
Synthesis of a naphthalenediimide-based cyclophane for controlling anion–arene interactions†
Yongjun
Li
*a,
Yingjie
Zhao
ab,
Runsheng
Jiang
ab,
Huibiao
Liu
a and
Yuliang
Li
*a
aBeijing National Laboratory for Molecular Science, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: liyj@iccas.ac.cn; ylli@iccas.ac.cn; Tel: +86-010-62587552
bGraduate University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 20th August 2014
Abstract
A cationic cyclophane based on a photoactive naphthalenediimide (NDI) moiety and cationic triazolium units has been prepared. This system was employed to control the interactions between anions and the NDI motif and tune the charge transfer properties of the NDI with different anions. High selectivity was observed for fluoride anions, which preferred to interact with the NDI through a SOMO–LUMO-based electronic transition to form NDI radical anions in solution. This cyclophane can crystallize to form a porous lattice which allowed for a gradual leaking of PF6− anions, with replacement by smaller-sized halides. Charge transfer complexes were formed upon replacing the non-nucleophilic hexafluorophosphate anions with halides in the molecular crystals.
Introduction
There is considerable interest in controlling the interactions between chromophores in supramolecular systems.1,2 Anions play important chemical and biological roles. There are three main types of anion–arene interaction which can be used to tune the photophysical properties of arenes.3 Cyclophanes are a large class of host molecules known to recognize a variety of guests ranging from inorganic and organic cations and anions to neutral molecules.4 Also in the recognition process all known modes of binding have been exploited, such as hydrogen bonding, donor–acceptor properties, cation–π interactions, and coordinate bonding.4,5 However, using a cyclophane to tune the anion–arene interactions and control the electron-transfer or charge-transfer process concerning an anion guest is rare.One way to accomplish such a task is to combine a proper photoactive moiety and known binding motifs into a single cyclophane.4 In this capacity, naphthalenediimide (NDI)6 was chosen because of its remarkable photochemical and electrochemical properties,7–9 and the electron deficient and aromatic nature of NDI is important for anion–π interactions3 to harness the charge-transfer properties. 1,2,3-Triazoles are new motifs that can participate in multiple noncovalent interactions.10–13 Halides were chosen as the anion species because halides as electrically charged donor units are known to easily form donor–acceptor complexes.14 In this paper, we first present the preparation of a cationic cyclophane based on a photoactive NDI moiety and cationic triazolium units by taking advantage of the ubiquitous Cu(I)-catalyzed “click chemistry”.15,16 Secondly, the anion-binding properties of this cationic macrocyclic host are employed to control the interactions between anions and the NDI motif and tune the properties of the NDI.
Results and discussion
