A family of [Ni8] cages templated by μ6-peroxide from dioxygen activation†
Alexandros
Perivolaris
a,
Constantinos C.
Stoumpos
a,
Jolanta
Karpinska
b,
Alan G.
Ryder
b,
Jamie M.
Frost
c,
Kevin
Mason
c,
Alessandro
Prescimone
c,
Alexandra M. Z.
Slawin
d,
Vadim G.
Kessler
e,
Jennifer S.
Mathieson
f,
Leroy
Cronin
f,
Euan K.
Brechin
*c and
Giannis S.
Papaefstathiou
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 157 71 Zografou, Greece. E-mail: gspapaef@chem.uoa.gr; Fax: +30 2107274782; Tel: +30 2107274840
bNanoscale Biophotonics Laboratory, School of Chemistry, National University of Ireland, Galway, Ireland
cEaStCHEM School of Chemistry, The University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: ebrechin@staffmail.ed.ac.uk; Fax: +44 (0)11 2754598; Tel: +44 (0)131 650 7545
dSchool of Chemistry, The University of St. Andrews, Purdie Building, St. Andrews, Fife KY16 9ST, UK
eDepartment of Chemistry, Swedish University of Agricultural Sciences, Box 7015, 750 07 Uppsala, Sweden
fWestCHEM, School of Chemistry, The University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK
First published on 13th May 2014
Abstract
A family of exceptionally thermally stable [Ni8] cages is reported, each being templated by a rare η3:η3:μ6-O22− species produced by dioxygen activation, where the reducing agent for the O2 reduction appears to be the ligand used in the reaction mixtures, which was found within the nickel cages in its oxidized form.
Introduction
Dioxygen activation has provided substantial impetus for important developments in several different fields. These include biomimetic and bioinorganic chemistry which aim to reveal the structures of the reactive intermediates at the active sites of metalloenzymes and give insights into the mechanistic details of dioxygen activation and oxygenation reactions,1–6 and catalysis since the metal–dioxygen intermediates have been proposed as active oxidants in C–H bond activation reactions.3It has been proposed that dioxygen activation first involves bonding of dioxygen at a reduced metal center to form metal-superoxo or metal-peroxo intermediates, followed by O⋯O bond cleavage leading to the formation of high-valent, metal-oxo species, which are responsible for the oxidation reaction of the substrates within the metalloenzyme.1–6 It is therefore believed that the presence of a redox-active metal center is a fundamental prerequisite for activating and reducing dioxygen.
Indeed, over the years, several high-valent metal-peroxo complexes, obtained by the action of dioxygen on a reduced metal center, have been structurally characterized by single-crystal X-ray crystallography.4 Examples include Ti(IV)-peroxo,4a V(V)-peroxo,4b Cr(IV)-peroxo,4c Mn(III)-peroxo,4d Mn(IV)-peroxo,4e Fe(III)-peroxo,4f Co(III)-peroxo4g–k and Cu(II)-peroxo4l,m complexes that were isolated by the action of dioxygen on Ti2(III), V(III), Cr(II), Mn(II), Mn(I), Fe(II), Co(II) and Cu(I) precursors, respectively.
Restricting further discussion to nickel–dioxygen species, there is only one Ni(II)-peroxo5a and one Ni(II)-superoxo5b compound, obtained by the oxygenation of a Ni0 and a NiI precursor, respectively, which have been structurally characterized. There are also a few reports of spectroscopically characterized nickel-peroxo and nickel-superoxo complexes obtained by the action of dioxygen on low-valent Ni0 and NiI precursors,2e,3a,6 while there are no structurally characterized Ni(II)-peroxo species obtained by the action of dioxygen on Ni(II) centers.
Since Ni(II) is inert toward O2, the most common approach to obtain Ni(II)-peroxo or Ni(II)-superoxo complexes is the use of H2O2.5b Indeed, there are a handful of structurally characterized Ni(II)-peroxo7a,b and Ni(II)-superoxo7c,d species obtained by the reaction of H2O2 with Ni(II) or Ni(III) precursors, respectively. It is worth noting that one Ni(II)-peroxo complex8 has been obtained by the chemical reduction of a Ni(II)-superoxo precursor,5b with the latter being originally synthesized by the reaction of a Ni(I) precursor with O2.
We herein report a family of [NiII8] cages templated by a rare η3:η3:μ6-O22− species produced by dioxygen reduction. Although we cannot ignore the presence of the Ni(II) ions, which might indeed be involved in the dioxygen activation, the reducing agent for the O2 reduction appears to be the ligand used in the reaction mixtures, which was found within the nickel cages in its oxidized form. Specifically, three octanuclear Ni(II) cages, namely [Ni8(O2)(abmo)6(MeCO2)2(MeO)6(MeOH)4]·4H2O (1·4H2O), [Ni8(O2)(abmo)6(PhCO2)2(MeO)6(MeOH)4]·2MeOH (2·2MeOH) and [Ni8(O2)(abmo)6(4ClPhCO2)2(MeO)6(MeOH)2(H2O)2] (3) (abmoH = α-benzilmonoxime, Scheme 1), were isolated from the reaction of nickel(II) carboxylates with α-benzoin oxime (aboH2, Scheme 1) in MeOH under aerobic conditions. Interestingly, abmoH is the oxidized form of aboH2. Direct evidence that the peroxide originates from the dioxygen reduction was provided by 18O2 labelling experiments in a deoxygenated reaction mixture of 2. To the best of our knowledge, complexes 1–3 represent the first examples of structurally characterized Ni(II)-peroxo complexes obtained by the action of dioxygen on Ni(II) precursors.
