
Zirconium trisulfide ultrathin nanosheets as efficient catalysts for water oxidation in both alkaline and neutral solutions
Junfeng
Xie†
ab,
Ruoxing
Wang†
b,
Jian
Bao
b,
Xiaodong
Zhang
b,
Hao
Zhang
b,
Shuang
Li
b and
Yi
Xie
*b
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan, Shandong 250014, People's Republic of China
bHefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, People's Republic of China. E-mail: yxie@ustc.edu.cn
First published on 3rd October 2014
Abstract
Developing highly efficient electrocatalysts for oxygen evolution reaction (OER) has emerged as one of the most important research hot spots in energy-related fields due to its critical role in coupling with hydrogen evolution reaction to enhance the total efficiency of water splitting. However, the well-established catalysts for water oxidation up to now are mainly effective in strongly alkaline solutions, thus hindering the practical application in total water splitting. Under this circumstance, herein, we put forward a novel oxygen-evolving electrocatalyst based on zirconium trisulfide (ZrS3) ultrathin nanosheets for the first time, achieving efficient water oxidation behavior in both alkaline and neutral solutions. With the merits of the abundant disulfide bonds on the surface of the ZrS3 ultrathin nanosheets, the OER performance exhibits great enhancement against the bulk material. An ultralow onset overpotential of 244 mV and a small Tafel slope of 45 mV decade−1 are achieved for the ZrS3 ultrathin nanosheets in strongly alkaline solution (pH = 14), while superior OER activity with low onset overpotential and small Tafel slope is also realized in weakly alkaline solutions and even under neutral conditions (pH = 6.9), suggesting the excellent water oxidation performance and showing broad applicable potential for total water splitting. The exploration of this novel catalyst will not only pave a new pathway in searching for advanced catalysts with high efficiency, but also offer an effective strategy in designing functional structures for other electrocatalysts.
Introduction
Electrochemical water splitting has been considered as an efficient and economic pathway to generate the clean fuel—hydrogen.1,2 In the past few years, the fundamental research on the materials that can catalyze electrochemical water splitting has been significantly developed.3,4 For the cathodic hydrogen evolution reaction (HER), a series of HER electrocatalysts, such as MoS2 and WS2,5–10 as well as several nitrides11–13 and phosphides,14,15 have been explored, exhibiting excellent activity and showing potential as alternatives to replace platinum. However, as a coupled half reaction for water splitting, the anodic oxygen evolution reaction (OER) associated with four-electron transfer needs to be paid extensive attention since its high kinetic barrier and ambiguous reaction dynamics severely restrict the total water splitting efficiency.3,16 Recently, a lot of metal oxides,17,18 hydroxides,19–21 and mixed compounds with a spinel structure22–25 have been identified as catalysts for water oxidation, i.e. oxygen evolution. However, the onset overpotential for these catalysts to drive the OER process is still not satisfied for practical water splitting. Besides, these catalysts are prone to undergo an additional intrinsic oxidation reaction near the OER onset, thus increasing the difficulty for analyzing the OER mechanism. Moreover, these catalysts are prone to drive water oxidation reaction in a strongly alkaline environment, which significantly limits the range of practical applications. Hence, developing efficient OER electrocatalysts with facile electrode reactions that can work under both alkaline and neutral conditions will be an effective approach to search for advanced water oxidation catalysts.Nowadays, transition metal chalcogenide (TMC)-based electrocatalysts have drawn increasing research attention due to their high efficiency for water oxidation. Recently, a novel CoSe2/nitrogen-doped graphene composite catalyst has been reported, exhibiting efficient water oxidation behavior in alkaline solution.26 Moreover, a series of sulfide-based electrocatalysts, such as Ni3S2/Ni foam,27 NiCo2S4,28 and Mn3O4/CoSe2,29 were also explored as efficient OER catalysts. However, the active sites of the aforementioned catalysts are mainly located at the multi-valence metal atoms, thus they cannot be served as the ideal materials to eliminate the influence of the intrinsic oxidation reaction. Under this circumstance, transition metal trisulfides attract our attention due to the existence of the unsaturated disulfide bonds that may act as the active sites for electrocatalytic processes. Based on the above analyses, herein, we put forward an efficient water oxidation catalyst based on the zirconium trisulfide (ZrS3) ultrathin nanosheets. With the merits of the unique crystal structure in which abundant unsaturated disulfide bonds exist on the surface of the nanosheets, the novel ZrS3-based nanosheet catalyst exhibits efficient OER activity in both alkaline and neutral environments. Furthermore, the two-dimensional (2D) ultrathin configuration of the as-synthesized nanosheets provides fast in-plane electrical conductivity as well as large surface area, which can boost the charge transfer process, increase the interactive area with the electrolyte, and further extensively facilitate the electrocatalytic processes.30–40 With the benefits of the above issues, ZrS3 ultrathin nanosheets exhibit a low onset overpotential of 244 mV in strongly alkaline solution (pH = 14). A small Tafel slope of 45 mV decade−1 is also achieved with no influence of other side reactions, demonstrating the excellent water oxidation activity. Moreover, superior OER activity with low onset overpotential and small Tafel slope is also realized in weakly alkaline solutions and even under neutral conditions (pH = 6.9), suggesting the broad applicable potential for practical water splitting. The exploration of this novel trisulfide-based catalyst will not only broaden our vision in searching for advanced catalysts with high efficiency, but also provide a new direction in designing functional structures for other existing electrocatalysts to improve their performance.