Synthesis and characterization of the NDI cyclophane
The cationic macrocycle nabp-Me22+·2PF6− (Scheme 1) was designed to provide a structure wherein the two triazolium linkers connecting the NDI to biphenylene were used as the anion binding motifs as well as the rotators. The rotation of the triazolium groups through the cavity can influence the distance of the complexed anion from the NDI, further tuning the electron-transfer or charge-transfer processes between the anions and the NDI. nabp was methylated with MeI in DMF solution and ion-exchanged into its PF6− salt nabp-Me22+·2PF6−. nabp-Me22+·2PF6− showed good solubility in polar solvents such as acetonitrile, acetone and DMF. In a similar way, nabp-benzyl2+·2PF6− was synthesized to enhance the solubility of the macrocycle for titration studies. nab-Me22+·2PF6− was synthesized as a reference compound to investigate the role of the cyclophane. nabp, nabp-Me22+·2PF6− and nabp-benzyl2+·2PF6− were fully characterized using their 1H NMR, 13C NMR and MS spectra.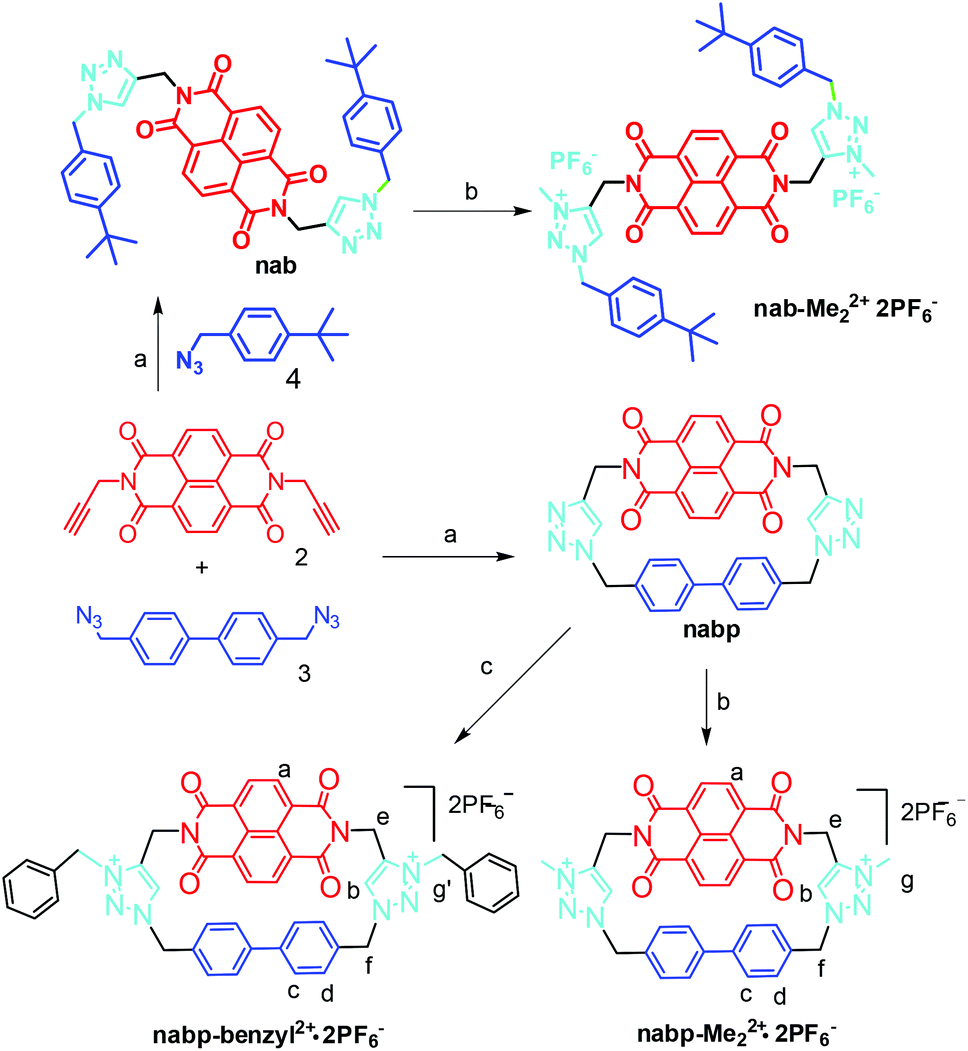 | ||
| Scheme 1 Reaction conditions: (a) Dropwise addition of 2 and 3 into CuI–DBU–PhMe–Ar over 10 h, then stirring for another 4 h, 70 °C, 50%. (b) MeI, CH2Cl2–DMF, 40 °C, 24 h; NH4PF6, acetone–H2O, 85%. (c) benzyl chloride, CH2Cl2–DMF, 40 °C, 48 h; NH4PF6, acetone–H2O, 73%. | ||
The NMR spectra of nabp at room temperature indicate that the biphenylene moiety rotates rapidly on the NMR time scale (Fig. S1†). However, upon decreasing the temperature from 298 to 228 K the dynamic processes are slowed, resulting in separation of the signals; the coalescence temperature is at about 298 K. At the same time, the triazole proton Hb and the biphenylene proton Hd (close to the triazole) shifted down-field with decreasing temperature, which is consistent with the shielding effect between the triazole and biphenylene groups when the movement of these groups is slowed.
X-ray diffraction analyses
The crystal and molecular structures of nabp-Me22+·2PF6− were determined using X-ray diffraction analyses.‡nabp-Me22+·2PF6− crystallizes in a monoclinic system with a P2(1)/c space group with four molecules in a unit cell (Table S1,†Fig. 1). The X-ray crystal structure confirmed that N-methylation preferentially occurred at N3 and N8, which is in agreement with the reactivity of the nitrogens in a triazole.17 The distance between the two triazole hydrogens is 7.816 Å. Single-crystal X-ray analyses revealed that the NDI and 4,4′-biphenylene moieties in nabp-Me22+·2PF6− are oriented face-to-face across the centre of the macrocycle (Fig. 1, bottom). The transannular (centroid–centroid) separation is 6.336 Å (C13–C26), which is slightly longer than that at the terminal nitrogen imide positions, which is 5.635 Å (C30–N1). In the unit cell, the triazolium plane is π–π stacked with the biphenylene group (Fig. 1 bottom, red).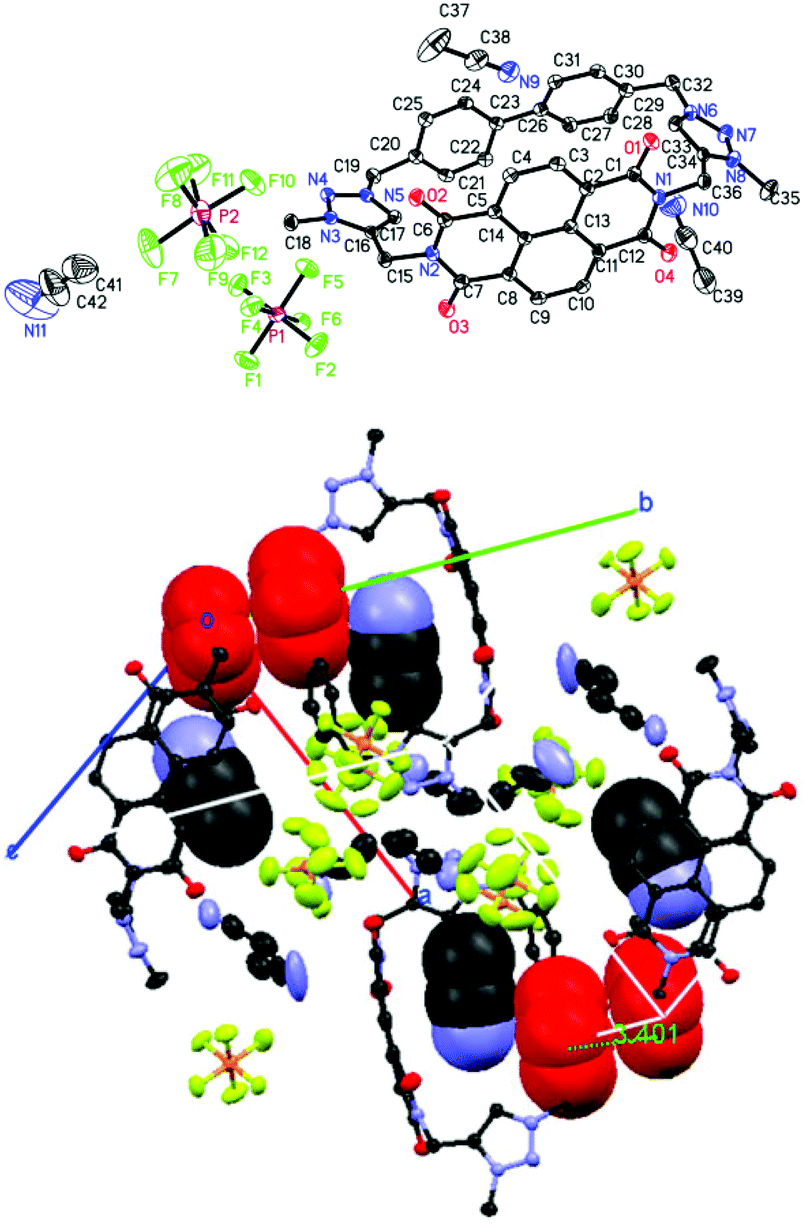 | ||