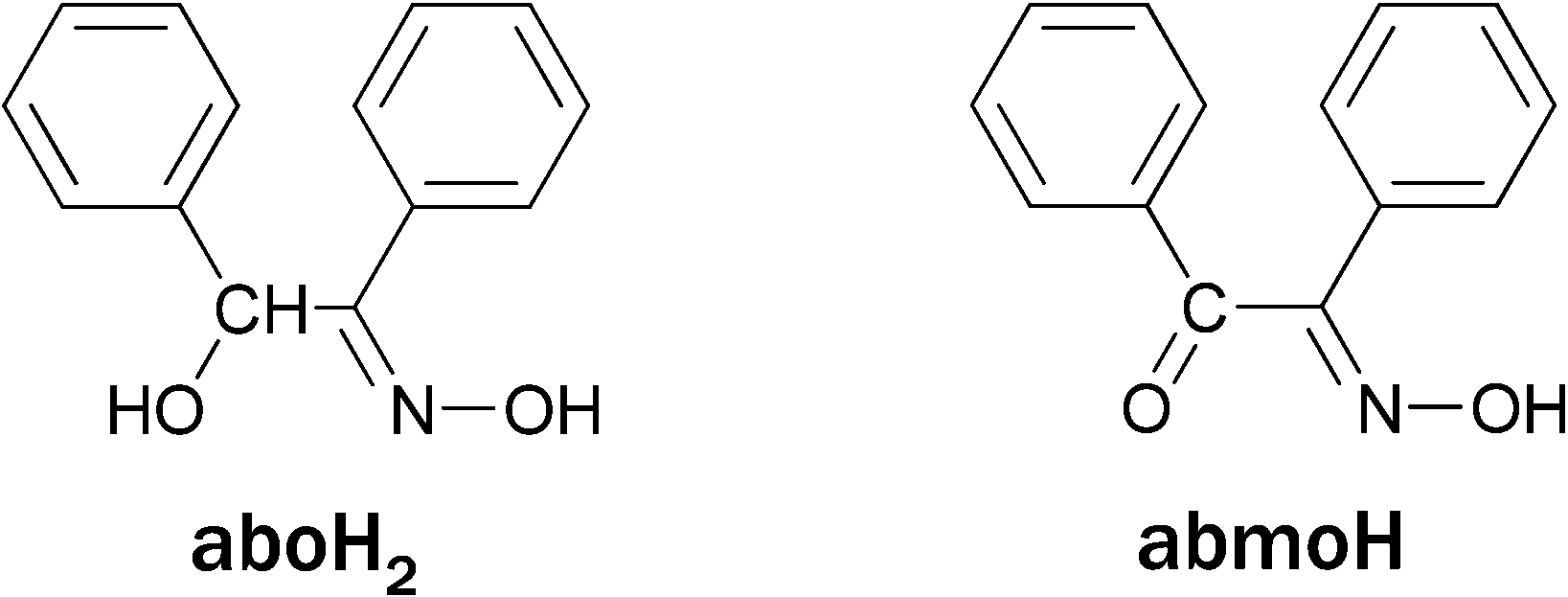 | ||
| Scheme 1 The structures of α-benzoin oxime (aboH2) and α-benzilmonoxime (abmoH). | ||
Such a dioxygen reduction where both the peroxide, O22− (i.e. the reduced species), and the oxidized species (i.e. the abmo−) are isolated and structurally characterized in the final product, and in which the metal ion appears not to change its oxidation state, has never been observed. This means that the electrons needed to reduce the dioxygen to O22− are provided by the organic ligand (i.e. not the metal ion). The significance of these findings is the provision of evidence that the dioxygen activation and its subsequent reduction may also depend on the organic surroundings of the metal-containing active site of metalloenzymes.1,2s–u We note that there are reports of structurally characterized metal-peroxo complexes obtained by dioxygen reduction where the metal center does not change the oxidation state, but in all cases the reducing agent was not identified.9 The same [Ni8] cages can be isolated from the reaction of nickel(II) carboxylates with abmoH, but only when H2O2 is added into the reaction mixtures. One side product, namely [Ni5(abo)2(aboH)6]·4MeOH·CH2Cl2 (4·4MeOH·CH2Cl2), was also isolated and characterized by X-ray crystallography. To the best of our knowledge, cages 1–3 represent the first polynuclear metal complexes which can be synthesized from different organic ligands (i.e. aboH2 and abmoH).
Experimental section
Materials and methods
Ni(MeCO2)2·4H2O, aboH2 (α-benzoin oxime) H2O2, Et3N, KBr, 18O2 (90%, 18O-enriched) and H218O2 (2–3% solution, 90% 18O-enriched) and all solvents were purchased from commercial sources and used as received. Ni(PhCO2)2·3H2O,10 Ni(4ClPhCO2)210 (4ClPhCO2H = 4-chloro-benzoic acid) and abmoH11 (α-benzilmonoxime) were prepared according to literature procedures. IR spectra were recorded as KBr pellets in the 4000–400 cm−1 range on a Shimadzu FT/IR IRAffinity-1 spectrometer. TGA data were collected with a Mettler Toledo TGA/DCS1 instrument in 40 μl Al pans under a N2 flow of 50 ml min−1. Small sample portions (∼1–2 mg) were used to avoid damage to the instrument due to the explosive nature of the peroxide complexes. Variable-temperature, solid-state direct current (dc) magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.Raman spectroscopy
Raman spectra were collected for all samples on two different Raman spectrometers, both with 785 nm excitation. The first system was a Raman WorkStation™ (Kaiser Optical Systems Inc., Ann Arbor, MI) over a 200–3000 cm−1 spectral range, with a resolution of ∼3–4 cm−1. This system was equipped with a −40 °C cooled CCD detector. The second system was a RamanStation spectrometer (AVALON Instruments Ltd, Belfast) equipped with a TE cooled (−90 °C) back thinned CCD detector. A laser power of ∼70 mW (at the sample) with an exposure time of 2 × 10 seconds was used and spectra were collected from 250 to 3311 cm−1 (at a resolution of 4 cm−1). For all data, exposure times (between 10 and 45 seconds) and accumulation numbers were varied in order to attain the optimal signal to noise (S/N) ratio in the Raman spectra, so that any shifts in the peroxo bonds could be accurately measured. The spectra were not corrected for instrument response.Mass spectrometry
Analysis was carried out using a Bruker Maxis Impact instrument with an electrospray (ESI-MS) ionisation source. The calibration solution used was an Agilent ESI-L low concentration tuning mix solution, Part No. G1969-85000, enabling calibration between approximately 50 m/z and 2000 m/z. Samples were dissolved in CH2Cl2/CH3OH at concentrations of 10−4 mol L−1 and introduced into the MS at a dry gas temperature of 180 °C. The ion polarity for all MS scans recorded was positive, with the voltage of the capillary tip set at 4000 V, end plate offset at −500 V, funnel 1 RF at 400 Vpp and funnel 2 RF at 400 Vpp, hexapole RF at 200 Vpp, ion energy at 5 eV, collision energy at 5 eV, collision cell RF at 1500 Vpp, transfer time at 120.0 μs and the pre-pulse storage time at 10.0 μs. Each spectrum was collected for 2 min.Synthesis
Results and discussion
Syntheses
The reaction of Ni(MeCO2)2·4H2O with aboH2 in MeOH and in the presence of triethylamine (Et3N) at room temperature results in a yellow-orange powder and a dark green-brown solution. Recrystallization of the yellow-orange powder revealed the pentanuclear complex 4, while slow evaporation of the dark green-brown solution afforded X-ray quality dark green crystals of 1. Similar reactions involving Ni(PhCO2)2·3H2O or Ni(4ClPhCO2)2 resulted in the same yellow-orange powder and X-ray quality dark green crystals of 2 and 3, respectively. The identity of the products is independent of the metal to ligand ratio. Performing the same reactions at elevated temperatures (i.e. above 50 °C) substantially reduces the yield of complex 4 and increases the yields of complexes 1–3. With the structures of complexes 1–3 known, we repeated the same reactions in the absence of O2: in this case only complex 4 forms and the solution does not turn dark green-brown, but retains its original green color. In order to provide direct evidence that the μ6-O22− originates from the reduction of dioxygen we synthesized the 18O-labelled