Dye-sensitized solar cells based on triazine-linked porphyrin dyads containing one or two carboxylic acid anchoring groups†
Galateia E.
Zervaki
a,
Panagiotis A.
Angaridis
a,
Emmanuel N.
Koukaras
b,
Ganesh D.
Sharma
*c and
Athanassios G.
Coutsolelos
*a
aLaboratory of Bioinorganic Chemistry, Department of Chemistry, University of Crete, Voutes Campus, P.O. Box 2208, 71003 Heraklion, Crete, Greece. E-mail: coutsole@chemistry.uoc.gr
bInstitute of Chemical Engineering Sciences, Foundation for Research & Technology, Hellas (FORTH/ICE-HT), Stadiou Str. Platani, Patras, 26504 Greece
cR&D Center for Engineering and Science, JEC Group of Colleges, Jaipur Engineering College, Kukas, Jaipur (Raj.) 303101, India. E-mail: gdsharma273@gmail.com
First published on 12th February 2014
Abstract
Two porphyrin dyads (5a and 5b) consisting of two meso aryl-substituted zinc-metallated porphyrin units, which are covalently linked through peripheral aryl-amino groups by a 1,3,5-triazine moiety, have been synthesized via stepwise substitution reactions of cyanuric chloride. Both porphyrin dyads 5a and 5b contain a carboxyphenyl meso substituent, while in the former the triazine ring is functionalized by a glycine group, and in the latter by a piperidine group. Photophysical and electrochemical studies of the two dyads, in combination with DFT theoretical calculations, suggest that there is negligible electronic interaction between the porphyrin units in the dyads’ ground states, but the frontier orbital energy levels are suitable for use as sensitizers in dye-sensitized solar cells (DSSCs). Solar cells sensitized by dyads 5a and 5b have been fabricated, and they were found to exhibit power conversion efficiencies (PCEs) of 5.28 and 3.50%, respectively, under illumination (AM1.5, 100 mW cm−2), and TiO2 films of 10 μm thickness. As revealed by the photovoltaic measurements (J–V curves) and the incident photon to current conversion efficiency (IPCE) spectra of the two solar cells, the higher PCE value of the 5a based solar cell is attributed to its enhanced short circuit current (Jsc), higher open circuit voltage (Voc), and fill factor (FF) values. Also, the 5a sensitized solar cell exhibits a larger dye loading value. This is attributed to the presence of two carboxylic acid anchoring groups in its molecular structure (compared to one carboxylic acid and one hindered piperidine-type anchoring group of 5b), which result in a more effective binding capacity onto the TiO2 film. Furthermore, electrochemical impedance spectra (EIS) demonstrated that the 5a based solar cell exhibits longer electron lifetime (τe) and more effective suppression of the recombination reactions of the injected electrons and the electrolyte.
Introduction
The depletion of the world's fossil fuel reserves, global warming, and rapidly growing energy demand render the quest for abundant, safe and renewable energy sources one of society's greatest technological challenges.1 In this respect, dye-sensitized solar cells (DSSCs) have attracted great academic and commercial interest over the last 20 years as a result of their low cost, ease of fabrication, and high power conversion efficiencies (PCEs) compared to conventional solar cells based on inorganic semiconducting materials.2,3 In 1991, O'Regan and Grätzel reported the first efficient DSSC composed of a ruthenium bipyridyl photosensitizer adsorbed on the surface of a nanocrystalline TiO2 film (photoanode), a counter electrode, and a redox electrolyte.2 Photoexcited electrons of the sensitizer are injected into the TiO2 conduction band (CB) and transferred through the counter electrode to the electrolyte, which regenerates the sensitizer to its original ground state. In an effort to improve the efficiency and durability of DSSC devices, a variety of sensitizers have been developed and tested.4 Ruthenium-polypyridyl complexes have been proved to be the most efficient sensitizers,5 giving devices with PCE values above 11%.6 However, their low molar extinction coefficients, instability, high cost and environmental issues limit their wide application. Recently, a variety of noble metal-free organic dyes have been designed, synthesized and utilized in DSSCs, resulting in solar cell efficiencies that exceed 10% (in some cases), rendering this type of molecules