| Fig. 1 ORTEP diagram and atomic labeling of the cyclophane nabp-Me22+·2PF6− (thermal ellipsoids are shown at 50% probability) (top). Side view and top view of nabp-Me22+·2PF6− in the unit cell (bottom); CH3CN (space filling) reside in each cyclophane and the π–π stacked triazolium and biphenylene are indicated (red, space filling). Torsion angle at the biphenyl linkage in the macrocycle is 18–19°. | ||
The packing diagram shows that nabp-Me22+·2PF6− is a cationic three-dimensional structure with infinite chains composed of alternating nabp-Me22+ boxes and PF6− anions (Fig. 2). nabp-Me22+·2PF6− contains channels that run along the length of the a or c axes (Fig. 2). In the direction of the a axis, one type of channel was formed from the cavity of the cyclophane, in which CH3CN molecules are inserted, with distances between the nitrogen of the acetonitrile and the centroids of the biphenyl linkage and the imide ring system of 3.25 and 3.06 Å, respectively. A distance of 2.536 Å between the nitrogen of the acetonitrile and the triazolium C–H was also observed. Another type of channel was formed between neighbour cyclophanes, which was driven by the electrostatic interaction between the cationic cyclophane framework and PF6− anions. In the X-ray crystal structure of nabp-Me22+·2PF6−, there are ordered and heavily disordered PF6− anions and acetonitrile molecules; the disordered species oscillated in the channel, supporting only weak electrostatic interactions between the cationic cyclophane and charge-balancing PF6− anions. This weak electrostatic interaction and the porous nature of the lattice may allow for a gradual leaking of the PF6− anions with replacement by smaller-sized halides. These features are important for the anion exchange that will be discussed below.
 | ||
| Fig. 2 Channels formed by the nabp-Me22+·2PF6− framework, running along the length of the a-axis (carbon: gray; phosphate: brown; fluoride: yellow; oxygen: red; nitrogen: blue; hydrogen is omitted for clarity). | ||
Cyclic voltammetry studies
Cyclic voltammetry gave Eer values of −0.932 and −1.425 V versus Fc+/Fc for nabp, and −0.82 V, −1.26 V, −2.132 V and −2.242 V versus Fc+/Fc for nabp-Me22+·2PF6− (Fig. 3). The first electron reduction potential (E1) of the NDI in nabp-Me22+·2PF6− is 0.112 V larger than that in nabp. These results indicated that the electron accepting ability of the NDI is influenced greatly by the nearby triazolium moieties, which electrostatically stabilize the NDI anion. The methylation further lowers the low-LUMO (LL) of the NDI compounds, which will help the electron-transfer from the anions to the NDI to form SOMO–LUMO-based electronic transitions.18 | ||
| Fig. 3 (a) Cyclic voltammetry (CV) of nabp and nabp-Me22+·2PF6− in DMF with a ferrocene/ferrocenium couple (Fc/Fc+) as the internal standard (nabp and nabp-Me22+·2PF6−, 0.1 mM; TBAPF6 = 0.1 M). | ||
Cyclophane–halide anion interactions in solution