Results and discussion
The zirconium trisulfide ultrathin nanosheets were fabricated for the first time via a modified liquid exfoliation process (see details in the Experimental section).41–43 X-ray diffraction (XRD) was applied to investigate the structural information of the products. As shown in Fig. 1A, all of the diffraction peaks for the bulk material are in good agreement with the standard pattern of the monoclinic zirconium trisulfide (JCPDS card no. 30-1498), revealing the high purity of the product. It is worth noting that, for the as-exfoliated zirconium trisulfide ultrathin nanosheets, the relative intensity of (00L) diffraction peaks is apparently enhanced, while the intensity of other peaks is eliminated, suggesting the preferential exposure of the basal planes which can be attributed to the successful exfoliation. Furthermore, no additional peak emerges after the liquid exfoliation process, confirming the structural stability of the ZrS3 ultrathin nanosheets. Moreover, Raman spectroscopy was performed to confirm the ZrS3 structure. As shown in Fig. 1B, all of the Raman peaks can be indexed as the characteristic Ag and Bg vibrational modes of the zirconium trisulfide lattice, thus verifying the existence of the ZrS3 structure.44 As a typical layered material with a van der Waals gap, an individual ZrS3 layer is a quadruple layer with four-atom thickness, in which sulfur atoms exist in two different forms with distinct coordinating environments – sulfur atoms in the −2 state (S2−) that bond with four zirconium atoms in the middle of the quadruple layer and the bridging disulfide bonds (S22−) on the basal surface of individual layers (Fig. 1C and D). Hence, the surface of the ZrS3 layers can be considered to be thoroughly built by the bridging S22− groups. The unique crystal structure with the basal surface that was completely built by unsaturated disulfide bonds may act as active sites for electrocatalytic reactions.45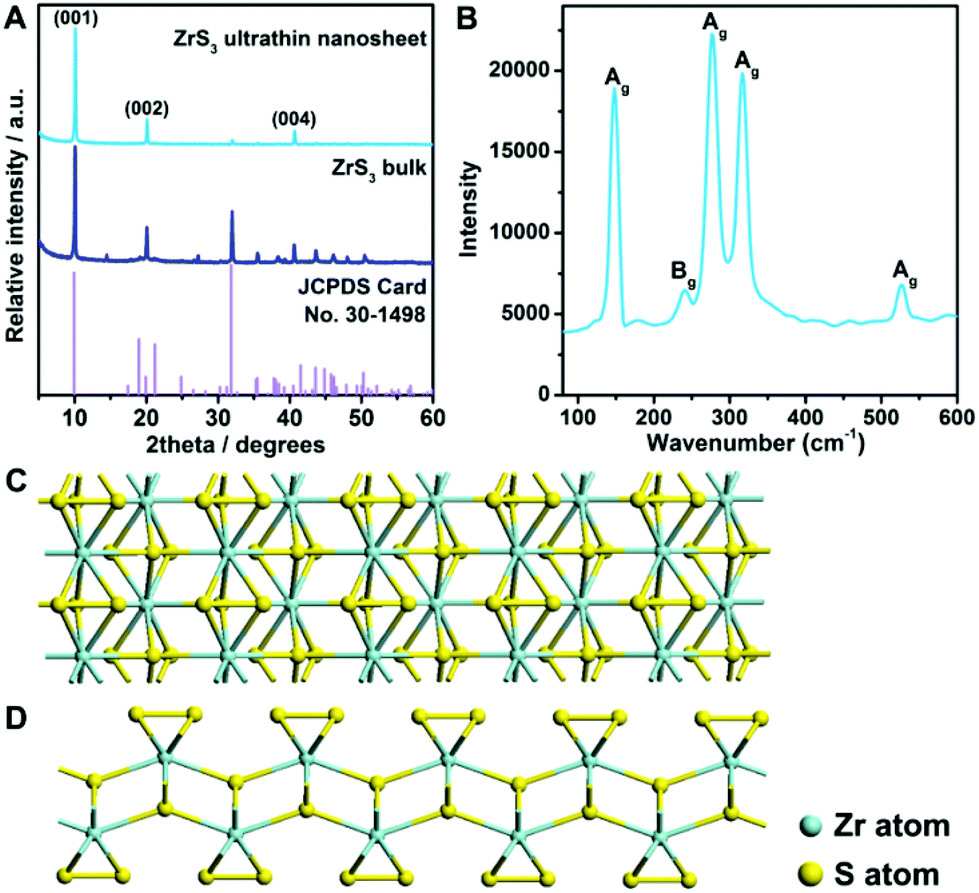 | ||
| Fig. 1 (A) XRD patterns of the ZrS3 ultrathin nanosheets and ZrS3 bulk with the reference of the standard pattern of zirconium trisulfide, from which the preferential exposure of the (00L) planes of the ultrathin nanosheets can be identified. (B) Raman spectrum showing the characteristic Ag and Bg vibrational modes of zirconium trisulfide lattice. (C) Top-view and (D) side-view of the crystal structure of an individual zirconium trisulfide layer. | ||
