could also form 1:1 complexes with host 11a in solution, and the association constants for the complexes 11a·G2, 11a·G3 and 11a·G4 were calculated to be 715(±29.3) M−1, 337(±36.7) M−1, and 213(±14.9) M−1, respectively.
:1 mixture of 11a and G1 in acetone-d6 showed a significant difference from those for 11a and G1. The spectrum exhibited only one set of different signals from those of the separated host and guest, which suggested that a new complex was formed, and the complexation between the host and the guest was a fast exchange process. The signals of aromatic protons H1 and H2 and methyl proton H3 of guest G1 shifted significantly upfield, which might be due to the strong shielding effect of the aromatic rings of 11a. Similarly, the signals of bridgehead protons He and Hf of 11a also shifted upfield, which was attributed to their positions in the shielding region of the bipyridinium rings of G1. Moreover, the aromatic protons Hi and Hc and the methylene protons Hg and Hh of 11a shifted downfield, which might be due to the charge transfer interaction between 11a and G1. These observations suggested that a stable complex between host 11a and guest G1 was formed. Furthermore, 1H NMR spectroscopic titrations afforded a quantitative estimate between 11a and G1 by monitoring of the changes of the chemical shift of the proton He in 11a. The results showed that a 1:1 complex between 11a and G1 was formed by a mole ratio plot14d,15 and the apparent association constant Ka,exp was calculated to be 505(±34.8) M−1.16 Similarly, guests G2–G4 could also form 1:1 complexes with host 11a in solution, and the association constants for the complexes 11a·G2, 11a·G3 and 11a·G4 were calculated to be 715(±29.3) M−1, 337(±36.7) M−1, and 213(±14.9) M−1, respectively.
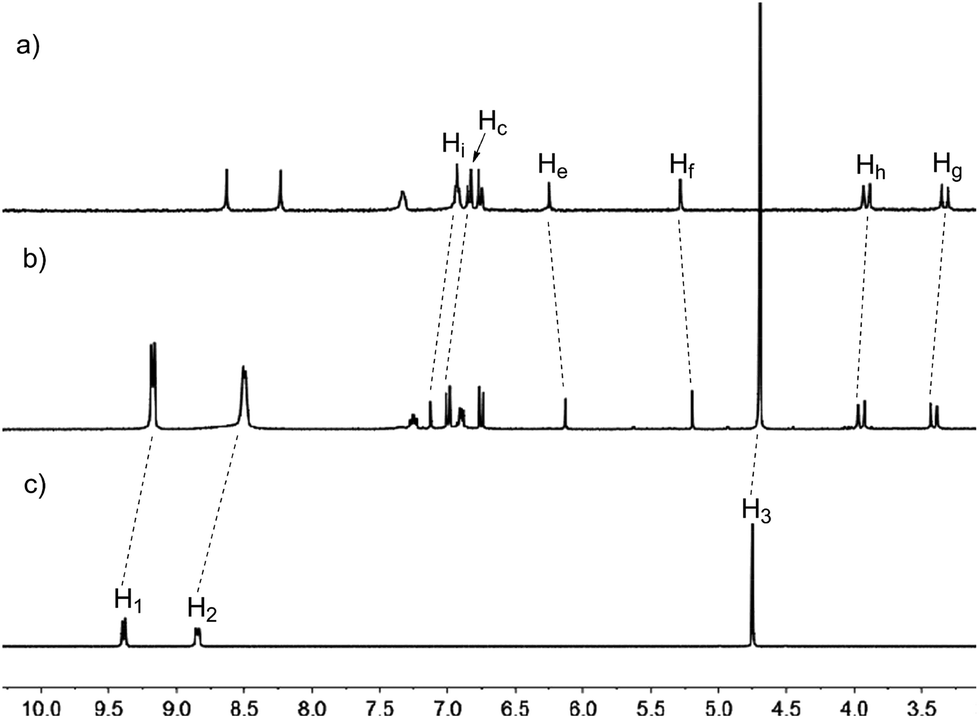 | ||
| Fig. 5 Partial 1H NMR spectra (300 MHz, acetone-d6, 298 K) of (a) free host 11a, (b) host 11a with 1.0 equiv. of G1 and (c) free guest G1. [11a]0 = 2.0 mM. | ||
The electrospray ionization mass spectrum (ESI-MS) provided further evidence for the formation of the complexes between 11a and guests G1–G4. As a result, the strong peaks at m/z 527.44, 555.66, 557.59 and 571.59 for [11a·G1-2PF6−]2+, [11a·G2-2PF6−]2+, [11a·G3-2PF6−]2+ and [11a·G4-2PF6−]2+, respectively, were observed,15 which indicated that stable complexes between host 11a and the guests were formed.