The fluoride anion induces the formation of a radical anion of NDI, which in turn displays various SOMO–LUMO electronic transitions. Upon the addition of fluoride, nabp-Me22+·2PF6− showed an instantaneous change from colorless to dark brown. UV-vis-NIR spectroscopy of nabp-Me22+·2PF6− shows peaks at 358 and 377 nm. In the presence of fluoride, a new set of intense and characteristic visible and near-infrared (NIR) absorption bands for the NDI radical anions appeared at 476, 608, 701 and 782 nm (Fig. 4a). These new peaks at 476–782 nm for nabp-Me22+·2PF6− with fluoride, and their relative intensity ratios, match perfectly with the signature peaks that are due to D0–Dn electron transfer in the radical anions of the NDI moieties.18 The other strong evidence for the NDI radical anions comes from the diagnostic EPR signal that is unique to the imide radical (g, 2.0056) of [nabp-Me22+]˙−, which is formed in the presence of fluoride (Fig. 4b).19 The fluoride ion was oxidized, as supported by the disappearance of the 19F NMR signal at 102 ppm upon the addition of one equivalent of nabp-Me2+·2PF6− (Fig. S13†).18b UV-vis-NIR titration using other halides (Cl−, Br− and I−) indicated that only F− produces the NDI radical anion species (Fig. S11†). nab-Me22+·2PF6− can also interact with F− and exhibits electron transfer behavior in the solution state to form an NDI radical anion, but the interaction is weaker than in the case of nabp-Me22+·2PF6−, as indicated by the fact that one equivalent of F− can interact with 80% of the nabp-Me22+·2PF6− molecules, while 30 equivalents of F− are needed to interact with 80% of the nab-Me22+·2PF6− molecules (Fig. S12†).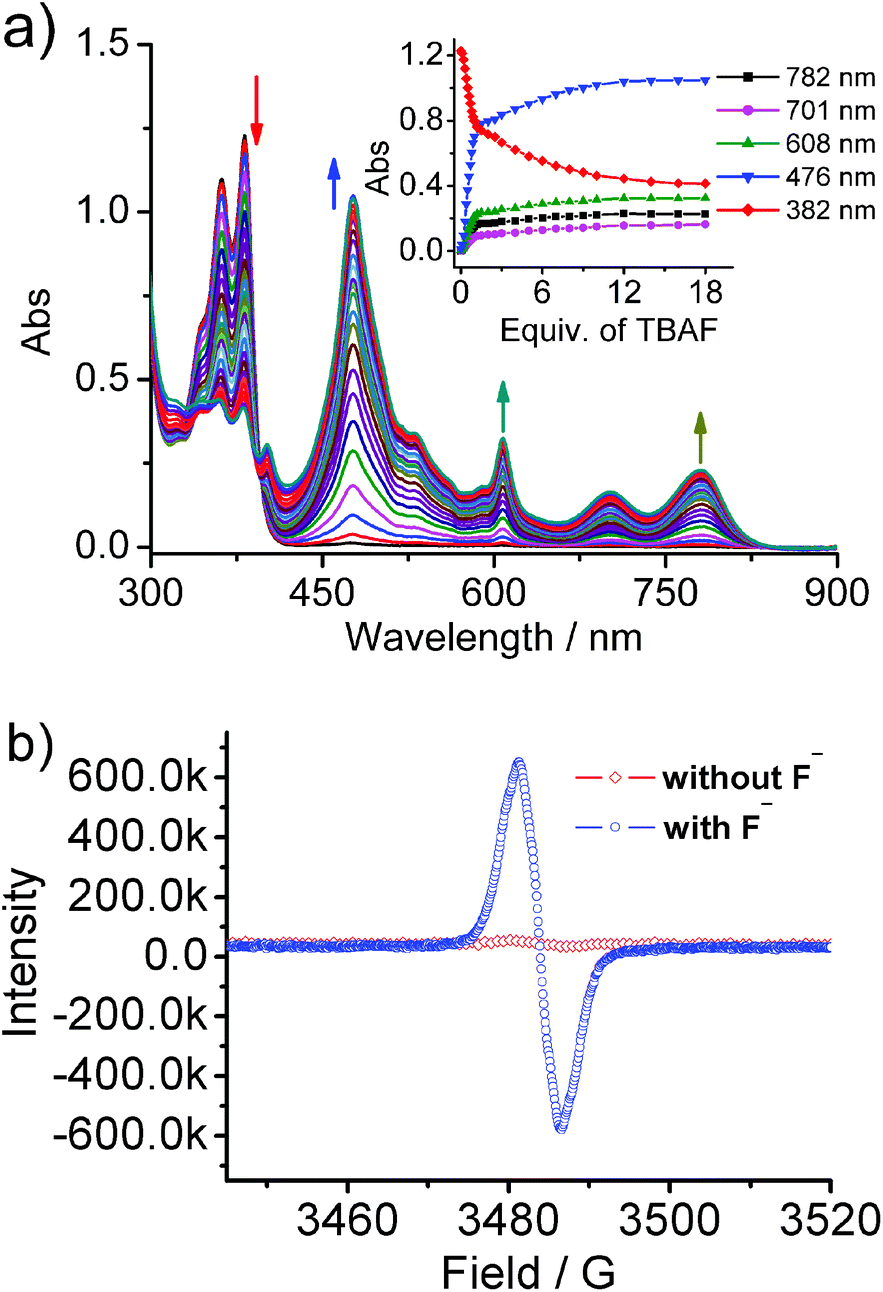 | ||
| Fig. 4 (a) UV-vis-NIR spectra showing the response of nabp-Me22+·2PF6− (5.0 × 10−5 M) upon incremental addition of F− (nabp-Me22+·2PF6− in DMF). Inset shows the response of the different optical channels as a function of F−. (b) ESR spectra of nabp-Me22+·2PF6− in the presence and absence of F−, in DMF (nabp-Me22+·2PF6−, 1.0 × 10−4 M). | ||
For the cyclophane system studied here, there are three main binding sites for anions, that is, the triazolium C–H for hydrogen bonding, the cationic triazolium ring for electrostatic interaction, and the electron-deficient NDI for anion–π interaction. The triazole-based C5–H hydrogen bonds are aided by the relatively large 5 Debye dipole,20,21 with its positive end directed almost in line with the C5–H bond. The anion binding properties of nabp-benzyl2+·2PF6− were investigated through titrations with tetrabutylammonium (TBA+) salts of Cl−, Br− and I− in CD3CN (Fig. S4–6†). The triazolium proton Hb and proton He of the methylene linked to the imide are shifted downfield upon addition of anions. The shift of the triazolium Hb could be attributed to the hydrogen bonding interaction contributed by the anions, while proton He interacts with the halides brought close by the triazolium. Furthermore, the remaining aromatic protons do not shift very much during the titration, suggesting that the triazolium serves a primary role in binding the halide anion. The shift of proton Hb decreased with increasing size of halide. For the methylene proton He and biphenylene proton Hd, similar shift trends were observed though to a smaller extent. On account of the flexibility in shape of this cyclophane (VT 1H NMR, Fig. S1†), there is an