complex 2 (2A) by the action of 18O2 in a de-oxygenated reaction mixture of 2. The 18O-labelled 2A was obtained and characterized by Raman spectroscopy. However, none of the bands in the recorded spectra were clearly identified as being sensitive to isotopic substitution of the peroxide. We therefore also performed ESI-MS on both unlabelled 2 and 18O-labelled 2A, where we observed a 4 unit shift correlating to the labelling of both O-atoms of the peroxo ligand. Having established the presence of the abmo− ligands within the cages of 1–3, we attempted to isolate these complexes starting from abmoH instead of aboH2. The solution does not turn dark green-brown until H2O2 is added to the reaction mixture in each case. As in the Ni(RCO2)2–aboH2–Et3N reaction system, the reactions of Ni(RCO2)2 with abmoH and H2O2 in MeOH in the presence of Et3N result in dark green-brown solutions from which complexes 1–3 were isolated and their structures were re-confirmed by single-crystal X-ray crystallography. The 18O-labelled 2A was also synthesized by adding H218O2 to a reaction mixture of Ni(PhCO2)2·3H2O–abmoH–Et3N.Description of structures
Although complexes 1–312 are not isostructural, their molecular structures are very similar. A labelled picture of the representative complex 1, whose structure will be discussed in detail, is shown in Fig. 1 (see ESI† for figures of complexes 2 and 3).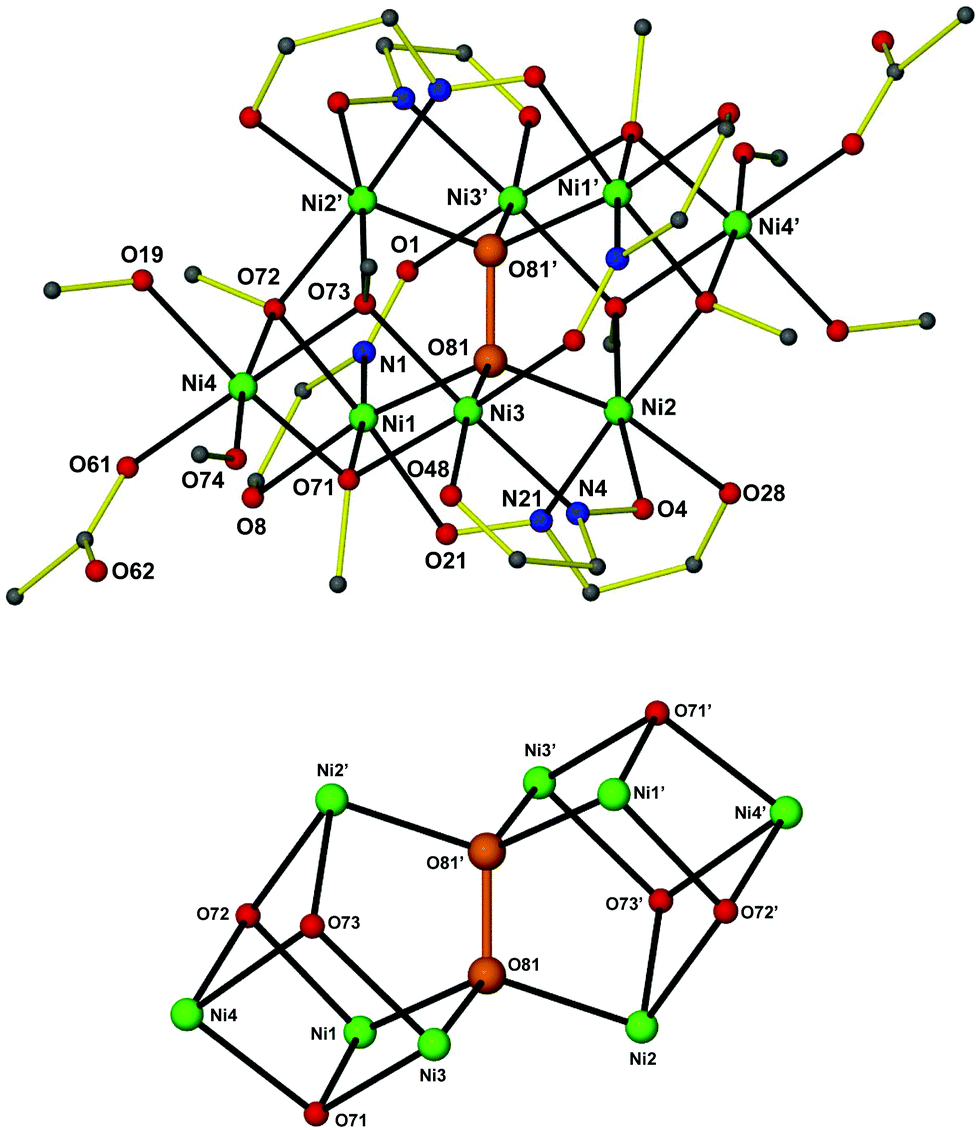 | ||
| Fig. 1 The structure of [Ni8(O2)(abmo)6(MeCO2)2(MeO)6(MeOH)4] 1 (top). All hydrogen atoms and most carbon atoms have been omitted for clarity. A view of the metal–oxygen core (bottom). Only the metal ions, the peroxide and the methoxide oxygen atoms are shown. Colour code: Ni green, C grey, N blue, O red. The η3:η3:μ6-O22− has been highlighted in orange. Symmetry code: (′) −x, 1 − y, −z. | ||
Complex 112 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The [Ni8] cage is situated at a center of inversion and consists of a Ni6(O2) core with staggered symmetry (D3d). The six NiII ions are situated at the apices of a distorted octahedron, with the Ni⋯Ni separations ranging from 3.362 to 4.856 Å. The octahedron is doubly face-capped by the remaining two NiII ions (Ni4 and Ni4′). All NiII ions are in distorted octahedral geometries. Six abmo− ligands bridge the central Ni6 core in a η1:η1:η1:μ-fashion, with each abmo− ligand chelating to one NiII ion through the carbonyl O atom and the oximato N atom and bridging to another through the deprotonated oximato O atom. Six μ3-MeO− ligands (three unique: O71, O72 and O73) bridge the peripheral, face-capping NiII ions to the central core. The octahedral environment around these peripheral NiII ions is completed by two MeOH molecules and a terminal MeCO2− ligand. Alternatively, the metal core of the octanuclear cage may be described as being composed of two “corner” sharing cubanes, with the shared “corner” being the μ6-O22− moiety (Fig. 1). The presence of abmo− corroborates the absence of the hydrogen atom at the carbonyl carbon atom and the short C⋯O distance (i.e. 1.243(7), 1.246(7) and 1.253(6) Å for each of the three abmo− ligands) and the presence of a C
. The [Ni8] cage is situated at a center of inversion and consists of a Ni6(O2) core with staggered symmetry (D3d). The six NiII ions are situated at the apices of a distorted octahedron, with the Ni⋯Ni separations ranging from 3.362 to 4.856 Å. The octahedron is doubly face-capped by the remaining two NiII ions (Ni4 and Ni4′). All NiII ions are in distorted octahedral geometries. Six abmo− ligands bridge the central Ni6 core in a η1:η1:η1:μ-fashion, with each abmo− ligand chelating to one NiII ion through the carbonyl O atom and the oximato N atom and bridging to another through the deprotonated oximato O atom. Six μ3-MeO− ligands (three unique: O71, O72 and O73) bridge the peripheral, face-capping NiII ions to the central core. The octahedral environment around these peripheral NiII ions is completed by two MeOH molecules and a terminal MeCO2− ligand. Alternatively, the metal core of the octanuclear cage may be described as being composed of two “corner” sharing cubanes, with the shared “corner” being the μ6-O22− moiety (Fig. 1). The presence of abmo− corroborates the absence of the hydrogen atom at the carbonyl carbon atom and the short C⋯O distance (i.e. 1.243(7), 1.246(7) and 1.253(6) Å for each of the three abmo− ligands) and the presence of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The η3:η3:μ6-O22− is fully surrounded by (six) NiII ions at the very center of the cage, and clearly templates the formation of the octanuclear cluster. It would appear highly unlikely that the octanuclear metal cage would form first, before O22− encapsulation. The O–O bond length in the peroxide is 1.487(4) Å [1.508 Å in 212 and 1.491 Å in 312] and is well within the limits of other reported peroxide bond lengths.1–9 The Ni–Operoxide bond lengths are 2.055(3) Å, 2.031(3) Å and 2.015(3) Å for Ni1–O81, Ni2–O81 and Ni3–O81, respectively, with the Ni–Operoxide–Ni angles being 114.61(13)°, 97.58(12)° and 113.09(13)° for the Ni1–O81–Ni2, Ni1–O81–Ni3 and Ni2–O81–Ni3 angles, respectively. The Ni–O81–O81′ angles are 111.47(19)°, 108.76(18)° and 111.03(19)° for Ni1, Ni2 and Ni3, respectively.