Synthesis and structure of [2]rotaxane
The complexation between 11a and the paraquat derivatives encouraged us to further design and synthesize a [2]rotaxane assembly. As shown in Scheme 3, macrocycle 11a and guest G5 were mixed in CH2Cl2–CH3CN (2:1, v/v) to afford a transparent yellow solution, which was then treated with 3,5-di-tert-butylbenzoic anhydride in the presence of a catalytic amount of tri(n-butyl)phosphine to give [2]rotaxane 15 in 28% yield. The fact that the [2]rotaxane survives column chromatography indicated that the 3,5-di-tert-butylbenzene could serve as an efficient bulky group to prevent deslippage of the rotaxane. The 1H NMR spectrum of 15 showed that the signals of the two terminal groups of the axis were different, which could be attributed to their different chemical environment in the rotaxane. Similar phenomena were also found for the other groups of the axis, which were consistent with the formation of the rotaxane. Moreover, the ESI-HRMS spectrum further supported the formation of the rotaxane, in which the signal at m/z 829.4333 corresponding to [15-2PF6−]2+ was observed.
 | ||
| Scheme 3 Synthesis of [2]rotaxane 15. | ||
Conclusion
In summary, a new kind of triptycene-derived calix[6]arene analogue has been synthesized by two different routes. It was found that the macrocycles all showed highly symmetric structures and fixed conformations in solution, and the macrocycles 8b, 9b, 10b and 14a could form tubular assemblies and 3D microporous architectures in the solid state. We also found that macrocycle 14a can encapsulate one chloroform molecule inside the cavity in the solid state, and macrocycle 11a could form stable complexes with paraquat derivatives with different functional groups in solution. Moreover, a [2]rotaxane assembly based on the calix[6]arene analogue was further obtained. We believe that these novel calix[6]arene analogues can find wide applications in molecular recognition and self-assembly, which are underway in our laboratory.Experimental
General methods
Melting points, taken on an electrothermal melting point apparatus, are uncorrected. 1H NMR and 13C NMR spectra were recorded on a DMX300 NMR. MALDI-TOF mass spectra were obtained on a BIFLEXIII mass spectrometer. Elemental analyses were performed by the Analytical Laboratory of Institute of Chemistry, CAS. 1,8-Dimethoxy-2,7-dihydroxymethyltriptycene 1 was prepared according to a literature procedure.10b Materials 2, 3, 4 and 12 obtained commercially were used without further purification.Synthesis of compound 5