evident energy cost that is associated with the loss of conformational flexibility of the receptor,22 and the anion binding affinity of this cyclophane is limited. nabp-benzyl2+·2PF6− showed binding constants of 125 M−1 for Cl−, 63 M−1 for Br− and 45 M−1 for I− (from the chemical shift of the triazolium CH, Fig. S6†). nabp-Me22+·2PF6− will precipitate with the addition of halides in normal solvents, thus the binding affinities were investigated in DMSO (Fig. S7–9†). nabp-Me22+·2PF6− showed binding constants of 43.7 M−1 for Cl−, 16.7 M−1 for Br− and 8.5 M−1 for I−. These results indicated that the triazolium C–H⋯X− decreased with increasing anion size, and the triazolium C–H prefers to stretch out to bind with anions with the cooperation of He and the biphenylene proton Hd. 1H NMR titration of F− showed a broadening of the spectrum of the receptor (Fig. S2†) which could be explained by the formation of the NDI radical anions.
Cyclophane–halide anion interactions in the solid state
A charge transfer complex is expected to form upon replacing the non-nucleophilic hexafluorophosphate anion with halides in the presence of a cationic moiety incorporating a strongly electron accepting naphthalene diimide moiety.23 The ability of the triazolium C–H⋯X− to rotate into the annulus when combined with the larger sizes of Cl−, Br− and I− inhibited the observation of an evident charge transfer from the halide to the NDI in solution. By constraining the movements of the molecules, the anion-triazolium ion-pair can make contact with the NDI closely, and charge transfer phenomena occur. For example, by simply immersing the as-synthesized nabp-Me22+·2PF6− crystals in 0.1 M TBAF, TBACl, TBABr or TBAI solution at room temperature for 3 days, the colours of the crystals become different to each other: pale yellow for F−, yellow for Cl−, red-brown for Br− and dark brown for I− (Fig. 5A–E). FTIR and PXRD (Fig. S15 and 16†) indicated that the replacement of the larger PF6− anions with the smaller halide anions occurs through a diffusion process at the solid–liquid interface.24nab-Me22+·2PF6− did not show this behavior (Fig. S17†).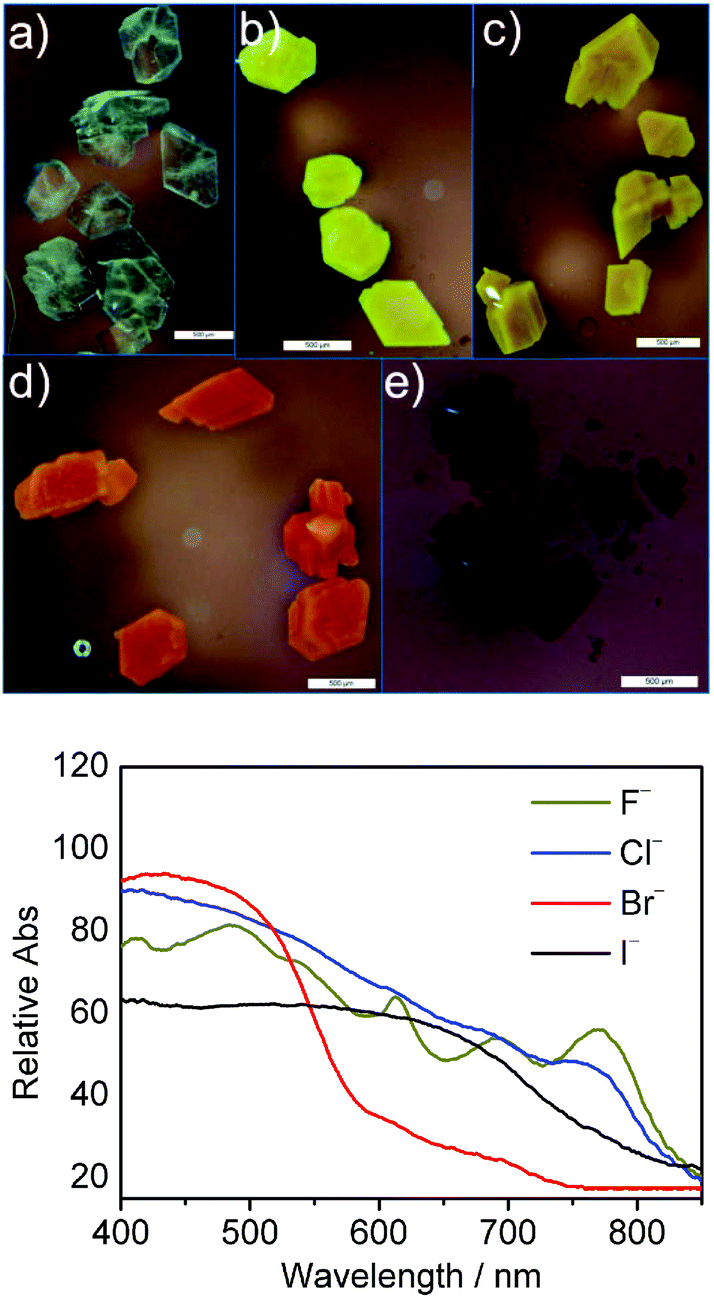 | ||
| Fig. 5 Optical micrographs of nabp-Me22+ crystals before (a) and after anion exchange with F− (b), Cl− (c), Br− (d) and I− (e) (scale bar: 500 μm). Reflection UV-vis spectra (bottom) of the complexes of nabp-Me22+ with F−, Cl−, Br− and I− (in KBr pellet). | ||
Reflection UV-vis spectra of the complexes of Br− and I− showed broad charge-transfer (CT) bands at 400–580 nm and 400–750 nm, respectively (Fig. 5 bottom). While for F− and Cl−, the color of the KBr pellet formed under high pressure is different from the anion-exchanged solid and the powder, the characteristic peaks for the radical anions of the NDI were observed. That is, under high pressure, F− and Cl− can form close contacts with the NDI moiety to give NDI radical anions; the formation of charge-transfer complexes is preferred after replacing the non-nucleophilic hexafluorophosphate anions in the molecular crystals with halides.