O bond. The η3:η3:μ6-O22− is fully surrounded by (six) NiII ions at the very center of the cage, and clearly templates the formation of the octanuclear cluster. It would appear highly unlikely that the octanuclear metal cage would form first, before O22− encapsulation. The O–O bond length in the peroxide is 1.487(4) Å [1.508 Å in 212 and 1.491 Å in 312] and is well within the limits of other reported peroxide bond lengths.1–9 The Ni–Operoxide bond lengths are 2.055(3) Å, 2.031(3) Å and 2.015(3) Å for Ni1–O81, Ni2–O81 and Ni3–O81, respectively, with the Ni–Operoxide–Ni angles being 114.61(13)°, 97.58(12)° and 113.09(13)° for the Ni1–O81–Ni2, Ni1–O81–Ni3 and Ni2–O81–Ni3 angles, respectively. The Ni–O81–O81′ angles are 111.47(19)°, 108.76(18)° and 111.03(19)° for Ni1, Ni2 and Ni3, respectively.
Complexes 1–3 join a handful of structurally characterized abmoH complexes13 and a very limited number of structurally characterized NiII-peroxo complexes.5a,7a,b,8 Indeed the related complex [Ni8(O2)(L)12](NO3)2 (5; LH = N-substituted 3-hydroxy-2-pyridinone) is the only other reported example containing the η3:η3:μ6-O22− ion.7a The formation of the latter cluster was achieved only when H2O2 was added into the reaction mixture. The central Ni6(O2) core in 5 is similar to that seen for 1–3, but with the two capping NiII ions residing in the faces of the octahedron lying in the same plane as the O22− ion, rather than in the perpendicular plane for 1–3. Other dissimilarities are the square pyramidal coordination geometry around the central NiII atoms and the absence of the MeO− bridges.
Complex 412 (Fig. 2) crystallizes in the triclinic space group P. The [Ni5] cluster is situated at an inversion center and consists of five coplanar NiII atoms, two abo2− and six aboH− ligands. The central NiII atom (Ni1) is in a square planar geometry, being chelated by the hydroxyl oxygen atoms and oximato nitrogen atoms from two η2:η1:η1:μ3 abo− ligands in a trans conformation. The hydroxyl O atom and the oximato O atom from both sides of the central Ni1 bridge Ni2 and its symmetry equivalent ion. Ni2 is in a distorted octahedral environment, being surrounded by a chelating aboH− ligand and the hydroxyl oxygen atoms from a cis-[Ni(aboH)2] subunit. The latter consists of a square planar NiII ion (Ni3) being chelated by two η1:η2:μ aboH− ligands through the hydroxyl oxygen atoms and the oximic nitrogen atoms in a cis configuration. Complex 4 is one of just a few structurally characterized aboH2 complexes.14
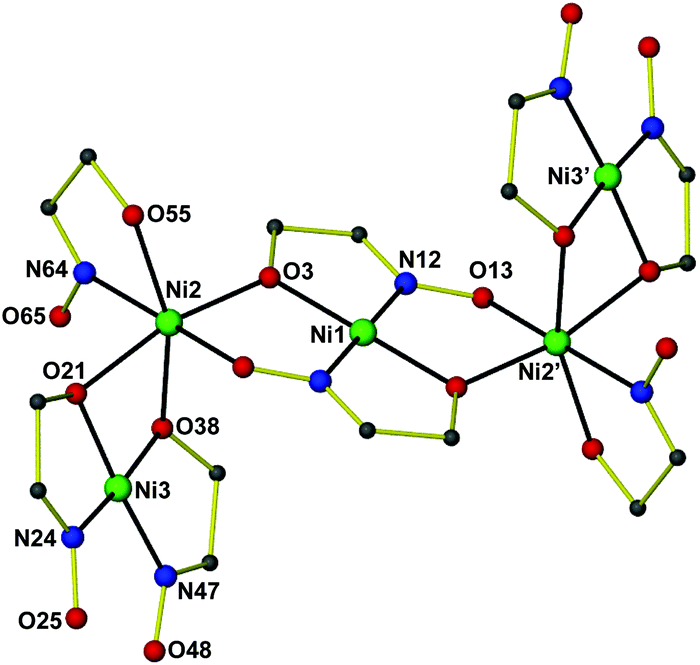 | ||
| Fig. 2 The structure of [Ni5(abo)2(aboH)6] 4. All hydrogen atoms and most carbon atoms have been omitted for clarity. Color code: Ni green, C grey, N blue, O red. Symmetry code: (′) 1 − x, −y, 1 − z. | ||
Raman spectroscopy
Raman spectra, for the comparison of the 16O (2·2MeOH) and 18O (2A·2MeOH) complexes, were collected for all samples on two different Raman spectrometers, both with 785 nm excitation. Although we used two different Raman spectrometers and varied both the exposure times and accumulation numbers in order to maximize the signal to noise (S/N) ratio in the Raman spectra, we did not observe any shifts in the respective spectra that could be ascribed conclusively to the peroxide stretching mode (Fig. S3 and S4 in the ESI†). Moreover, no Raman band was found to be sensitive to isotopic substitution with 18O in the known [Ni8(O2)(L)12](NO3)2 cluster.7a It should be noted that the Raman spectra were relatively noisy and if a weak peroxide band was present it was most likely buried in the noise signal.Mass spectrometry
Both the unlabelled (2) and 18O-labelled (2A) complexes were observed as both 1+ and 2+ species where one or both benzoate ligands were lost. A shift of 4 units was observed in the spectrum of 2A compared to the spectrum of 2, with this shift correlating to the labelling of both O atoms of the peroxo ligand (see Fig. 3 and Tables 1 and 2 for mass spectra and assignments).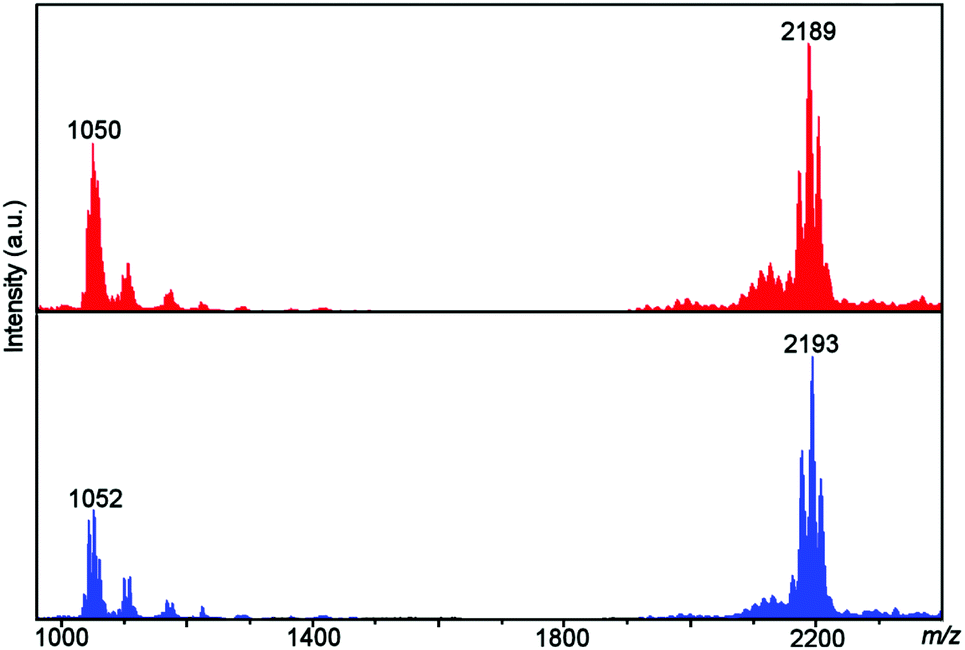 | ||