A mixture of 1,8-dimethoxy-2,7-dihydroxymethyltriptycene 1 (374 mg, 1 mmol), 1,3-dimethoxy-2-methylbenzene 2 (1.52 g, 10 mmol) and boron trifluoride etherate (0.13 mL, 1 mmol) in dry CH2Cl2 (50 mL) was stirred at room temperature for 2 h. The resulting mixture was concentrated and chromatographed on a silica gel column with CH2Cl2–EtOAc as the eluent to give the pure product 5 (488 mg, 76%) as a white solid. Mp: 105–106 °C. 1H NMR (300 MHz, CDCl3): δ 2.14 (s, 6H), 3.63 (s, 6H), 3.75 (s, 6H), 3.85 (s, 6H), 3.89 (s, 2H), 3.90 (s, 2H), 5.32 (s, 1H), 6.16 (s, 1H), 6.50 (d, J = 9.0 Hz, 2H), 6.63 (d, J = 9.0 Hz, 2H), 6.77 (d, J = 6.0 Hz, 2H), 6.98–7.03 (m, 4H), 7.33–7.36 (m, 1H), 7.41–7.44 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 9.2, 29.1, 42.6, 53.9, 55.6, 60.5, 62.0, 106.0, 119.4, 119.5, 123.5, 123.7, 125.1, 125.2, 125.8, 127.1, 127.8, 131.4, 137.0, 144.8, 146.1, 146.4, 153.6, 157.2, 157.3. MALDI TOF-MS: m/z 665.3 [M + Na]+, 681.2 [M + K]+. Anal. Calcd for C42H42O6: C 78.48, H 6.59; found: C 78.14, H 6.62.Synthesis of compound 6
Starting from 1 (374 mg, 1 mmol) and 1,2,3-trimethoxybenzene 3 (1.68 g, 10 mmol), following the same procedure for the synthesis of 5, compound 6 (478 mg, 71%) was obtained as a white solid. Mp: 92–94 °C. 1H NMR (300 MHz, CDCl3): δ 3.75 (s, 6H), 3.79 (s, 6H), 3.85–3.86 (m, 16H), 5.33 (s, 1H), 6.16 (s, 1H), 6.53 (d, J = 9.0 Hz, 2H), 6.64–6.68 (m, 4H), 7.00–7.05 (m, 4H), 7.34–7.37 (m, 1H), 7.42–7.45 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 29.1, 42.6, 53.8, 56.0, 60.7, 60.8, 62.1, 107.1, 119.4, 123.6, 123.7, 124.6, 125.1, 125.2, 126.8, 127.0, 131.2, 137.0, 142.2, 144.7, 146.1, 146.3, 151.8, 152.1, 153.5. MALDI TOF-MS: m/z 697.7 [M + Na]+. Anal. Calcd for C42H42O8: C 74.76, H 6.27; found: C 74.88, H 6.24.Synthesis of compound 7
Starting from 1 (374 mg, 1 mmol) and 1,3-dimethoxy-2-bromobenzene 4 (2.17 g, 10 mmol), following the same procedure for the synthesis of 5, compound 7 (525 mg, 68%) was obtained as a white solid. Mp: 141–143 °C. 1H NMR (300 MHz, CDCl3): δ 3.76 (s, 6H), 3.83 (s, 12H), 3.92 (s, 2H), 3.94 (s, 2H), 5.35 (s, 1H), 6.14 (s, 1H), 6.56 (d, J = 9.0 Hz, 2H), 6.65 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 7.00–7.06 (m, 4H), 7.35–7.38 (m, 1H), 7.42–7.45 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 29.3, 42.6, 53.8, 56.4, 60.7, 62.1, 107.1, 107.5, 119.5, 123.6, 123.7, 125.2, 125.3, 127.2, 127.8, 129.6, 130.6, 137.1, 144.5, 146.2, 146.3, 153.5, 155.6, 156.2. MALDI TOF-MS: m/z 795.3 [M + Na]+. Anal. Calcd for C40H36Br2O6·1.8C6H12: C 66.03, H 6.28; found: C 66.42, H 5.81.Synthesis of compounds 8a and 8b
To a solution of boron trifluoride etherate (0.26 mL, 2 mmol) in CH2Cl2 (50 mL) was slowly added a solution of 5 (642 mg, 1 mmol) and 1 (374 mg, 1 mmol) in CH2Cl2 (50 mL) at room temperature under argon atmosphere. After that, the mixture was stirred for an additional 12 h. The solution was evaporated in vacuo, and the residue was separated by column chromatography over 200–300 mesh silica gel with petroleum ether–CH2Cl2–EtOAc as the eluent to give compound 8a (284 mg, 29%) and 8b (225 mg, 23%) as white solids. 