Conclusions
In summary, a cationic cyclophane based on a photoactive NDI moiety and cationic triazolium units has been prepared and was employed to control the interactions between anions and the NDI motif. The fluoride anion induces the formation of the radical anion of NDI, which in turn displays various SOMO–LUMO electronic transitions. The formation of charge-transfer complexes between NDI and Cl−, Br− or I− constrains the movement of the molecules in the solid state. Visible charge-transfer absorptions for the charge transfer complexes of the halides in the molecular crystals encourage their use in the design of functional molecular crystals and materials for the study of photoinduced electron transfer and energy conversion for application in the field of molecular electronics.Experimental section
General considerations
All reagents were obtained from commercial suppliers and used as received unless otherwise noted. Column chromatography was performed on silica gel (160–200 mesh), and thin-layer chromatography (TLC) was performed on precoated silica gel plates and observed under UV light. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker Avance DPS-400 and Bruker Avance DPS-600 spectrometers at room temperature (298 K). Chemical shifts were referenced to the residual solvent peaks. Matrix-assisted laser desorption/ionization reflectron time-of-flight (MALDI-TOF) mass spectrometry was performed on a Bruker Biflex III mass spectrometer. Electronic absorption spectra were measured using a JASCO V-579 spectrophotometer. The single crystal X-ray diffraction data were collected using a Rigaku Saturn X-ray diffractometer with graphite-monochromator Mo-Kα radiation (λ = 0.71073 Å) at 173 K. Intensities were corrected for absorption effects using the multi-scan technique SADABS (Siemens Area Detector Absorption Corrections). The structures were solved using direct methods and refined by a full-matrix least-squares technique based on F2 using the SHELXL 97 program (Sheldrick, 1997). Compounds 2 and 3 were synthesized in accordance with literature procedures.25,26Synthesis of compound nabp
DBU (4.0 mmol, 0.7 mL) was added to toluene (200 mL). The solution was degassed with argon for 30 minutes and heated to 70 °C while flushing with argon. At 70 °C, CuI (0.04 mmol, 6.6 mg) was added to the mixture. A solution of 2 (171 mg, 0.5 mmol) and 3 (132 mg, 0.5 mmol) in toluene (60 mL) was added to the solution slowly over 10 h and stirred for another 6 h under argon. The mixture was concentrated in vacuo. The product was purified via column chromatography (SiO2, CHCl3–methanol 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to afford nabp (151 mg, 50% yield) as a pale yellow solid. 1H NMR (400 MHz, d6-DMSO) δ = 8.62 (s, 4 H), 7.58 (s, 2 H), 7.17 (d, 4 H, J = 8.2 Hz), 7.12 (d, 4 H, J = 8.2 Hz), 5.53 (s, 4 H), 5.33 (s, 4 H). MS (MALDI-TOF): m/z: calcd for C34H23N8O4 [M + H]+: 607.2; found: 607.4. Elemental analysis calcd (%) for C34H22N8O4: C 67.32, H 3.66, N 18.47; found: C 67.26, H 3.71, N 18.39.
:3) to afford nabp (151 mg, 50% yield) as a pale yellow solid. 1H NMR (400 MHz, d6-DMSO) δ = 8.62 (s, 4 H), 7.58 (s, 2 H), 7.17 (d, 4 H, J = 8.2 Hz), 7.12 (d, 4 H, J = 8.2 Hz), 5.53 (s, 4 H), 5.33 (s, 4 H). MS (MALDI-TOF): m/z: calcd for C34H23N8O4 [M + H]+: 607.2; found: 607.4. Elemental analysis calcd (%) for C34H22N8O4: C 67.32, H 3.66, N 18.47; found: C 67.26, H 3.71, N 18.39.