| Fig. 3 ESI-MS of 2 (top, red) and 2A (bottom, blue) showing the observation of the unlabelled and 18O labelled [Ni8] complexes. | ||
| m/z | Formula |
|---|---|
| 1034 | [Ni8(O2)(abmo)6(MeO)6(H2O)2]2+ |
| 1041 | [Ni8(O2)(abmo)6(MeO)6(H2O)(MeOH)]2+ |
| 1050 | [Ni8(O2)(abmo)6(MeO)6(H2O)2(MeOH)]2+ |
| 1059 | [Ni8(O2)(abmo)6(MeO)6(H2O)3(MeOH)]2+ |
| 2171 | [Ni8(O2)(abmo)6(PhCO2)(MeO)6(H2O)]+ |
| 2189 | [Ni8(O2)(abmo)6(PhCO2)(MeO)6(H2O)2]+ |
| 2203 | [Ni8(O2)(abmo)6(PhCO2)(MeO)6(H2O)(MeOH)]+ |
| m/z | Formula |
|---|---|
| 1043 | [Ni8(18O2)(abmo)6(MeO)6(H2O)(MeOH)]2+ |
| 1052 | [Ni8(18O2)(abmo)6(MeO)6(H2O)2(MeOH)]2+ |
| 1061 | [Ni8(18O2)(abmo)6(MeO)6(H2O)3(MeOH)]2+ |
| 2175 | [Ni8(18O2)(abmo)6(PhCO2)(MeO)6(H2O)]+ |
| 2193 | [Ni8(18O2)(abmo)6(PhCO2)(MeO)6(H2O)2]+ |
| 2207 | [Ni8(18O2)(abmo)6(PhCO2)(MeO)6(H2O)(MeOH)]+ |
Magnetic properties
Variable-temperature magnetic susceptibility data for the representative cage 1 were recorded between 250 and 5 K in an applied field of 1.0 kG. A plot of χMT versus T for 1 is shown in Fig. 4. The χMT product for complex 1 decreases upon cooling from a value of 7.88 cm3 K mol−1 at 250 K to a value of 0.76 cm3 K mol−1 at 5 K. The shape of the curve as well as the low value of the χMT product at 5 K suggest the presence of dominant antiferromagnetic exchange interactions and a diamagnetic spin ground state. The structural complexity of the molecule (there are many different exchange interaction pathways present) precludes any detailed quantitative analysis. However, in order to qualitatively estimate the magnitude of the exchange, the experimental data were satisfactorily fitted using a simple one exchange parameter (J) model by employing spin Hamiltonian (1). The best fit parameter was J = –5.02 cm−1 with a fixed g = 2.20.| Ĥ = −2J(Ŝ1·Ŝ2 + Ŝ1·Ŝ3 + Ŝ1·Ŝ4 + Ŝ1·Ŝ5 + Ŝ1·Ŝ6 + Ŝ1·Ŝ8 + Ŝ2·Ŝ3 + Ŝ2·Ŝ4 + Ŝ2·Ŝ5 + Ŝ2·Ŝ6 + Ŝ2·Ŝ8 + Ŝ3·Ŝ4 + Ŝ4·Ŝ5 + Ŝ4·Ŝ6 + Ŝ4·Ŝ8 + Ŝ5·Ŝ6 + Ŝ5·Ŝ7 +Ŝ5·Ŝ8 + Ŝ6·Ŝ7 + Ŝ6·Ŝ8 + Ŝ7·Ŝ8) | (1) |
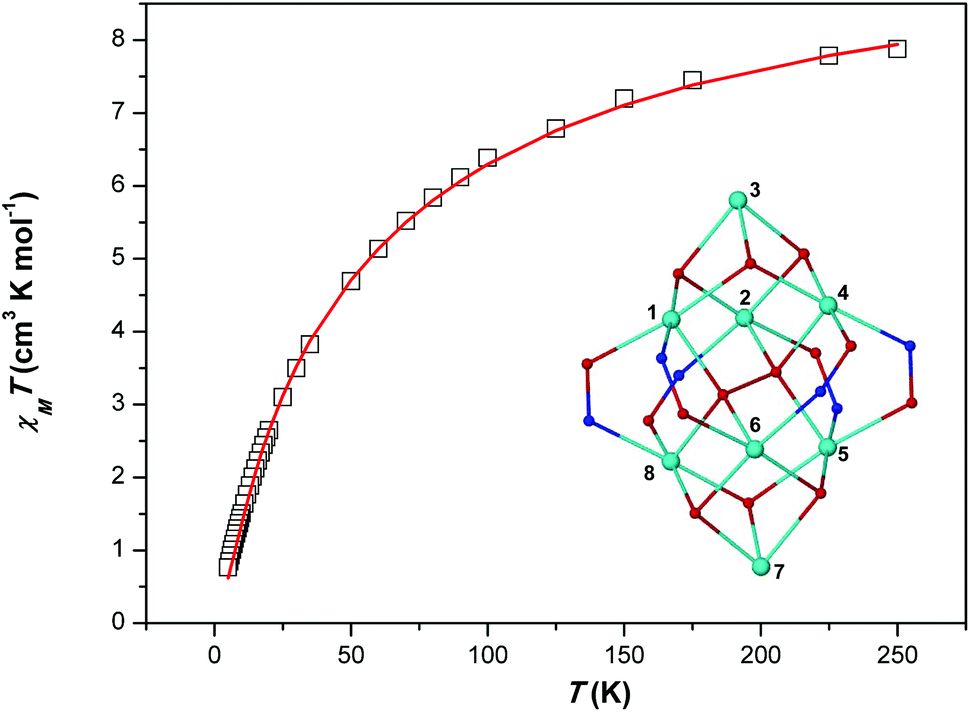 | ||
| Fig. 4 χ M T vs. T (□) plot for complex 1. The solid red line represents the fit of the experimental data to spin-Hamiltonian (1) and the model shown in the inset – see text for details. | ||
Thermal properties
Complexes 1–3 are stable in the solid state, can be handled in air and stored at room temperature as powdered samples or as single-crystals within their mother liquor for a period of over two years. Thermogravimetric analysis (TGA, Fig. 5) reveals that the three [Ni8] cages are stable up to 200 °C. This is an abnormal behaviour for peroxide complexes7a and is attributed to the isolation of the μ6-O22− within the cores of the [Ni8] cages.15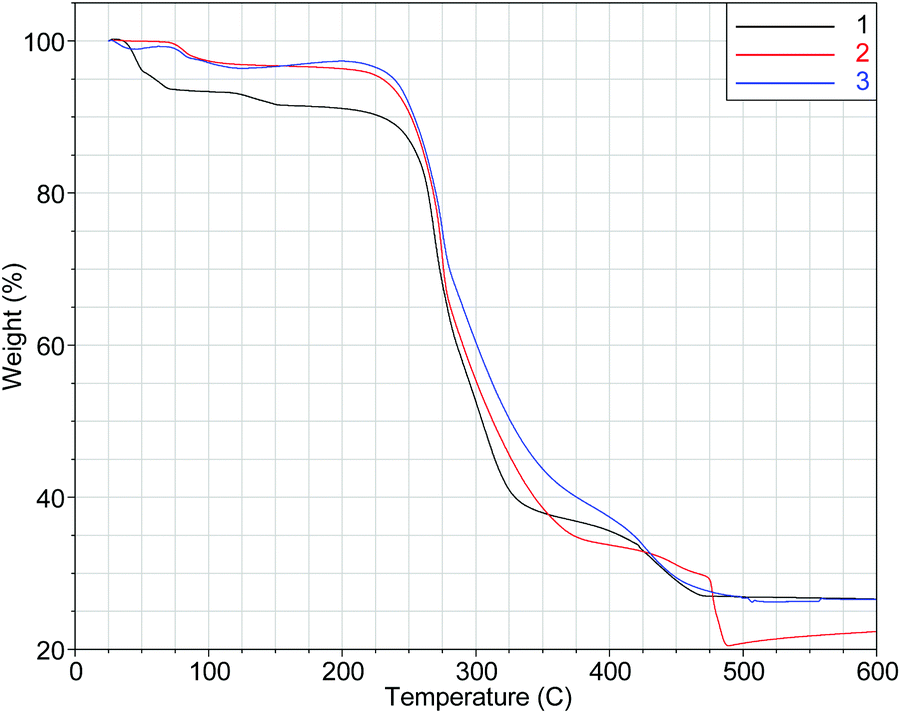 | ||
| Fig. 5 TGA plots in the 25–600 °C range of 1–3. | ||
Conclusions
In summary, we have reported the synthesis and X-ray characterization of a family of octanuclear NiII cages which are templated by a rare η3:η3:μ6-O22− species produced by dioxygen activation. This is the first time the O22− (i.e. the reduced species) and the reducing agent for the O2 reduction – which is not the metal ion (i.e. the organic ligand found within the nickel cages in its oxidized form) – were isolated and characterized by X-ray crystallography within the same compound(s). Cages 1–3 also represent unique paradigms of O2 activation and reduction by NiII reaction blends, which means that the electrons needed for reducing the dioxygen into the O22− are not provided by the metal ion but by the organic ligand2s–u (i.e. aboH2) which was found in its oxidized form (i.e. abmoH) within the cages. An alternative route providing access to the same [Ni8] cages involves the use of the oxidized form of the ligand (i.e. abmoH) along with H2O2. The octanuclear cages are the first examples of polynuclear metal complexes which can be made from different organic ligands (i.e. aboH2 and abmoH), and are