8a: Mp >300 °C. 1H NMR (300 MHz, CDCl3): δ 2.25 (s, 6H), 3.64 (s, 12H), 3.72 (d, J = 15.0 Hz, 4H), 3.89 (s, 12H), 3.92 (d, J = 15.0 Hz, 4H), 5.25 (s, 2H), 6.10 (s, 2H), 6.35 (d, J = 6.0 Hz, 4H), 6.45 (s, 2H), 6.89 (d, J = 6.0 Hz, 4H), 6.97–7.02 (m, 4H), 7.26–7.33 (m, 2H), 7.42–7.45 (m, 2H).13C NMR (75 MHz, CDCl3): δ 9.9, 29.0, 42.5, 53.7, 60.6, 61.8, 119.4, 123.4, 123.7, 123.8, 125.1, 126.6, 128.4, 130.7, 130.9, 136.6, 144.7, 145.9, 146.3, 153.5, 156.3. MALDI TOF-MS: m/z 1003.7 [M + Na]+, 1019.7 [M + K]+. Anal. Calcd for C66H60O8·2CH2Cl2: C 70.96, H 5.60; found: C 70.84, H 5.86. 8b: Mp >300 °C. 1H NMR (300 MHz, CDCl3): δ 2.16 (s, 6H), 3.56 (s, 12H), 3.64 (s, 12H), 3.76 (s, 4H), 5.23 (s, 2H), 5.99 (s, 2H), 6.36 (s, 2H), 6.48 (d, J = 7.5 Hz, 4H), 6.91–6.94 (m, 8H), 7.24–7.27 (m, 2H), 7.32–7.34 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 8.8, 28.3, 41.4, 52.8, 59.4, 60.6, 118.3, 122.5, 122.6, 122.7, 124.1, 125.9, 127.0, 128.3, 129.2, 135.8, 143.6, 145.1, 145.3, 152.7, 155.0. MALDI TOF-MS: m/z 1003.2 [M + Na]+, 1019.2 [M + K]+. Anal. Calcd for C66H60O8·2CH2Cl2: C 70.96, H 5.60; found: C 71.06, H 5.81.Synthesis of compounds 9a and 9b
Starting from 1 (374 mg, 1 mmol) and 6 (674 mg, 1 mmol), following the same procedure for the synthesis of 8, compounds 9a (212 mg, 21%) and 9b (182 mg, 18%) were obtained as white solids. 9a: Mp >300 °C. 1H NMR (300 MHz, CDCl3): δ 3.57 (d, J = 15 Hz, 4H), 3.69 (s, 12H), 3.81 (s, 18H), 3.83 (d, J = 15 Hz, 4H), 5.18 (s, 2H), 6.04 (s, 2H), 6.22 (s, 2H), 6.34 (d, J = 7.2 Hz, 4H), 6.84 (d, J = 7.2 Hz, 4H), 6.92–6.94 (m, 4H), 7.25–7.26 (m, 2H), 7.35–7.36 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 28.6, 41.5, 52.7, 59.6, 59.8, 60.8, 118.4, 122.4, 122.6, 124.1, 125.6, 125.8, 127.3, 129.5, 135.7, 143.6, 144.9, 145.0, 145.3, 149.5, 152.6. MALDI TOF-MS: m/z 1035.8 [M + Na]+, 1051.8 [M + K]+. Anal. Calcd for C66H60O10·1.1CHCl3: C 70.42, H 5.38; found: C 70.40, H 5.64. 9b: Mp >300 °C. 1H NMR (300 MHz, CDCl3): δ 3.62 (s, 12H), 3.70 (s, 12H), 3.72 (s, 6H), 3.82 (s, 4H), 5.24 (s, 2H), 5.98 (s, 2H), 6.19 (s, 2H), 6.49 (d, J = 6 Hz, 4H), 6.89–6.95 (m, 8H), 7.23–7.26 (m, 2H), 7.31–7.32 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 28.4, 41.4, 52.8, 59.6, 59.7, 60.6, 118.3, 122.5, 122.6, 124.1, 124.5, 126.0, 127.0, 128.9, 135.8, 143.6, 144.8, 145.1, 145.3, 149.3, 152.7. MALDI TOF-MS: m/z 1012.7 [M]+, 1035.7 [M + Na]+, 1051.7 [M + K]+. Anal. Calcd for C66H60O10·2H2O: C 75.55, H 6.15; found: C 75.47, H 6.20.Synthesis of compounds 10a and 10b
