
Formation of glutathione sulfenate and sulfinate complexes by an organoiridium(III) anticancer complex†
Zhe
Liu‡
and
Peter J.
Sadler
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk
First published on 24th September 2014
Abstract
The organoiridium(III) anticancer complex [(η5-Cpxbiph)Ir(phpy)py] (Cpxbiph = biphenyltetramethylcyclopentadienyl, phpy = phenylpyridine, py = pyridine) unexpectedly reacts with the tripeptide glutathione in the presence of coenzyme NADH to give glutathione sulfenate (Ir–S(O)G) and sulfinate (Ir–S(O)2G) complexes. These new adducts may play a role in the anticancer activity of such organoiridium complexes.
The platinum drug cis-[PtCl2(NH3)2] (cisplatin) and the related complexes carboplatin and oxaliplatin have made a major contribution to cancer chemotherapy in the last 30 years.1 Most attention in relation to their mechanism of action has focussed on DNA as the target.2 Recently interest in platinum binding to peptides and proteins has emerged in order to understand not only off-target effects which cause toxicity, but also platinum transport.3 The tripeptide GSH (γ-L-Glu-L-Cys-Gly) is highly abundant in cells and is thought to be involved in platinum detoxification.4 In common with many other transition metal ions, PtII, a ‘soft’ metal ion, binds strongly to thiolate sulphur, a ‘soft’ ligand. GSH is an important antioxidant, preventing damage to cellular components caused by reactive oxygen species.5 Thus, reactions of metal complexes with GSH can affect the redox state of cells.6
We have previously reported that, unlike platinum drugs, some half-sandwich RuII arene anticancer complexes form glutathione complexes that rapidly undergo oxidation at the bound thiolate sulfur to give unusual sulfenato and sulfinato adducts.7 Curiously this appears to promote subsequent reactions with DNA. The sulfenato ligand can be displaced by the DNA base guanine.8 Oxidation of bound thiolate sulfur can also occur when the cysteine thiolate is in a protein (e.g. albumin, glutathione transferase) although the local reactivity of cysteine residues depends on their environment and the nature of the arene.9 This poses the question as to whether thiol-mediated redox reactions could play a role in the mechanism of action of other half-sandwich transition metal complexes.
Organometallic cyclopentadienyl IrIII complexes exhibit promising anticancer activity.10 We have reported previously the formation of the Ir-GSH adduct [(η5-Cpxbiph)Ir(phpy)(SG)]− (Ir–SG, Cpxbiph = biphenyltetramethylcyclopentadienyl, phpy = C^N-chelated phenylpyridine ligand), Chart 1, by substitution of the chlorido and pyridine (py) ligands in the anticancer complexes [(η5-Cpxbiph)Ir(phpy)Cl] (Ir–Cl)11 and [(η5-Cpxbiph)Ir(phpy)(py)]+ (Ir–py) with glutathione, reactions which may influence their potency.12 In the work reported here we have investigated whether Ir–SG is unreactive, as might be expected for a third row transition metal with a sulfur ligand, which might mean that this adduct would be a ‘dead-end’ product in cells with little involvement in anticancer activity, or perhaps can undergo oxidation at sulfur in a similar manner to RuII arene complexes.