To survey the morphological and chemical information of the as-exfoliated product, transmission electron microscopy (TEM) and elemental analyses were applied. As shown in Fig. 2A, the TEM image clearly reveals the nanosheet morphology, and the homogeneous contrast against the carbon sheet suggests the ultrathin nature of the nanosheet. As can be seen, the lateral size of the ZrS3 ultrathin nanosheet is in the range of several hundred nanometers. The high-resolution transmission electron microscopy (HRTEM) image indicates that the interplanar spacing of the basal surface of the nanosheet is 2.54 Å, which is consistent with the distance between individual (200) planes (Fig. 2B). Furthermore, from the HRTEM analysis, the high crystallinity of the ZrS3 ultrathin nanosheets can be confirmed, which ensures the electrochemical stability during the long-period catalytic processes. Energy-dispersive spectroscopy (EDS) elemental mapping analyses were carried out in the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) mode, from which the homogeneous distribution of zirconium and sulfur on the whole nanosheet can be revealed (Fig. 2C). Furthermore, the chemical states of sulfur were identified by X-ray photoelectron spectroscopy (XPS). As can be seen from Fig. 2D, the S 2p peaks can be divided and indexed as S2− and bridging S22− ligands, confirming the existence of the ZrS3 structure. The surface of the ZrS3 ultrathin nanosheets was thoroughly built by the disulfide bonds, thus ensuring an extremely high exposure of unsaturated sulfur atoms and further providing an ideal material prototype to investigate the role that disulfide bonds play during the electrocatalytic process.
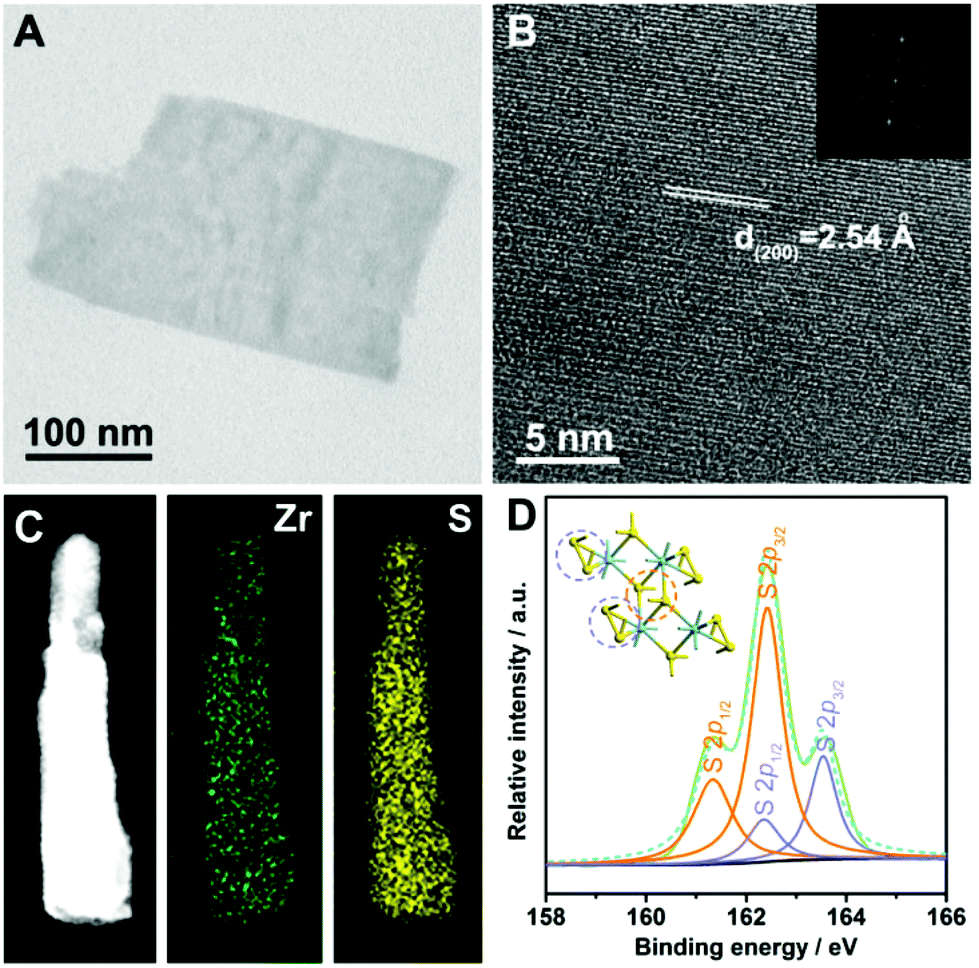 | ||
| Fig. 2 (A) TEM image of an individual ZrS3 ultrathin nanosheet. (B) HRTEM image of the basal surface of the ZrS3 ultrathin nanosheets and the corresponding FFT pattern. (C) HAADF-STEM image of the ZrS3 ultrathin nanosheet and the corresponding elemental mapping images for zirconium and sulfur, respectively. (D) XPS spectrum (blue dashed line) and the fitted curves (green solid line) reveal the binding energies of the sulfur in two forms, where the orange curves represent the S2− ligands and the purple curves represent the bridging S22− ligands. Inset: structural model of zirconium trisulfide showing the corresponding coordinating environments of S2− ligands (highlighted in orange circle) and the bridging S22− ligands (highlighted in purple circles), respectively. | ||
In order to investigate the oxygen-evolving performance of zirconium trisulfide, electrochemical measurements of both ZrS3 bulk and ultrathin nanosheets were performed in 1 M KOH solution. By assembling the as-exfoliated ZrS3 ultrathin nanosheets into thin films on the conducting indium tin oxide (ITO) substrates, the working electrode with an active area of 1 cm2 can be prepared. As shown in the polarization curves in Fig. 