Starting from 1 (374 mg, 1 mmol) and 7 (772 mg, 1 mmol), following the same procedure for the synthesis of 8, compounds 10a (222 mg, 20%) and 10b (167 mg, 15%) were obtained as white solids. Compound 10a: Mp: 274–275 °C. 1H NMR (300 MHz, CDCl3): δ 3.66 (s, 12H), 3.70 (d, J = 21 Hz, 4H), 3.81 (s, 12H), 3.85 (d, J = 21 Hz, 4H), 5.20 (s, 2H), 6.03 (s, 2H), 6.30 (d, J = 7.5 Hz, 4H), 6.49 (s, 2H), 6.85 (d, J = 7.5 Hz, 4H), 6.92–6.95 (m, 4H), 7.26–7.27 (m, 2H), 7.35–7.37 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 29.7, 42.5, 53.7, 61.0, 61.8, 112.6, 119.4, 123.4, 123.7, 125.2, 126.6, 130.2, 130.3, 132.4, 136.7, 144.5, 146.2, 153.5, 154.9. MALDI TOF-MS: m/z 1133.7 [M + Na]+, 1149.7 [M + K]+. Anal. Calcd for C64H54Br2O8·C6H12·3H2O: C 67.31, H 5.81; found: C 67.51, H 6.42. 10b: Mp: 232–234 °C. 1H NMR (300 MHz, CDCl3): δ 3.69 (s, 12H), 3.72 (s, 12H), 3.83–3.86 (m, 8H), 5.33 (s, 2H), 6.05 (s, 2H), 6.55–6.58 (m, 6H), 6.98–7.04 (m, 8H), 7.33–7.36 (m, 2H), 7.39–7.42 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 28.9, 41.4, 52.8, 59.7, 60.5, 111.6, 118.5, 122.5, 122.7, 124.2, 126.0, 128.5, 128.9, 130.2, 135.9, 143.4, 145.1, 145.4, 152.7, 153.6. MALDI TOF-MS: m/z 1133.8 [M + Na]+, 1149.8 [M + K]+. Anal. Calcd for C64H54Br2O8·C6H12·3H2O: C 67.31, H 5.81; found: C 67.05, H 6.51.Synthesis of compounds 11a and 11b
To a solution of 8 (98 mg, 0.1 mmol) in dichloromethane (10 mL), boron tribromide (0.1 mL, 1 mmol) in dichloromethane (5 mL) was slowly added at 0 °C. After the mixture was stirred at room temperature overnight, 2 N HCl was then added to quench the reaction at 0 °C. A white solid was precipitated, filtrated and washed with water to give a crude product, which was then submitted to column chromatography with petroleum ether–EtOAc–CH3OH (10:10:1, v/v/v) as the eluent to give 11a (75 mg, 86%) and 11b (72 mg, 83%) as white solids. Compound 11a: Mp >300 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.09 (s, 6H), 3.54 (d, J = 15 Hz, 4H), 3.69 (d, J = 15 Hz, 4H), 5.24 (s, 2H), 6.22 (s, 2H), 6.31 (d, J = 7.5 Hz, 4H), 6.62 (s, 2H), 6.67 (d, J = 7.5 Hz, 4H), 6.93–6.95 (m, 4H), 7.31–7.36 (m, 4H), 8.28 (br, s, 4H), 8.84 (br, s, 4H). 13C NMR (75 MHz, DMSO-d6): δ 10.1, 29.0, 52.6, 112.1, 115.1, 119.4, 123.1, 123.3, 124.5, 125.1, 126.6, 129.4, 130.7, 144.9, 145.4, 146.6, 148.6, 151.0. MALDI TOF-MS: m/z 891.3 [M + Na]+, 907.3 [M + K]+. Anal. Calcd for C58H44O8·2.9CH2Cl2: C 65.59, H 4.50; found: C 65.57, H 4.46. 11b: Mp >300 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.05 (s, 6H), 3.44 (d, J = 18 Hz, 4H), 3.70 (d, J = 18 Hz, 4H), 5.35 (s, 2H), 5.79 (s, 2H), 6.28 (s, 2H), 6.34 (d, J = 7.5 Hz, 4H), 6.84 (d, J = 7.5 Hz, 4H), 6.90–6.93 (m, 4H), 7.32–7.34 (m, 4H), 7.88 (br, s, 4H), 8.26 (br, s, 4H). 13C NMR (75 MHz, DMSO-d6): δ 9.8, 29.6, 52.9, 111.7, 115.4, 117.4, 123.2, 123.4, 124.3, 124.4, 125.0, 126.0, 126.2, 132.1, 145.1, 145.7, 146.9, 148.9, 151.9. MALDI TOF-MS: m/z 891.8 [M + Na]+, 907.7 [M + K]+. Anal. Calcd for C58H44O8·2.9CH2Cl2: C 65.59, H 4.50; found: C 65.47, H 4.52.