exceptionally thermally stable (up to 200 °C).15 They also increase the number of structurally characterized NiII-peroxo compounds from four5a,7a,b,8 to seven. The vibrational modes of the μ6-O22− were not identified by Raman spectroscopy since no band was sensitive to isotopic substitution with 18O; similarly no Raman band was found to be sensitive to isotopic substitution with 18O in the known [Ni8(μ6-O2)(L)12](NO3)2 cluster.7a In contrast, a shift of 4 units was observed in the ESI-MS spectrum of the 18O-labelled 2A compared to the spectrum of unlabelled 2. This shift correlates to the labelling of both O atoms of the peroxo ligand, thus providing direct evidence that the peroxide originates from the reduction of dioxygen. Magnetic susceptibility revealed relatively weak antiferromagnetic exchange interactions between neighboring NiII ions, resulting in a diamagnetic spin ground state. The side product, a [Ni5] cluster, was also isolated and characterized by X-ray crystallography. We are currently exploring the coordination chemistry of aboH2 and abmoH with other 3d and 4f ions while examining the chemical reactivity of the [Ni8] cages.Acknowledgements
This research has been co-financed by the European Union (European Social Fund – ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) – the Research Funding Program: “THALES. Investing in knowledge society” through the European Social Fund. The Bodossaki Foundation is also acknowledged for donating the TGA instrument to the Dept. of Chemistry of UoA.Notes and references
- (a) For a special issue on dioxygen activation by metalloenzymes and models, see: W. Nam (ed.), Acc. Chem. Res., 2007, 40, 465–634; ; (b) For a forum on dioxygen activation and reduction, see: W. B. Tolman and E. I. Solomon, Inorg. Chem., 2010, 49, 3555–3556 Search PubMed.
- (a) J. S. Valentine, Chem. Rev., 1973, 73, 235–245 Search PubMed; (b) L. Vaska, Acc. Chem. Res., 1976, 9, 175–183 Search PubMed; (c) J. Reim, R. Werner, W. Haase and B. Krebs, Chem. – Eur. J., 1998, 4, 289–298 Search PubMed; (d) C. J. Cramer, W. B. Tolman, K. H. Theopold and A. L. Rheingold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3635–3640 Search PubMed; (e) M. T. Kieber-Emmons, R. Schenker, G. P. Yap, T. C. Brunold and C. G. Riordan, Angew. Chem., Int. Ed., 2004, 43, 6716–6718 Search PubMed; (f) S. Hikichi, H. Okuda, Y. Ohzu and M. Akita, Angew. Chem., Int. Ed., 2009, 48, 188–191 Search PubMed; (g) J. G. Liu, T. Ohta, S. Yamaguchi, T. Ogura, S. Sakamoto, Y. Maeda and Y. Naruta, Angew. Chem., Int. Ed., 2009, 48, 9262–9267 Search PubMed; (h) P. L. Holland, Dalton Trans., 2010, 39, 5415–5425 Search PubMed; (i) R. Sarangi, J. Cho, W. Nam and E. I. Solomon, Inorg. Chem., 2011, 50, 614–620 Search PubMed; (j) S. Yao, C. Herwig, Y. Xiong, A. Company, E. Bill, C. Limberg and M. Driess, Angew. Chem., Int. Ed., 2010, 49, 7054–7058 Search PubMed; (k) A. D. Bochevarov, J. Li, W. J. Song, R. A. Friesner and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 7384–7397 Search PubMed; (l) J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502–505 Search PubMed; (m) Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258–9261 Search PubMed; (n) E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian and J. S. Woertink, Faraday Discuss., 2011, 148, 11 Search PubMed; (o) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 Search PubMed; (p) L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 899–910 Search PubMed; (q) S. Maji, J. C. Lee, Y. J. Lu, C. L. Chen, M. C. Hung, P. P. Chen, S. S. Yu and S. I. Chan, Chem. – Eur. J., 2012, 18, 3955–3968 Search PubMed; (r) S. Yao and M. Driess, Acc. Chem. Res., 2012, 45, 276–287 Search PubMed; (s) G. Dong, S. Shaik and W. Lai, Chem. Sci., 2013, 4, 3624–3635 Search PubMed; (t) G. Dong and W. Lai, J. Phys. Chem. B, 2014, 118, 1791–1798 Search PubMed; (u) A. J. Fielding, J. D. Lipscomb and L. Que, J. Biol. Inorg. Chem., 2014 DOI:10.1007/s00775-014-1122-9 , in press.