3A, the ZrS3 ultrathin nanosheets exhibit an ultralow OER onset overpotential of 244 mV (∼500 mV vs. Ag/AgCl electrode), beyond which the anodic current density rises rapidly under more positive potentials. This low onset overpotential for the ZrS3 ultrathin nanosheets shows a 456 mV decrease than that of the bulk counterpart (∼700 mV), and exhibits tremendous advances compared with other electrocatalysts for water oxidation, such as Co3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22–24 and Ni(OH)2,20 suggesting the excellent catalytic activity. It is worth noting that no additional oxidation peak exists in the whole range of the polarization curves for both bulk and ultrathin nanosheet catalysts. That is to say, unlike the traditional water oxidation catalysts, ZrS3-based catalysts undergo only one electrochemical reaction in the whole potential range, suggesting a higher efficiency for oxygen evolution reaction. Moreover, the absence of the intrinsic oxidation peaks can lead to a more accurate determination of the onset overpotential, thus providing more valuable information for the study of the oxygen-evolving mechanism. Furthermore, the ultrathin nanosheets possess a much better processability compared with the bulk material, thus resulting in faster interlayer electron transport to boost the oxygen evolution reaction. Considering the fact that the surface of the ZrS3 ultrathin nanosheets is wholly built by the unsaturated disulfide bonds, it is reasonable to believe that the abundant disulfide bonds may act as the active sites to drive the oxygen evolution reaction.
22–24 and Ni(OH)2,20 suggesting the excellent catalytic activity. It is worth noting that no additional oxidation peak exists in the whole range of the polarization curves for both bulk and ultrathin nanosheet catalysts. That is to say, unlike the traditional water oxidation catalysts, ZrS3-based catalysts undergo only one electrochemical reaction in the whole potential range, suggesting a higher efficiency for oxygen evolution reaction. Moreover, the absence of the intrinsic oxidation peaks can lead to a more accurate determination of the onset overpotential, thus providing more valuable information for the study of the oxygen-evolving mechanism. Furthermore, the ultrathin nanosheets possess a much better processability compared with the bulk material, thus resulting in faster interlayer electron transport to boost the oxygen evolution reaction. Considering the fact that the surface of the ZrS3 ultrathin nanosheets is wholly built by the unsaturated disulfide bonds, it is reasonable to believe that the abundant disulfide bonds may act as the active sites to drive the oxygen evolution reaction.