Synthesis of compound 13
A mixture of 1,8-dimethoxy-2,7-dihydroxymethyltriptycene 12 (374 mg, 1 mmol), 2-methylresorcinarene (1.24 g, 10 mmol) and a catalytic amount of p-toluenesulfonic acid (19 mg, 0.1 mmol) in dry CH3CN (50 mL) was heated at reflux for 4 h. The resulting mixture was cooled and concentrated. The residue was chromatographed on a silica gel column with petroleum ether–EtOAc (1:1, v/v) as the eluent to give the product 13 (445 mg, 76%) as a white solid. Mp: 135–138 °C. 1H NMR (300 MHz, CDCl3): δ 2.04 (s, 6H), 3.71 (s, 4H), 4.04 (s, 6H), 4.69 (br, s, 2H), 5.31 (s, 1H), 6.10 (s, 1H), 6.26 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 6.0 Hz, 2H), 6.88 (d, J = 6.0 Hz, 2H), 6.98–7.01 (m, 2H), 7.06 (d, J = 9.0 Hz, 2H), 7.31–7.34 (m, 1H), 7.35 (br, s, 2H), 7.42–7.45 (m, 1H). 13C NMR (75 MHz, CDCl3): 8.4, 30.8, 42.6, 53.6, 63.3, 107.0, 111.5, 118.9, 120.9, 123.7, 123.8, 125.4, 125.6, 127.1, 127.3, 130.4, 136.9, 143.6, 145.6, 146.6, 151.2, 153.0, 153.6. MALDI TOF-MS: m/z 586.3 [M]+, 609.3 [M + Na]+. Anal. Calcd for C38H34O6·0.2H2O: C 77.32, H 5.87; found: C 77.22, H 5.99.
Synthesis of compounds 14a and 14b
To a solution of a catalytic amount of p-toluenesulfonic acid (19 mg, 0.1 mmol) in o-dichlorobenzene (50 mL) was slowly added a solution of 13 (586 mg, 1 mmol) and 1 (374 mg, 1 mmol) in o-dichlorobenzene (50 mL) at 100 °C under argon atmosphere. After that, the mixture was heated at 100 °C for an additional 12 h. The reaction mixture was concentrated in vacuo, and the residue was separated by column chromatography over 200–300 mesh silica gel with petroleum ether–EtOAc (2:1, v/v) as the eluent to give 14a (139 mg, 15%) and 14b (102 mg, 11%) as white solids. 14a: Mp >300 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.06 (s, 6H), 3.46 (d, J = 15.0 Hz, 4H), 3.84 (s, 12H), 3.86 (d, J = 15.0 Hz, 4H), 5.39 (s, 2H), 6.02 (s, 2H), 6.12 (d, J = 7.5 Hz, 4H), 6.21 (s, 2H), 6.89 (d, J = 7.5 Hz, 4H), 6.97–7.04 (m, 4H), 7.37–7.40 (m, 2H), 7.54–7.56 (m, 2H), 7.94 (br, s, 4H). 13C NMR (75 MHz, DMSO-d6): δ 10.0, 28.5, 41.7, 52.1, 61.4, 111.9, 117.7, 118.9, 123.4, 123.5, 125.0, 125.6, 129.3, 131.4, 136.2, 144.3, 145.3, 146.0, 152.1, 153.0. MALDI TOF-MS: m/z 947.3 [M + Na]+, 963.3 [M + K]+. Anal. Calcd for C62H52O8·2H2O: C 77.48, H 5.87; found: C 77.34, H 6.07. 14b: Mp >300 °C. 1H NMR (300 MHz, CDCl3): δ 1.87 (s, 6H), 3.56 (d, J = 12.0 Hz, 4H), 3.81 (s, 12H), 3.83 (d, J = 12.0 Hz, 4H), 5.32 (s, 2H), 5.76 (s, 2H), 6.91 (s, 2H), 7.00–7.09 (m, 12H), 7.36–7.39 (m, 4H). 13C NMR (75 MHz, CDCl3): δ 8.7, 33.6, 42.6, 53.8, 61.3, 114.1, 118.8, 119.6, 122.8, 124.0, 125.2, 125.3, 126.8, 128.5, 131.0, 137.2, 144.4, 146.4, 146.5, 151.6, 153.4. MALDI TOF-MS: m/z 947.7 [M + Na]+, 963.7 [M + K]+. Anal. Calcd for C62H52O8·0.7CH2Cl2: C 76.49, H 5.47; found: C 76.45, H 5.66.