Synthesis of compound nabp-Me22+·(PF6)2−
Compound nabp (121 mg, 0.2 mmol) in 2 mL DMF and MeI (2 mL) was heated at 40 °C overnight, and the mixture was poured into 50 mL CH2Cl2 to obtain a precipitate, which was washed with CH2Cl2. The solid was suspended in acetone and NH4PF6 (5 equivalents) was added; the mixture became a clear solution. After removal of the solvent, the solid residue was washed with water then dried in vacuo to obtain nabp-Me22+·(PF6)2− (165 mg, 85%). 1H NMR (400 MHz, CD3CN) δ = 8.65 (s, 4 H), 7.95 (s, 2 H), 7.35 (d, 4 H, J = 8.3 Hz), 7.25 (d, 4 H, J = 8.3 Hz), 5.67 (s, 4 H), 5.49 (s, 4 H), 4.40 (s, 6 H). 13C NMR (100 MHz, CD3CN, δ, ppm): 163.3, 141.9, 140.9, 133.1, 132.1, 130.6, 130.0, 128.0, 127.4, 127.3, 57.9, 39.4, 33.2. MS (MALDI-TOF): m/z: calcd for C36H28N8O4 [M − 2PF6]2+: 636.2; found: 636.4. Elemental analysis calcd (%) for C36H28F12N8O4P2: C 46.66, H 3.05, N 12.09; found: C 46.71, H 3.02, N 12.04.Synthesis of compound nabp-benzyl2+·(PF6)2−
Compound nabp-benzyl2+·(PF6)2− (87 mg, 73%) was obtained using benzyl bromide in the similar way to nabp-Me22+·(PF6)2−. 1H NMR (400 MHz, CD3CN) δ = 8.64 (s, 4 H), 7.83 (s, 2 H), 7.46 (m, 10 H), 7.39 (d, 4 H, J = 7.9 Hz), 7.29 (d, 4 H, J = 7.9 Hz), 5.97 (s, 4 H), 5.69 (s, 4 H), 5.48 (s, 4 H). 13C NMR (100 MHz, CD3CN) δ = 163.60, 141.92, 141.38, 132.97, 132.50, 131.31, 130.91, 130.77, 130.63, 130.43, 129.92, 128.48, 127.69, 58.62, 56.79, 33.85. MS (MALDI-TOF): m/z: calcd for C48H36N8O4 [M − 2PF6]2+: 788.3; found: 788.4. Elemental analysis calcd (%) for C48H36F12N8O4P2: C 53.44, H 3.36, N 10.39; found: C 53.37, H 3.31, N 10.33.Synthesis of compound nab
Compound 2 (171 mg, 0.5 mmol), 4 (190 mg, 1.0 mmol) and DBU (4.0 mmol, 0.7 mL) were added to toluene (500 mL). The solution was degassed with argon for 30 minutes and heated to 70 °C while flushing with argon. At 70 °C, CuI (0.04 mmol, 6.6 mg) was added to the mixture, which was stirred for 12 h under argon. The mixture was concentrated in vacuo. The product was purified via column chromatography (SiO2, CHCl3–methanol 100:3) to afford nab (270 mg, 75% yield) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.73 (s, 4 H), 7.59 (s, 2 H), 7.36 (d, J = 7.9 Hz, 4 H), 7.20 (d, J = 7.9 Hz, 4 H), 5.46 (d, J = 12.0 Hz, 8 H), 1.29 (s, 18 H). 13C NMR (100 MHz, CDCl3) δ 162.45, 151.91, 142.97, 131.38, 131.17, 127.98, 126.75, 126.59, 126.01, 123.19, 53.93, 35.56, 34.62, 31.23. MS (MALDI-TOF): m/z: calcd for C42H40N8O4 [M]+: 720.3; found: 720.4. Elemental analysis calcd (%) for C42H40N8O4: C, 69.98; H, 5.59; N, 15.55; found: C 69.94, H 5.53, N 15.48.
Synthesis of compound nab-Me22+·(PF6)2−
Compound nab (144 mg, 0.2 mmol) in 2 mL DMF and MeI (2 mL) was heated at 40 °C overnight. The mixture was concentrated in vacuo and washed with CH2Cl2. The solid was suspended in acetone and NH4PF6 (5 equivalents) was added. The mixture became a clear solution. After removal of the solvent, the solid residue was washed with water then dried in vacuo to obtain nab-Me22+·(PF6)2− (165 mg, 80%). 1H NMR (400 MHz, d6-DMSO) δ 9.14 (s, 2 H), 8.74 (s, 4 H), 7.42 (q, J = 8.3 Hz, 8 H), 5.80 (s, 4 H), 5.49 (s, 4 H), 4.41 (s, 6 H), 1.24 (s, 18 H). 13C NMR (100 MHz, DMSO) δ 163.15, 152.36, 139.60, 131.64, 131.14, 130.50, 129.23, 127.00, 126.76, 126.31, 56.17, 39.04, 34.86, 32.98, 31.40. MS (MALDI-TOF): m/z: calcd for C44H46N8O4 [M − 2PF6]2+: 750.4; found: 750.5. Elemental analysis calcd (%) for C44H46F12N8O4P2: C 50.77, H 4.45, N 10.77; found: C 50.80, H 4.41, N 10.70.Acknowledgements
This work was supported by the National Nature Science Foundation of China (21031006, 91227113 and 21322301), the National Basic Research 973 Program of China (2011CB932302, 2012CB932900), and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDA09020302).Notes and references
- (a) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed; (b) J. Kärnbratt, M. Hammarson, S. Li, H. L. Anderson, B. Albinsson and J. Andréasson, Angew. Chem., Int. Ed., 2010, 49, 1854–1857 CrossRef PubMed.