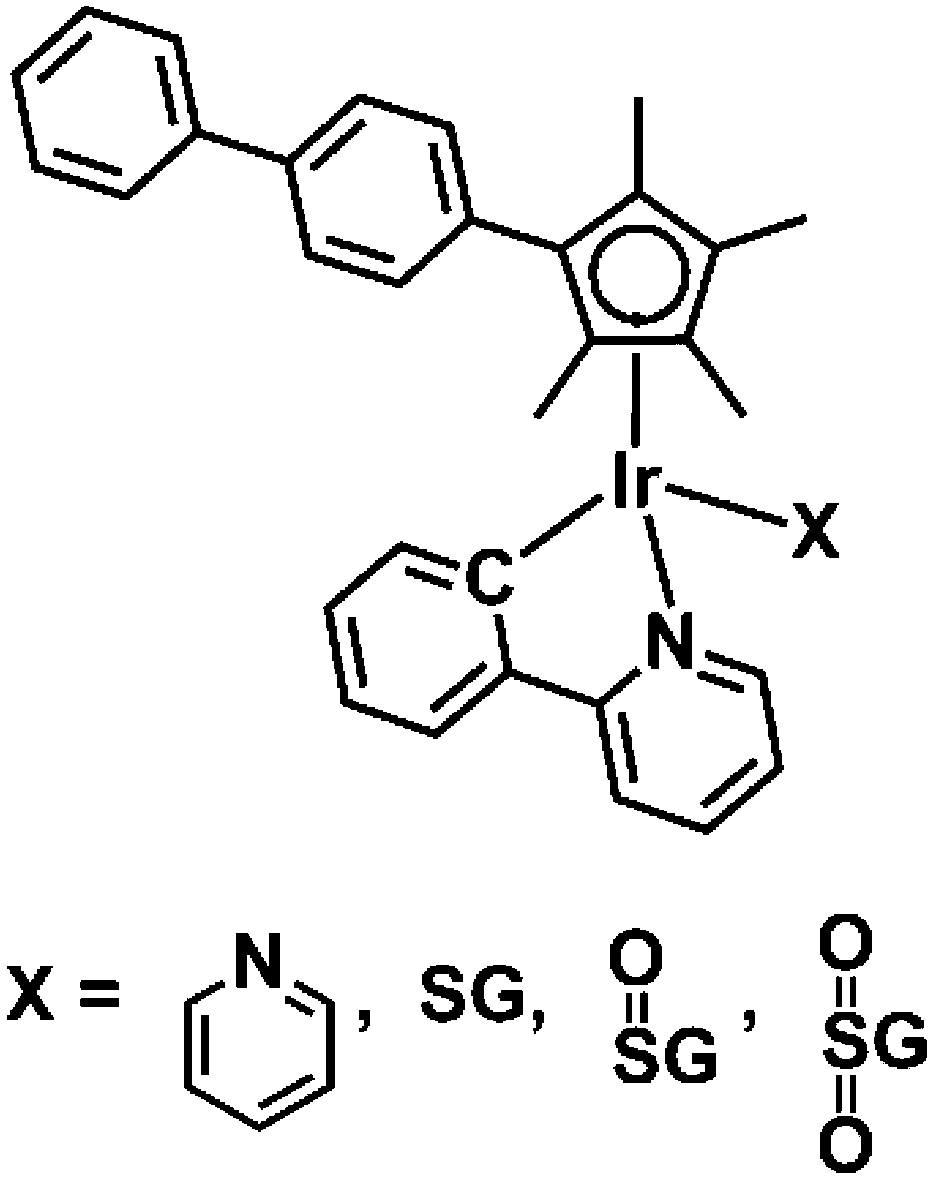 | ||
| Chart 1 Iridium complexes studied in this work. | ||
First we investigated the formation and stability of the iridium glutathione adducts. To a solution of complex Ir–py, 3.5 mol equiv. GSH was added. After 12 h, 95% of Ir–py had reacted with formation of complex [(η5-Cpxbiph)Ir(phpy)(SG)]− (Ir–SG).12 There was little change to the solution after further 18 h. When the above reaction was studied in the presence of 104 mM NaCl (mimicking the chloride concentration in blood plasma), 90% of the {Ir(η5-Cpxbiph)} fragment (which is readily monitored by the sharp 1H NMR singlets for the Cp methyl groups between 1.5 and 2.0 ppm) was still in the form of Ir–SG and only 10% was present as Ir–Cl or Ir–py after 17 h (Fig. S1†), indicating the high stability of Ir–SG.
However, when coenzyme NADH (2 mol equiv.) was added to a solution containing 95% Ir–SG and 5% Ir–py, surprisingly oxidation of NADH to NAD+ was observed (Fig. 1, Table 1, Reaction 1). Notably this led to ca. 34% of Ir–SG being oxidized to the sulfenato complex [(η5-Cpxbiph)Ir(phpy)(S(O)G]− (Ir–S(O)G). Complex Ir–S(O)G gave two sets of peaks for the Cpxbiph methyl protons at 1.93, 1.88, 1.84, and 1.69 ppm and 1.92, 1.88, 1.82 and 1.69 ppm in a ratio ca. 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Fig. 1C, assignable to two diastereomers; one diastereomer may be preferentially stabilized by H-bonding. The formation of Ir–S(O)G was confirmed by ESI-MS analysis giving a singly-charged ion peak centred at m/z 943.3, assignable to {(η5-Cpxbiph)Ir(phpy)(S(O)G) + 2H}+ (calcd m/z 943.2, Fig. 2).