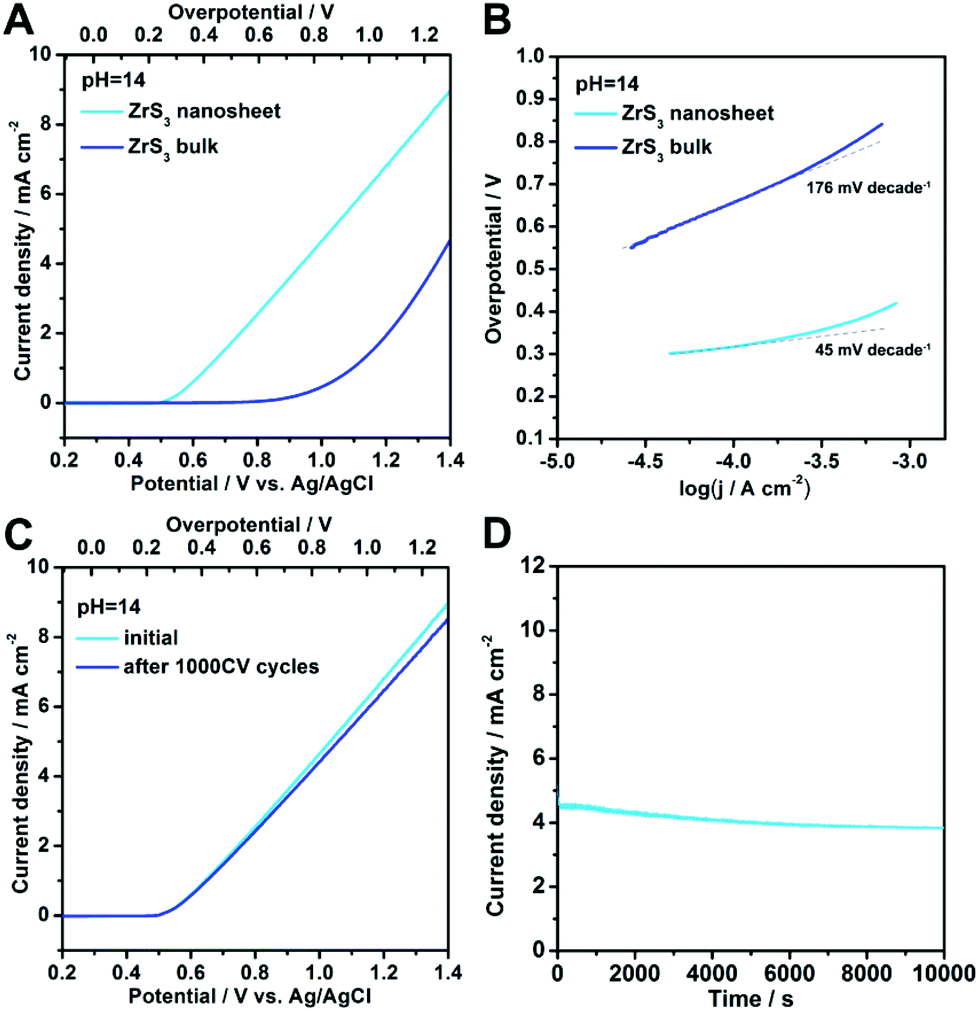 | ||
| Fig. 3 (A) Polarization curves and (B) the corresponding Tafel plots of ZrS3 ultrathin nanosheets and the bulk counterpart, suggesting the magnificent enhancement of OER activity after liquid exfoliation. (C) Polarization curves of the ZrS3 ultrathin nanosheets revealing negligible degradation of the water oxidation activity even after 1000 CV cycles. (D) Time dependence of the anodic current density at a static overpotential of 800 mV. | ||
To obtain more insight into the OER process for ZrS3-based catalysts, Tafel plots for both ultrathin nanosheets and the bulk counterpart were investigated. As shown in Fig. 3B, the ZrS3 ultrathin nanosheets exhibit a small Tafel slope of 45 mV decade−1, which is much smaller than that of the bulk material (176 mV decade−1), revealing the excellent OER activity. It is worth noting that the smaller Tafel slope for ZrS3 ultrathin nanosheets is more advantageous for practical applications, since it will lead to a faster increment of anodic current density with increasing overpotential. Moreover, with the merits of the absence of the intrinsic oxidation reactions near the OER onset, the analysis of the Tafel slope for ZrS3-based catalysts is more reliable and valuable than that of the transition metal (hydr)oxide-based catalysts, since the Tafel slope for such traditional catalysts is a combined result that reflects not only the OER process but also the intrinsic oxidation reaction, while the Tafel slope for ZrS3-based catalysts is only relevant to the OER process. Hence, the study of the Tafel slope for ZrS3-based catalysts is much more valuable for understanding the OER mechanism.