Synthesis of [2]rotaxane 15
Under an argon atmosphere, a mixture of 11a (30 mg, 0.03 mmol) and G5 (22 mg, 0.03 mmol) in a solution of dry CH2Cl2 (4 mL) and CH3CN (2 mL) was stirred vigorously at r.t. for 5 h. After that, 3,5-di-tert-butylbenzoic anhydride (54 mg, 0.12 mmol) and (n-Bu)3P (3 mg, 0.015 mmol) were added, and the mixture was stirred for another 12 h. The reaction mixture was then concentrated under reduced pressure to give a residue, which was purified by column chromatography over silica gel with CH2Cl2–CH3OH = 6:1 (v/v) as eluent to give 15 (19 mg, 28%) as a yellow solid. Mp: 181–183 °C. 1H NMR (300 MHz, DMSO-d6): δ 1.09–1.36 (m, 44H, two sets of s at 1.29 and 1.30, total 36H for –C(CH3), 8H for –CH2CH2CH2CH2–), 1.50–1.73 (m, 8H, –CH2CH2CH2–), 1.99 (s, 6H, Ar–CH3), 3.47 (d, J = 15 Hz, 4H, Ar–CH2–Ar), 3.71 (d, J = 15 Hz, 4H, Ar–CH2–Ar), 4.38 (t, J = 6 Hz, 2H, –CH2CH2OH), 4.49 (t, J = 6 Hz, 2H, –CH2CH2OH), 4.89 (s, 2H, –C5H4N+CH2CH2–), 5.26–5.35 (m, 4H, 2H for Ar3–CH, 2H for –C5H5NCH2CH2–), 6.19 (s, 2H, Ar3–CH), 6.33 (s, 2H, ArH), 6.36 (d, J = 6 Hz, 4H, ArH), 6.50 (d, J = 6 Hz, 4H, ArH), 6.60–6.68 (m, 2H, ArH), 6.80–6.72 (m, 4H, ArH), 7.06–7.12 (m, 2H, ArH), 7.17–7.28 (m, 6H, 4H for –C5H4N+–, 2H for (t-Bu)2–ArH), 7.67–7.77 (m, 8H, 4H for –C5H4N+–, 4H for (t-Bu)2–ArH), 7.84 (s, 2H, Ar–OH), 7.94 (s, 2H, Ar–OH). ESI-HRMS: calcd for [15-2PF6−]2+: 829.4337; found: 829.4333.
Acknowledgements
We thank the National Natural Science Foundation of China (91127009, 21332008), and the National Basic Research Program (2011CB932501) for financial support.Notes and references
-
(a)
Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer, Academic Publishers, Amsterdam, Holland, 2001 Search PubMed
; (b) Calixarenes Revisited Monographs in Supramolecular Chemistry, ed. C. D. Gutsche and J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1998 Search PubMed
- T. Pierro, C. Gaeta, C. Talotta, A. Casapullo and P. Neri, Org. Lett., 2011, 13, 2650–2653 CrossRef CAS PubMed
- C. Gaeta, C. Talotta, S. Mirra, L. Margarucci, A. Casapullo and P. Neri, Org. Lett., 2013, 15, 116–119 CrossRef CAS PubMed
-
(a) J. L. Atwood, S. J. Dalgarno, M. J. Hardie and C. L. Raston, Chem. Commun., 2005, 337–339 RSC
-
(a) J. L. Atwood, D. L. Clark, R. K. Juneja, G. W. Orr, K. D. Robinson and R. L. Vincent, J. Am. Chem. Soc., 1992, 114, 7558–7559 CrossRef CAS
-
(a) X. Zeng, D. Coquière, A. Alenda, E. Garrier, T. Prangè, Y. Li, O. Reinaud and I. Jabin, Chem.–Eur. J., 2006, 12, 6393–6402 CrossRef CAS PubMed
-
(a) A. Arduini, R. Ferdani, A. Pochini and A. Secchi, Angew. Chem., Int. Ed., 2000, 39, 3453–3456 CrossRef CAS
-
(a) G. Izzet, J. Zeitouny, H. Akdas-Killig, Y. Frapart, S. Menage, B. Douziech, I. Jabin and O. Reinaud, J. Am. Chem. Soc., 2008, 130, 9514–9523 CrossRef CAS PubMed
-
(a) C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC
-
(a) X.-H. Tian, X. Hao and C.-F. Chen, Chem. Commun., 2009, 6771–6773 RSC
- X.-H. Xian and C.-F. Chen, Org. Lett., 2010, 12, 524–527 CrossRef PubMed
- M. Xue and C.-F. Chen, Chem. Commun., 2008, 6128–6130 RSC
-
(a) T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed
-
(a) C. Zhang and C.-F. Chen, J. Org. Chem., 2007, 72, 3880–3888 CrossRef CAS PubMed
- See ESI† for details.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of new compounds. Variable-temperature 1H NMR experiments. X-ray crystal data and crystal packing. Determination of the association constants. ESI-MS spectra of the complexes. CCDC 931895–931898, 931783. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00055a |
| This journal is © the Partner Organisations 2014 |