:1, Fig. 1C, assignable to two diastereomers; one diastereomer may be preferentially stabilized by H-bonding. The formation of Ir–S(O)G was confirmed by ESI-MS analysis giving a singly-charged ion peak centred at m/z 943.3, assignable to {(η5-Cpxbiph)Ir(phpy)(S(O)G) + 2H}+ (calcd m/z 943.2, Fig. 2).
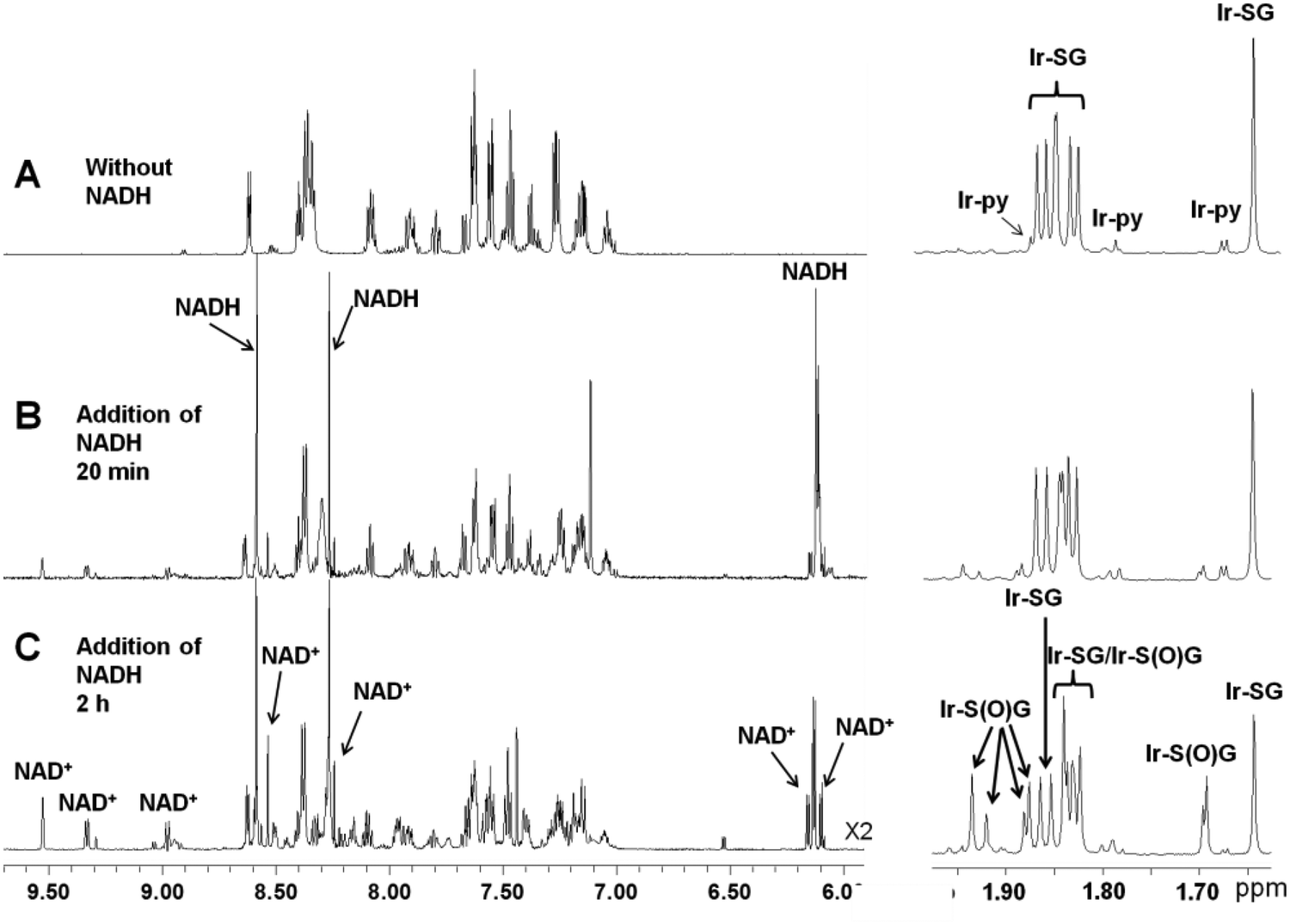 | ||
| Fig. 1 1H NMR spectra for the reaction of NADH with Ir–SG, showing formation of NAD+ and sulfenato complex [(η5-Cpxbiph)Ir(phpy)(S(O)G)]− (Ir–S(O)G). (A) Formation of Ir–SG from reaction of Ir–py (0.6 mM) with GSH (3.5 mol equiv.) in 30% MeOD-d4–70% PBS H2O buffer; (B) 20 min after addition of NADH (2 mol equiv.) to the above equilibrium solution; (C) Formation of NAD+ and Ir–S(O)G, 2 h after addition of NADH. Peak assignments are shown. | ||
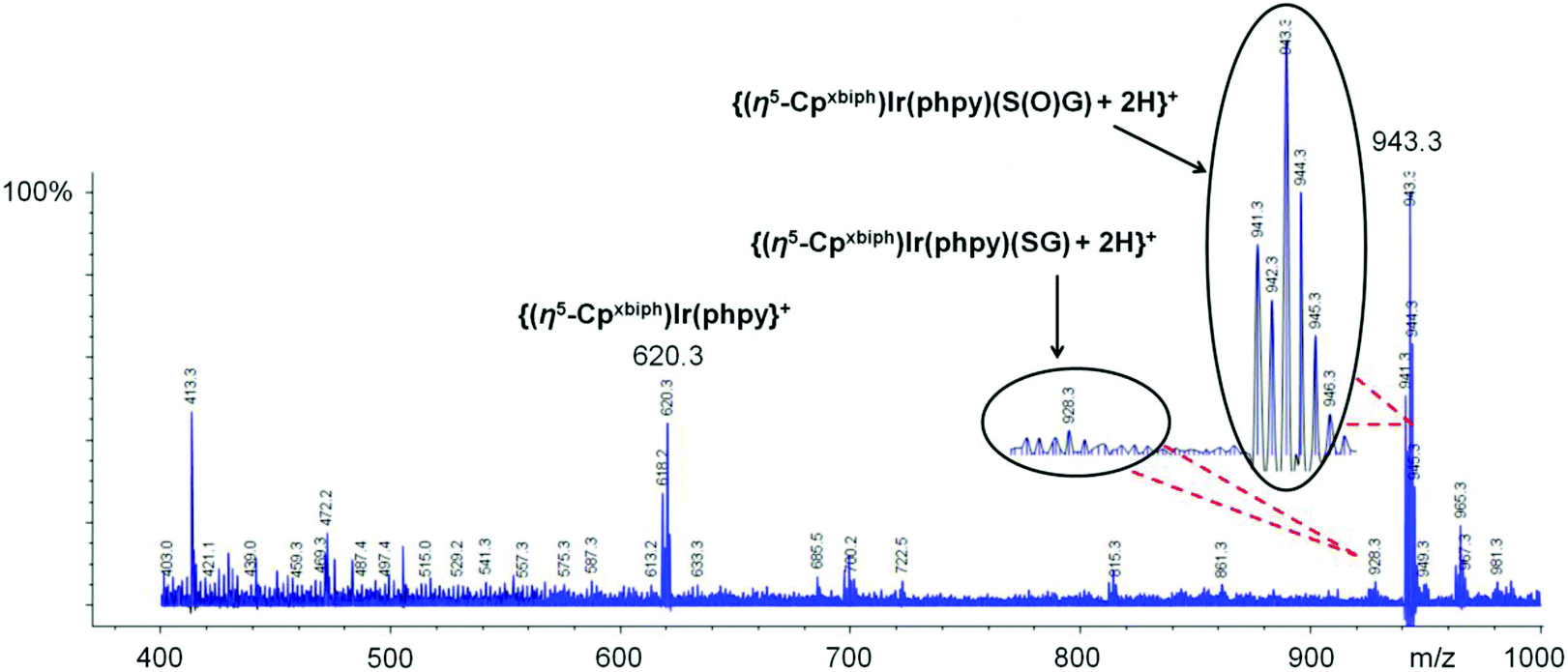 | ||
| Fig. 2 ESI-MS for an equilibrium solution containing Ir–SG and NADH in 30% MeOD-d4–70% PBS H2O buffer (v/v) 298 K. The peak at m/z 943.3 is assignable to the glutathione sulfenato complex {(η5-Cpxbiph)Ir(phpy)(S(O)G) + 2H}+ (calcd m/z 943.2). | ||
| Reaction | Buffer | Atmosphere | Products |
|---|---|---|---|
| 1 | Yes | Air | Ir–S(O)G, NAD+ |
| 2 | No | Air | Ir–S(O)G, NAD+ |
| 3 | Yes | N2 | Ir–SG, NAD+ |