Apart from the evaluation of the OER activity, electrochemical stability is another significant criterion to assess an oxygen-evolving electrocatalyst. In order to survey the electrochemical stability of the ZrS3-based catalysts, the long-term cycling test and continuous OER process at a static overpotential were conducted. As shown in Fig. 3C, the polarization curve of the ZrS3 ultrathin nanosheets shows negligible degradation even after 1000 voltammetric cycles, suggesting the superior electrochemical stability of the ZrS3 ultrathin nanosheets for long-period OER catalysis. Furthermore, when an overpotential of 800 mV was applied, a continuous OER process occurred to generate molecular O2. As can be seen from Fig. 3D, the anodic current density exhibits only slight decrement even after a long period of 10000 seconds. Of note, the slight degradation of the anodic current density might be caused by the consumption of OH− or the remaining of O2 bubbles on the surface of the catalyst that hindered the reaction. Hence, the electrochemical stability under continuous OER catalysis is also verified. The outstanding features like high catalytic activity as well as excellent stability indicate that the ZrS3 ultrathin nanosheet is an advanced electrocatalyst for water oxidation reaction.
Beside the performance in a strongly alkaline environment, the OER activity at various pH values including weakly alkaline conditions and even neutral solution is also significant for practical applications.46–49 The polarization curves in different environments (pH = 6.9, 9.2 and 13) are shown in Fig. 4A–C, respectively. As can be seen, the ZrS3 ultrathin nanosheets exhibit higher catalytic activity compared with the bulk material at all of these pH values, confirming the excellent OER performance of the nanosheet catalyst. The onset overpotentials for the ZrS3 ultrathin nanosheets are 610 mV, 420 mV and 280 mV for pH = 6.9, 9.2 and 13, respectively. To the best of our knowledge, the onset overpotential in neutral solution (pH = 6.9) is among the best records for various water oxidation catalysts, demonstrating the superior OER activity of the ZrS3 ultrathin nanosheets under neutral conditions.46–49 Furthermore, the Tafel plots obtained in different environments are shown in Fig. 4D, from which the Tafel slopes can be determined as 102 mV decade−1, 74 mV decade−1 and 51 mV decade−1 for pH = 6.9, 9.2 and 13, respectively, indicating the excellent activity for water oxidation. The high OER activity, small Tafel slope and excellent performance in a neutral environment make this new catalyst a promising candidate for water oxidation in the future.
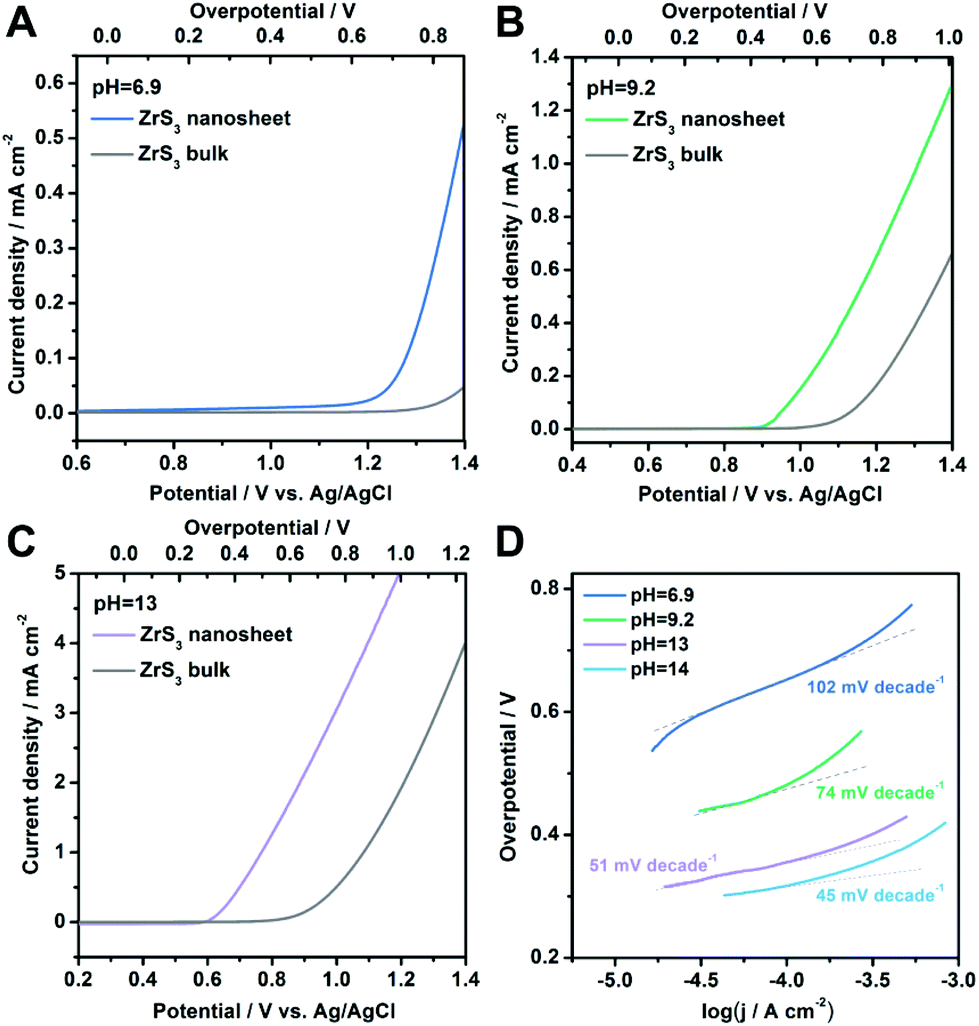 | ||
| Fig. 4 (A–C) Polarization curves of ZrS3 ultrathin nanosheets and the bulk material at various pH values. (D) Corresponding Tafel plots under the same conditions. | ||
Conclusions
In summary, ZrS3 ultrathin nanosheets with high exposure of disulfide bonds were successfully prepared by liquid exfoliation. With the benefits of the existence of abundant unsaturated disulfide bonds, the ZrS3 ultrathin nanosheets exhibit an ultralow onset overpotential of 244 mV and a small Tafel slope of 45 mV decade−1 for OER, demonstrating excellent water oxidation activity. Moreover, superior electrochemical stability was also achieved, making this novel material an efficient catalyst for OER catalysis in an alkaline environment. The superior OER performance for the ZrS3 ultrathin nanosheets may arise from the abundant disulfide bonds on the surface of the nanosheets, which will provide a hopeful direction to design advanced OER catalysts. Besides, excellent catalytic activity of ZrS3 ultrathin nanosheets is also achieved at various pH values, ranging from the neutral solution to alkaline conditions, which is significant for practical applications. The relationship between the structural features and the electrocatalytic performance revealing from this work will benefit the design and exploration of novel electrocatalysts in the near future.Experimental section
Materials