- (a) Y. S. Nam, T. Shin, H. Park, A. P. Magyar, K. Choi, G. Fantner, K. A. Nelson and A. M. Belcher, J. Am. Chem. Soc., 2010, 132, 1462–1463 CrossRef CAS PubMed; (b) J. M. Haider and Z. Pikramenou, Chem. Soc. Rev., 2005, 34, 120–132 RSC; (c) Y. J. Li, T. F. Liu, H. B. Liu, M.-Z. Tian and Y. L. Li, Acc. Chem. Res., 2014, 47, 1186–1198 CrossRef CAS PubMed.
- (a) B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428 RSC; (b) J. J. Lu and J. K. Kochi, Cryst. Growth Des., 2009, 9, 291–296 CrossRef CAS; (c) V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128(46), 14788–14789 CrossRef CAS PubMed; (d) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533–538 CrossRef CAS PubMed; (e) S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS PubMed; (f) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- F. Diederich, Cyclophanes; Monographs in Supramolecular Chemistry, The Royal Society of Chemistry, Cambridge, U.K., 1991 Search PubMed.
- (a) J. Schulz and F. Vogtle, Top. Curr. Chem., 1994, 172, 41–86 CrossRef CAS; (b) A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed.
- S. V. Bhosale, C. H. Jania and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- S. Bhosale, A. L. Sission, P. Talukdar, A. Furstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Roger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS PubMed.
- (a) F. B. L. Cougnon, N. A. Jenkins, G. Dan Pantoş and J. K. M. Sander, Angew. Chem., Int. Ed., 2011, 51(6), 1443–1447 CrossRef PubMed; (b) S. Gabutti, S. Schaffner, M. Neuburger, M. Fischer, G. Schafer and M. Mayor, Org. Biomol. Chem., 2009, 7, 3222–3229 RSC.
- G. P. Wiederrecht, B. A. Yoon and M. R. Wasielewski, Science, 1995, 270, 1794–1797 CAS.
- (a) M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC; (b) H. Y. Zheng, W. D. Zhou, J. Lv, X. D. Yin, Y. J. Li, H. B. Liu and Y. L. Li, Chem. – Eur. J., 2009, 15, 13253–13262 CrossRef CAS PubMed; (c) Y.-J. Li, L. Xu, W.-L. Yang, H.-B. Liu, S.-W. Lai, C.-M. Che and Y.-L. Li, Chem. – Eur. J., 2012, 18, 4782–4790 CrossRef CAS PubMed; (d) A. Kumar and P. S. Pandey, Org. Lett., 2008, 10, 165–168 CrossRef CAS PubMed; (e) B. Schulze, C. Friebe, M. D. Hager, W. Gunther, U. Kohn, B. O. Jahn, H. Gorls and U. S. Schubert, Org. Lett., 2010, 12, 2710–2713 CrossRef CAS PubMed.
- (a) Y. J. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed; (b) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; (c) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed.
- R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS PubMed.
- (a) Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650–4652 CrossRef CAS PubMed; (b) O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2007, 129, 48–58 CrossRef CAS PubMed; (c) G. Aragay, A. Frontera, V. Lloveras, J. Vidal-Gancedo and P. Ballester, J. Am. Chem. Soc., 2013, 135, 2620–2627 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- Y. Liu, X. Y. Zhang, L. M. Klivansky and G. Koshkakaryan, Chem. Commun., 2007, 4773–4775 RSC.
- (a) M. R. Ajayakumar and P. Mukhopadhyay, Org. Lett., 2010, 12, 2646–2649 CrossRef CAS PubMed; (b) S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS PubMed.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- H. Schneider, K. M. Vogelhuber, F. Schinle and J. M. Weber, J. Am. Chem. Soc., 2007, 129, 13022–13026 CrossRef CAS PubMed.
- M. H. Palmer, R. H. Findlay and A. J. Gaskell, J. Chem. Soc., Perkin Trans. 2, 1974, 420–428 RSC.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 CrossRef PubMed.
- S. V. Lindeman, J. Hecht and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 11597–11606 CrossRef CAS PubMed.
- (a) H. G. Fei, D. L. Rogow and S. R. Oliver, J. Am. Chem. Soc., 2010, 132, 7202–7209 CrossRef CAS PubMed; (b) A. N. Khlobystov, N. R. Champness, C. J. Roberts, S. J. B. Tendler, C. Thompson and M. Schroder, CrystEngComm, 2002, 4, 426–431 RSC.
- J. R. Thomas, X. J. Liu and P. J. Hergenrother, J. Am. Chem. Soc., 2005, 127, 12434–12435 CrossRef CAS PubMed.
- G. Koshkakaryan, L. M. Klivansky, D. Cao, M. Snauko, S. J. Teat, J. O. Struppe and Y. Liu, J. Am. Chem. Soc., 2009, 131, 2078–2079 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Compound characterization data, NMR spectra, NMR titration isotherms, FTIR and PXRD spectra, and X-ray crystallographic data. CCDC 1013693. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00095a |
| ‡ Crystallographic data for nabp-Me22+·2PF6−: C42H37F12N11O4P2, Mr = 1049.77, monoclinic, space group = P2(1)/c, a = 13.697(2), b = 23.188(3), c = 14.177(2) Å, α = 90°, β = 95.173(2)°, γ = 90°, T = 173 K, Z = 4, μ = 0.204 mm−1, RF(RwF) = 0.0804 (0.1939) for 30215 observed independent reflections. |
| This journal is © the Partner Organisations 2014 |