We showed previously that complex Ir–py can catalytically oxidize NADH to NAD+ by hydride transfer from NADH to IrIII generating hydrido complexes (Ir–H) which can transfer the hydride to oxygen to form H2O2.12 The production of H2O2 was confirmed by the appearance of a light blue colour on an H2O2 detection stick (Fig. 3), showing that ca. 60 μM of H2O2 was present after 48 h. The role of H2O2 in the oxidation of Ir–SG was further confirmed by addition of catalase which decomposes H2O2 to water and oxygen. As expected, no sulfenato complex Ir–S(O)G was observed after 4 h of reaction of Ir–SG with NADH in the presence of catalase, confirming that H2O2 is responsible for oxidizing Ir–SG to Ir–S(O)G.
 | ||
| Fig. 3 Detection of hydrogen peroxide. (A) To a solution of Ir–SG (produced by reaction of 0.6 mM Ir–py with 3.5 mol equiv. GSH in 30% MeOD-d4–70% PBS H2O buffer (v/v) at 310 K), 2 mol equiv. NADH were added. (B) After 48 h, H2O2 (about 60 μM) was detected by quantofix peroxide test sticks. | ||
In the present case, the active hydrido species is probably formed from the small amounts of Ir–py (ca. 5% of Ir) present in solution in equilibrium with the more stable complex Ir–SG. The reaction appears to be catalytic, which is consistent with our previous results.12 The sulfenato complex Ir–S(O)G has not been previously reported, and the only other known Ir sulfenato complex appears to be [IrCl2(CH3SO)(CO)L2] (L = P(C6H5)3 or P(C6H5)2CH3) prepared by the reaction of CH3S(O)Cl with IrCl(CO)L2.13