1-Cyclohexyl-2-pyrrolidinone (99%) was supplied by Alfa Aesar, and the other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd. All of the chemicals were used as received without further purification.Synthesis of the ZrS3 bulk material
The ZrS3 bulk material was synthesized according to a method reported previously.50 In detail, ZrS3 bulk material was prepared by reacting 1 mmol of zirconium powder (0.0912 g) with 3 mmol of sulfur powder (0.0962 g) in a vacuum-sealed quartz tube with a diameter of 5 mm and a length of 10 cm. Then the quartz tube with the mixed precursors was heated in a horizontal tube furnace at 1000 °C for 24 h to ensure the growth of the large-sized bulk material. Caution: High-temperature reaction should be approached with reasonable care. The heating and cooling rates were set as 2 °C min−1. After the high-temperature reaction, the system was allowed to cool down to room temperature. Finally, the orange bulk material with a large size was obtained.Preparation of the ZrS3 ultrathin nanosheets
The ZrS3 ultrathin nanosheets were prepared by a modified liquid exfoliation process. Typically, 60 mg of ZrS3 bulk material was loaded in a Teflon cup with a capacity of 23 mL, and 8 mL n-propylamine was added. Then the dispersion was tightly sealed in a stainless steel autoclave after stirring for 30 min and then was heated at 120 °C for 3 days. After the solvothermal treatment, the reaction system was cooled down to room temperature naturally. The obtained product was then washed with water and absolute ethanol several times. After this procedure, 30 mg of the as-treated product, 10 mL of absolute ethanol and 10 mL of 1-cyclohexyl-2-pyrrolidinone were loaded into a weighing bottle with a capacity of 20 mL. After ultrasound treatment for 12 h to ensure the complete exfoliation, the supernatant was collected by centrifugation, washed with absolute ethanol and acetone, and dried at 60 °C under vacuum.Characterization
X-ray diffraction (XRD) was performed on a Philips X'Pert Pro Super diffractometer with Cu Kα radiation (λ = 1.54178 Å). Raman spectra were recorded on a Renishaw RM3000 Micro-Raman system with a 514.5 nm Ar laser. Transmission electron microscopy (TEM) was carried out on a JEM-2100F field emission electron microscope at an acceleration voltage of 200 kV. High-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding energy-dispersive spectroscopy (EDS) mapping analyses were performed on a JEOL JEM-ARF200F TEM/STEM with a spherical aberration corrector. X-ray photoelectron spectra (XPS) were acquired on a Thermo ESCALAB250 with Al Kα as the excitation source.Electrochemical measurements
All of the electrochemical measurements were performed in a three-electrode system on an electrochemical workstation (CHI660B). Typically, 4 mg of the catalyst (nanosheets or bulk) and 10 μL of Nafion solution (Sigma Aldrich, ∼5% in a mixture of lower aliphatic alcohols and water) were dispersed in 0.2 mL water–isopropanol solution with a volume ratio of 3:1 by sonicating for 1 h to form a homogeneous ink. Then 0.1 mL homogeneous solution was diluted with water and loaded onto an indium tin oxide (ITO) conducting substrate as a thin-film working electrode. Linear sweep voltammetry with a scan rate of 5 mV s−1 was conducted using the Ag/AgCl (in 3 M KCl solution) electrode as the reference electrode, and a platinum sheet electrode as the counter electrode to obtain the polarization curves. In order to investigate the OER activity at various pH values, mixed phosphate buffer solution (pH = 6.9), sodium borate buffer solution (pH = 9.2), 0.10 M KOH solution (pH = 13) and 1 M KOH solution (pH = 14) were applied as the electrolytes, respectively. All of the electrolytes were degassed by bubbling pure oxygen for 30 min before the electrochemical measurements. Cyclic voltammetry (CV) was conducted between 0 and 1.4 V vs. Ag/AgCl electrode at 50 mV s−1 to investigate the cycling stability.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (11079004, 21331005, and 11321503), the Chinese Academy of Science (XDB01020300), and the Fundamental Research Funds for the Central University (WK2060190032).Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- T. E. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed.