- (a) R. Latifi, L. Tahsini, D. Kumar, G. N. Sastry, W. Nam and S. P. de Visser, Chem. Commun., 2011, 47, 10674–10676 Search PubMed; (b) M. Zhou and R. H. Crabtree, Chem. Soc. Rev., 2011, 40, 1875–1884 Search PubMed; (c) Y. Xiong, S. Yao, R. Muller, M. Kaupp and M. Driess, Nat. Chem., 2010, 2, 577–580 Search PubMed; (d) C. Citek, C. T. Lyons, E. C. Wasinger and T. D. Stack, Nat. Chem., 2012, 4, 317–322 Search PubMed; (e) L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. Stack, Science, 2005, 308, 1890–1892 Search PubMed; (f) J. Reedijk, Science, 2005, 308, 1876–1877 Search PubMed; (g) M. Rolff, J. N. Hamann and F. Tuczek, Angew. Chem., Int. Ed., 2011, 50, 6924–6927 Search PubMed.
- (a) P. Jeske, G. Haselhorst, T. Weyhermueller, K. Wieghardt and B. Nuber, Inorg. Chem., 1994, 33, 2462–2471 Search PubMed; (b) A. F. Cozzolino, D. Tofan, C. C. Cummins, M. Temprado, T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2012, 134, 18249–18252 Search PubMed; (c) A. Yokoyama, J. Eun Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269–15272 Search PubMed; (d) M. K. Coggins, X. Sun, Y. Kwak, E. I. Solomon, E. Rybak-Akimova and J. A. Kovacs, J. Am. Chem. Soc., 2013, 135, 5631–5640 Search PubMed; (e) C.-M. Lee, C.-H. Chuo, C.-H. Chen, C.-C. Hu, M.-H. Chiang, Y.-J. Tseng, C.-H. Hu and G.-H. Lee, Angew. Chem., Int. Ed., 2012, 51, 5427–5430 Search PubMed; (f) K. Kim and S. J. Lippard, J. Am. Chem. Soc., 1996, 118, 4914–4915 Search PubMed; (g) E. Bouwman and W. L. Driessen, J. Am. Chem. Soc., 1988, 110, 4440–4441 Search PubMed; (h) S. Schmidt, F. W. Heinemann and A. Grohmann, Eur. J. Inorg. Chem., 2000, 1657–1667 Search PubMed; (i) A. L. Gavrilova, C. Jin Qin, R. D. Sommer, A. L. Rheingold and B. Bosnich, J. Am. Chem. Soc., 2002, 124, 1714–1722 Search PubMed; (j) D. B. Leznoff, M. J. Katz, L. K. L. Cheng, N. D. Draper and R. J. Batchelor, J. Mol. Struct., 2006, 796, 223–229 Search PubMed; (k) G. Givaja, M. Volpe, M. A. Edwards, A. J. Blake, C. Wilson, M. Schröder and J. B. Love, Angew. Chem., Int. Ed., 2007, 46, 584–586 Search PubMed; (l) M. Kodera, K. Katayama, Y. Tachi, K. Kano, S. Hirota, S. Fujinami and M. Suzuki, J. Am. Chem. Soc., 1999, 121, 11006–11007 Search PubMed; (m) F. Meyer and H. Pritzkow, Angew. Chem., Int. Ed., 2000, 39, 2112–2115 Search PubMed.
- (a) M. Matsumoto and K. Nakatsu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2711–2713 Search PubMed; (b) S. Yao, E. Bill, C. Milsmann, K. Wieghardt and M. Driess, Angew. Chem., Int. Ed., 2008, 47, 7110–7113 Search PubMed.
- (a) G. Wilke, H. Schott and P. Heimbach, Angew. Chem., Int. Ed. Engl., 1967, 6, 92–93 Search PubMed; (b) S. Otsuka, A. Nakamura and Y. Tatsuno, J. Am. Chem. Soc., 1969, 91, 6994–6999 Search PubMed; (c) K. Fujita, R. Schenker, W. Gu, T. C. Brunold, S. P. Cramer and C. G. Riordan, Inorg. Chem., 2004, 43, 3324–3326 Search PubMed; (d) M. T. Kieber-Emmons, J. Annaraj, M. Sook Seo, K. M. Van Heuvelen, T. Tosha, T. Kitagawa, T. C. Brunold, W. Nam and C. G. Riordan, J. Am. Chem. Soc., 2006, 128, 14230–14231 Search PubMed; (e) M. T. Kieber-Emmons and C. G. Riordan, Acc. Chem. Res., 2007, 40, 618–625 Search PubMed; (f) A. Company, S. Yao, K. Ray and M. Driess, Chem. – Eur. J., 2010, 16, 9669–9675 Search PubMed.
- (a) E. J. Brown, A. K. Duhme-Klair, M. I. Elliott, J. E. Thomas-Oates, P. L. Timmins and P. H. Walton, Angew. Chem., Int. Ed., 2005, 44, 1392–1395 Search PubMed; (b) J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568–572 Search PubMed; (c) K. Shiren, S. Ogo, S. Fujinami, H. Hayashi, M. Suzuki, A. Uehara, Y. Watanabe and Y. Moro-oka, J. Am. Chem. Soc., 2000, 122, 254–262 Search PubMed; (d) J. Cho, H. Furutachi, S. Fujinami, T. Tosha, H. Ohtsu, O. Ikeda, A. Suzuki, M. Nomura, T. Uruga, H. Tanida, T. Kawai, K. Tanaka, T. Kitagawa and M. Suzuki, Inorg. Chem., 2000, 46, 2873–2885 Search PubMed.
- S. Yao, Y. Xiong, M. Vogt, H. Grutzmacher, C. Herwig, C. Limberg and M. Driess, Angew. Chem., Int. Ed., 2009, 48, 8107–8110 Search PubMed.
- (a) J. Auld, A. C. Jones, A. B. Leese, B. Cockayne, P. J. Wright, P. O'Brien and M. Motevalli, J. Mater. Chem., 1993, 3, 1203–1208 Search PubMed; (b) N. Hovnaninian, J. Galloy and P. Miele, Polyhedron, 1995, 14, 297–300 Search PubMed; (c) A. Sofetis, F. Fotopoulou, C. P. Raptopoulou, Th. F. Zafiropoulos, S. P. Perlepes and N. Klouras, Polyhedron, 2009, 28, 3356–3360 Search PubMed; (d) E. I. Tolis, M. Helliwell, S. Langley, J. Raftery and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 3804–3808 Search PubMed.