The reaction of NADH with Ir–SG was also studied in unbuffered aqueous solution, in air (Reaction 2), and under a nitrogen atmosphere in buffered solution (Reaction 3), Table 1. Importantly, no Ir–S(O)G was observed in the absence of NADH. However, addition of NADH to solutions under the conditions of Reaction 2 led to the generation of Ir–S(O)G, as for Reaction 1. In contrast, no Ir–S(O)G was formed after addition of NADH in Reaction 3 which was carried out under N2 (Fig. S2†). These results indicate that: (1) buffer has little effect on the species formed, (2) Ir–S(O)G is formed only in the presence of O2, indicating that the oxygen atom in Ir–S(O)G originates from O2via the formation of H2O2, and (3) NADH plays a crucial role in the reaction, involving hydride transfer from NADH to Ir and finally to O2.12
To confirm that H2O2 can directly oxidize Ir–SG to Ir–S(O)G, the oxidation of Ir–SG (produced by reaction of Ir–py with GSH) by H2O2 (10 mol equiv.) was studied at 310 K (Fig. 4). Ir–SG was oxidized to the sulfenato complex [(η5-Cpxbiph)Ir(phpy)(S(O)G)]− (Ir–S(O)G, Fig. 4B), and Ir–S(O)G was further oxidized to the sulfinato complex [(η5-Cpxbiph)Ir(phpy)(S(O)2G)]− (Ir–S(O)2G) after 24 h (Fig. 4C). These results were confirmed by ESI-MS analysis showing a singly-charged ion peak at m/z 943.2 (Fig. 4B), assignable to the glutathione sulfenato complex {(η5-Cpxbiph)Ir(phpy)(S(O)G) + 2H}+ (calcd m/z 943.2), and a singly charged ion peak centered at m/z 981.1 (Fig. 4C), assignable to the sulfinato complex {(η5-Cpxbiph)Ir(phpy)(S(O)2G) + H + Na}+ (calcd m/z 981.2).