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- X. Sun, J. Dai, Y. Guo, C. Wu, F. Hu, J. Zhao, X. Zeng and Y. Xie, Nanoscale, 2014, 6, 8359–8367 RSC.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014 10.1039/c4sc02019g.
- W.-F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403–409 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 Search PubMed.
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516–4521 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- A. Zhao, J. Masa, W. Xia, A. Maljusch, M.-G. Willinger, G. Clavel, K. Xie, R. Schlögl, W. Schuhmann and M. Muhler, J. Am. Chem. Soc., 2014, 136, 7551–7554 CrossRef CAS PubMed.
- M.-R. Gao, X. Cao, Q. Gao, Y.-F. Xu, Y.-R. Zheng, J. Jiang and S.-H. Yu, ACS Nano, 2014, 8, 3970–3978 CrossRef CAS PubMed.
- W. Zhou, X.-J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- Z. Zhang, X. Wang, G. Cui, A. Zhang, X. Zhou, H. Xu and L. Gu, Nanoscale, 2014, 6, 3540–3544 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang, Y.-R. Zheng and S.-H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- X. Zhang and Y. Xie, Chem. Soc. Rev., 2013, 42, 8187–8199 RSC.
- Y. Sun, S. Gao and Y. Xie, Chem. Soc. Rev., 2014, 43, 530–546 RSC.
- J. Xie, S. Li, R. Wang, H. Zhang and Y. Xie, Chem. Sci., 2014, 5, 1328–1335 RSC.
- J. Xie, X. Sun, N. Zhang, K. Xu, M. Zhou and Y. Xie, Nano Energy, 2013, 2, 65–74 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, J. Zhao, B. Pan, M. Kong, J. Chen and Y. Xie, J. Am. Chem. Soc., 2012, 134, 11908–11911 CrossRef CAS PubMed.
- K. Xu, P. Chen, X. Li, C. Wu, Y. Guo, J. Zhao, X. Wu and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 10477–10481 CrossRef CAS PubMed.
- J. Feng, X. Sun, C. Wu, L. Peng, C. Lin, S. Hu, J. Yang and Y. Xie, J. Am. Chem. Soc., 2011, 133, 17832–17838 CrossRef CAS PubMed.
- J. Feng, L. Peng, C. Wu, X. Sun, S. Hu, C. Lin, J. Dai, J. Yang and Y. Xie, Adv. Mater., 2012, 24, 1969–1974 CrossRef CAS PubMed.
- Y. Sun, J. Zhu, L. Bai, Q. Li, X. Zhang, W. Tong and Y. Xie, Inorg. Chem. Front., 2014, 1, 58–64 RSC.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1420 CrossRef CAS.
- K.-G. Zhou, N.-N. Mao, H.-X. Wang, Y. Peng and H.-L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed.
- A. Zwick, M. A. Renucci and A. Kjekshus, J. Phys. C: Solid State Phys., 1980, 13, 5603 CrossRef CAS.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Nat. Commun., 2014, 5, 4256 Search PubMed.
- K. S. Joya, Y. F. Joya and H. J. M. de Groot, Adv. Energy Mater., 2014, 4 DOI:10.1002/aenm.201301929.
- Y. Zhang, B. Cui, Z. Qin, H. Lin and J. Li, Nanoscale, 2013, 5, 6826–6833 RSC.
- L. Li, X. Fang, T. Zhai, M. Liao, U. K. Gautam, X. Wu, Y. Koide, Y. Bando and D. Golberg, Adv. Mater., 2010, 22, 4151–4156 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2014 |