- J. Catterick and P. Thornton, J. Chem. Soc., Dalton Trans., 1975, 233–238 Search PubMed.
- T. W. J. Taylor and M. S. Marks, J. Chem. Soc., 1930, 2302–2307 Search PubMed.
- Crystal data for 1·4H2O: C98H108N6Ni8O32, M = 2351.58, triclinic, a = 13.291(2) Å, b = 13.838(2) Å, c = 16.545(4) Å, α = 108.734(3)°, β = 105.788(3)°, γ = 101.737(2)°, V = 2628.1(8) Å3, T = 293(2) K, space group P, Z = 1, MoKα, 17
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 894 reflections measured, 11581 independent reflections (Rint = 0.0352). The final R1 values were 0.0542 (I > 2σ(I)). The final wR(F2) values were 0.1391 (I > 2σ(I)). The final R1 values were 0.0933 (all data). The final wR(F2) values were 0.1603 (all data). The goodness of fit on F2 was 1.015. CCDC 894474. Crystal data for 2·2MeOH: C110H112N6Ni8O30, M = 2467.74, monoclinic, a = 13.259(2) Å, b = 28.761(5) Å, c = 15.273(3) Å, α = 90.00°, β = 103.829(2)°, γ = 90.00°, V = 5655.0(16) Å3, T = 296(2) K, space group P2(1)/c, Z = 2, MoKα, 21909 reflections measured, 8862 independent reflections (Rint = 0.0920). The final R1 values were 0.0631 (I > 2σ(I)). The final wR(F2) values were 0.1463 (I > 2σ(I)). The final R1 values were 0.1197 (all data). The final wR(F2) values were 0.1743 (all data). The goodness of fit on F2 was 0.996. CCDC 894475. Crystal data for 3: C106H98Cl2N6Ni8O28, M = 2444.48, triclinic, a = 13.372(10) Å, b = 14.701(11) Å, c = 15.110(11) Å, α = 94.446(6)°, β = 108.768(13)°, γ = 104.964(10)°, V = 2675(3) Å3, T = 93(2) K, space group P, Z = 1, MoKα, 26876 reflections measured, 9695 independent reflections (Rint = 0.0381). The final R1 values were 0.0601 (I > 2σ(I)). The final wR(F2) values were 0.1644 (I > 2σ(I)). The final R1 values were 0.0623 (all data). The final wR(F2) values were 0.1669 (all data). The goodness of fit on F2 was 1.075. CCDC 894476. Crystal data for 4·4MeOH·CH2Cl2: C117H112Cl2N8Ni5O20, M = 2314.67, triclinic, a = 12.8976(6) Å, b = 13.0924(6) Å, c = 16.4651(6) Å, α = 98.686(3)°, β = 99.661(3)°, γ = 90.904(3)°, V = 2707.0(2) Å3, T = 150.0 K, space group P, Z = 1, Cu Kα, 54794 reflections measured, 9656 independent reflections (Rint = 0.0484). The final R1 values were 0.0686 (I > 2σ(I)). The final wR(F2) values were 0.1827 (I > 2σ(I)). The final R1 values were 0.0829 (all data). The final wR(F2) values were 0.2087 (all data). The goodness of fit on F2 was 0.9640. CCDC 894477.
894 reflections measured, 11581 independent reflections (Rint = 0.0352). The final R1 values were 0.0542 (I > 2σ(I)). The final wR(F2) values were 0.1391 (I > 2σ(I)). The final R1 values were 0.0933 (all data). The final wR(F2) values were 0.1603 (all data). The goodness of fit on F2 was 1.015. CCDC 894474. Crystal data for 2·2MeOH: C110H112N6Ni8O30, M = 2467.74, monoclinic, a = 13.259(2) Å, b = 28.761(5) Å, c = 15.273(3) Å, α = 90.00°, β = 103.829(2)°, γ = 90.00°, V = 5655.0(16) Å3, T = 296(2) K, space group P2(1)/c, Z = 2, MoKα, 21909 reflections measured, 8862 independent reflections (Rint = 0.0920). The final R1 values were 0.0631 (I > 2σ(I)). The final wR(F2) values were 0.1463 (I > 2σ(I)). The final R1 values were 0.1197 (all data). The final wR(F2) values were 0.1743 (all data). The goodness of fit on F2 was 0.996. CCDC 894475. Crystal data for 3: C106H98Cl2N6Ni8O28, M = 2444.48, triclinic, a = 13.372(10) Å, b = 14.701(11) Å, c = 15.110(11) Å, α = 94.446(6)°, β = 108.768(13)°, γ = 104.964(10)°, V = 2675(3) Å3, T = 93(2) K, space group P, Z = 1, MoKα, 26876 reflections measured, 9695 independent reflections (Rint = 0.0381). The final R1 values were 0.0601 (I > 2σ(I)). The final wR(F2) values were 0.1644 (I > 2σ(I)). The final R1 values were 0.0623 (all data). The final wR(F2) values were 0.1669 (all data). The goodness of fit on F2 was 1.075. CCDC 894476. Crystal data for 4·4MeOH·CH2Cl2: C117H112Cl2N8Ni5O20, M = 2314.67, triclinic, a = 12.8976(6) Å, b = 13.0924(6) Å, c = 16.4651(6) Å, α = 98.686(3)°, β = 99.661(3)°, γ = 90.904(3)°, V = 2707.0(2) Å3, T = 150.0 K, space group P, Z = 1, Cu Kα, 54794 reflections measured, 9656 independent reflections (Rint = 0.0484). The final R1 values were 0.0686 (I > 2σ(I)). The final wR(F2) values were 0.1827 (I > 2σ(I)). The final R1 values were 0.0829 (all data). The final wR(F2) values were 0.2087 (all data). The goodness of fit on F2 was 0.9640. CCDC 894477. - (a) V. V. Sharutin, O. V. Molokova, O. K. Sharutina, T. I. Akimova, A. V. Gerasimenko and M. A. Pushilin, Russ. J. Coord. Chem., 2004, 30, 559–565 Search PubMed; (b) E. Soleimani, Rev. Chim., 2009, 60, 484–487 Search PubMed.
- (a) G. Karotsis, C. Stoumpos, A. Collins, F. White, S. Parsons, A. M. Slawin, G. S. Papaefstathiou and E. K. Brechin, Dalton Trans., 2009, 3388–3390 Search PubMed; (b) E. S. Koumousi, M. J. Manos, C. Lampropoulos, A. J. Tasiopoulos, W. Wernsdorfer, G. Christou and T. C. Stamatatos, Inorg. Chem., 2010, 49, 3077–3079 Search PubMed; (c) S. Liu, H. Zhu and J. Zubieta, Polyhedron, 1989, 8, 2473–2480 Search PubMed; (d) T. C. Stamatatos, G. Vlahopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer, G. Christou and S. P. Perlepes, Eur. J. Inorg. Chem., 2012, 3121–3131 Search PubMed; (e) G. C. Vlahopoulou, T. C. Stamatatos, V. Psycharis, S. P. Perlepes and G. Christou, Dalton Trans., 2009, 3646–3649 Search PubMed.
- N. Lopez, D. J. Graham, R. McGuire Jr., G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. CCDC 894474–894477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00048j |
| This journal is © the Partner Organisations 2014 |