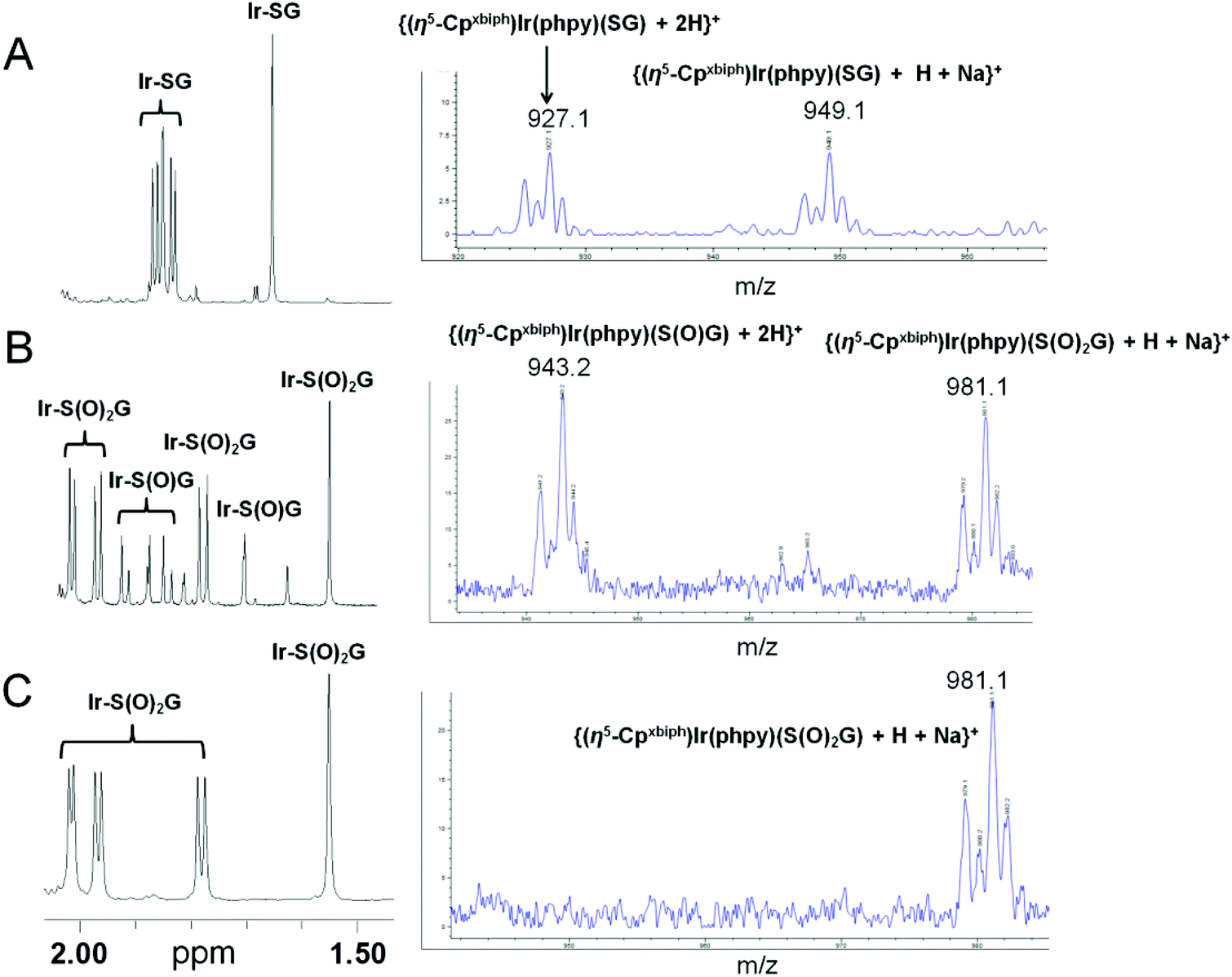 | ||
| Fig. 4 1H NMR (methyl region, left) and ESI-MS spectra (right) showing the oxidation of Ir–SG by H2O2 in 30% MeOD-d4–70% PBS D2O buffer (v/v, pH* = 7.4) at 310 K, with formation of the sulfenato complex [(η5-Cpxbiph)Ir(phpy)(S(O)G)]− (Ir–S(O)G) and sulfinato complex [(η5-Cpxbiph)Ir(phpy)(S(O)2G)]− (Ir–S(O)2G). (A) Formation of Ir–SG produced by reaction of Ir–py (1 mM) with GSH (3.5 mM); (B) 15 h after addition of H2O2 (10 mol equiv.) to the above equilibrium solution, Ir–S(O)G and Ir–S(O)2G were detected; (C) after 24 h, all Ir–S(O)G was further oxidized to Ir–S(O)2G. The sample for ESI-MS was prepared in 30% MeOH–70% PBS H2O buffer. | ||
In summary, we have shown that the iridium glutathione adduct Ir–SG formed by substitution of the chlorido or pyridine ligands in half-sandwich C,N-chelated IrIII biphenyltetramethylcyclopentadienyl by S-bound glutathione may be not a dead-end product if formed in cells from these anticancer agents. Complex Ir–SG can be readily oxidized to the sulfenato complex Ir–S(O)G and further to the sulfinate complex 1-S(O)2G by H2O2 produced via hydride transfer from coenzyme NADH to iridium and then to O2, Fig. 5.
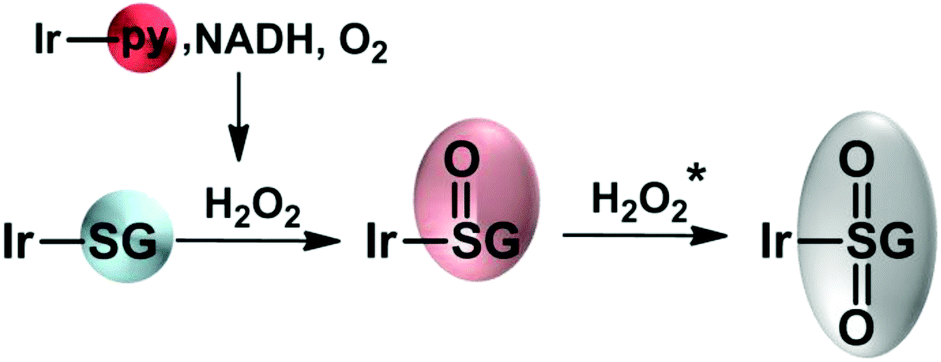 | ||
| Fig. 5 Possible reaction pathway for the oxidation of Ir–SG. *1-S(O2)G is formed when an excess of H2O2 is added. | ||
In future studies it will be interesting to investigate the possible role of these sulfenate and sulfinate complexes in the biological activity of iridium cyclopentadienyl complexes.
Acknowledgements
We thank the ERC (grant no. 247450), Science City (AWM/ERDF), and EPSRC for support, and members of EC COST Action CM1105 for stimulating discussions.Notes and references
- (a) L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed; (b) T. Boulikas, A. Pantos, E. Bellis and P. Christofis, Cancer Ther., 2007, 5, 537–583 Search PubMed.
- (a) P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed; (b) E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- (a) F. Arnesano and G. Natile, Pure Appl. Chem., 2008, 80, 2715–2725 CrossRef CAS; (b) W. Hu, Q. Luo, K. Wu, X. Li, F. Wang, Y. Chen, X. Ma, J. Wang, J. Liu, S. Xiong and P. J. Sadler, Chem. Commun., 2011, 47, 6006–6008 RSC; (c) H. Li, Y. Zhao, H. I. Phillips, Y. Qi, T. Y. Lin, P. J. Sadler and P. B. O'Connor, Anal. Chem., 2011, 83, 5369–5376 CrossRef CAS PubMed.
- (a) D. Dolphin, O. Avramović and R. Poulson, Glutathione: chemical, biochemical, and medical aspects, Wiley, New York, 1989 Search PubMed; (b) C. Hwang, A. J. Sinskey and H. F. Lodish, Science, 1992, 257, 1496–1502 CAS; (c) A. Corazza, I. Harvey and P. J. Sadler, Eur. J. Biochem., 1996, 236, 697–705 CAS; (d) K. S. Bibhesh, Asian J. Chem., 2005, 17, 1–32 Search PubMed; (e) K. Zhang, P. Mack and K. P. Wong, Int. J. Oncol., 1998, 12, 871–882 CAS.
- H. Sies, Free Radicals Biol. Med., 1999, 27, 916–921 CrossRef CAS.
- (a) M. Valko, H. Morris and M. T. D. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS; (b) I. Romero-Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS PubMed.
- F. Wang, J. Xu, K. Wu, S. K. Weidt, C. L. Mackay, P. R. R. Langridge-Smith and P. J. Sadler, Dalton Trans., 2013, 42, 3188–3195 RSC.
- W. Hu, Q. Luo, X. Ma, K. Wu, J. Liu, Y. Chen, S. Xiong, J. Wang, P. J. Sadler and F. Wang, Chem. – Eur. J., 2009, 15, 6586–6594 CrossRef CAS PubMed.
- (a) Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed; (b) S. Wirth, C. Rohbogner, M. Cieslak, J. Kazmierczak-Baranska, S. Donevski, B. Nawrot and I.-P. Lorenz, J. Biol. Inorg. Chem., 2010, 15, 429–440 CrossRef CAS PubMed; (c) A. Casini, F. Edafe, M. Erlandsson, L. Gonsalvi, A. Ciancetta, N. Re, A. Ienco, L. Messori, M. Peruzzini and P. J. Dyson, Dalton Trans., 2010, 39, 5556–5563 RSC; (d) M. Gras, B. Therrien, G. Süss-Fink, A. Casini, F. Edafe and P. J. Dyson, J. Organomet. Chem., 2010, 695, 1119–1125 CrossRef CAS PubMed; (e) J. Ruiz, V. Rodriguez, N. Cutillas, K. G. Samper, M. Capdevila, O. Palacios and A. Espinosa, Dalton Trans., 2012, 41, 12847–12856 RSC; (f) S. J. Lucas, R. M. Lord, R. L. Wilson, R. M. Phillips, V. Sridharan and P. C. McGowan, Dalton Trans., 2012, 41, 13800–13802 RSC; (g) R. Payne, P. Govender, B. Therrien, C. M. Clavel, P. J. Dyson and G. S. Smith, J. Organomet. Chem., 2013, 729, 20–27 CrossRef CAS PubMed; (h) S. Schäfer and W. S. Sheldrick, J. Organomet. Chem., 2007, 692, 1300–1309 CrossRef PubMed; (i) M. Ali Nazif, J.-A. Bangert, I. Ott, R. Gust, R. Stoll and W. S. Sheldrick, J. Inorg. Biochem., 2009, 103, 1405–1414 CrossRef CAS PubMed; (j) G. Gupta, A. Garci, B. S. Murray, P. J. Dyson, G. Fabre, P. Trouillas, F. Giannini, J. Furrer, G. Suss-Fink and B. Therrien, Dalton Trans., 2013, 42, 15457–15463 RSC; (k) K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069–12083 RSC; (l) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- Z. Liu, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Organometallics, 2011, 30, 4702–4710 CrossRef CAS.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- T. A. George and D. D. Watkins, Inorg. Chem., 1973, 12, 398–402 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qi00098f |
| ‡ Current address: Department of Chemistry, University of Basel, Spitalstrasse 51, 4056 Basel, Switzerland. |
| This journal is © the Partner Organisations 2014 |