strong competitors to the ruthenium-based sensitizers.7A class of metal-free organic dyes that have stimulated significant interest as sensitizers in DSSCs is porphyrins,8 due to their light harvesting potential and their physicochemical properties. They exhibit absorptions in the 400–450 nm and the 500–700 nm regions with high and moderate molar extinction coefficients, respectively. In addition, their structures can be functionalized by rational design and synthesis, allowing fine tuning of their photophysical and electrochemical properties.9 Recently, Diau, Grätzel and co-workers reported a donor-π-acceptor (D-π-A) zinc-porphyrin, substituted at meso-positions with long alkoxyl-phenyl groups, ethynyl bridging groups, and amino groups, and a carboxylic acid group for anchoring to TiO2 that resulted in a new benchmark in DSSCs with an overall PCE value of 12.3%.10 However, the synthesis of porphyrin derivatives of this type is very demanding, requiring several steps, including catalytic Sonogashira and Buchwald–Hartwig cross-coupling reactions, which makes their commercial application infeasible. Moreover, their long-term stability is a major concern, due to the instability of the bridging and anchoring groups. In addition, the relatively narrow absorbing range of porphyrins and their weak absorptions in the green and red regions of the solar spectrum limit their DSSC performance.
In order to further improve the efficiency of porphyrin-based solar cells, one has to extend the light harvesting ability of porphyrin dyes to the near-infrared region. This can be accomplished by covalently linking porphyrin macrocycles with other chromophores or other porphyrin units, either through suitable π-conjugated groups or directly. Indeed, porphyrin arrays linked by ethynyl groups through their meso-positions exhibit strong electronic coupling between porphyrin units, resulting in splitting of their Soret bands and broadening of their Q bands.11 In addition, porphyrin dyads linked directly through their meso positions exhibit slightly higher light harvesting efficiencies than that of the corresponding single porphyrins units,12 while doubly and triply fused porphyrin arrays show wide absorption covering the visible and near IR regions.13 Utilization of porphyrin assemblies of the above-mentioned types in DSSCs can potentially result in enhanced photovoltaic performances. For example, a DSSC sensitized by an ethynyl-bridged carbazole–porphyrin–porphyrin triad showed efficient light to current conversion throughout the visible and near IR regions resulting in a PCE value of 5.21%.14
A π-conjugated group that has been recently employed as a linker in the synthesis of metal-free organic dyes for DSSC and other photoconductive materials applications is 1,3,5-triazine.15 The structural, chemical and electronic properties of this unit allow the synthesis of complex π-conjugated multi-chromophoric dyes through simple organic reactions16 that offer improved light harvesting ability, as well as improved electron injection and transportation rates.17 Triazine-bridged porphyrin arrays have been used as electron donors in bulk heterojunction (BHJ) solar cells and were found to enhance electron extraction efficiency, as in the case of the triangular shaped, triazine-bridged porphyrin triad reported by Luechai et al.18 In the area of DSSCs, You et al. reported a DSSC sensitized by a triazine-bridged D-π-A metal-free organic dye that resulted in a PCE value of 3.63%.19 In our earlier work, we reported two triazine-bridged porphyrin dyads with carboxylic acid anchoring groups on the triazine ring, which were used in DSSCs achieving PCE values of 3.61 and 4.46%.20
Herein, we present the syntheses of two new triazine-bridged porphyrin dyads, 5a and 5b, the former with two carboxylic acid anchoring groups for attachment to the TiO2 surface, and the latter with one carboxylic acid anchoring group and a piperidine binding site, which has the potential to act as an additional anchoring group (Scheme 1). These dyes were used as a sensitizer in DSSCs, which resulted in PCE values of 5.28 and 3.50%, respectively. The higher PCE value of the former solar cell is attributed to its higher photovoltaic parameters, higher dye loading value, longer electron lifetime, and stronger recombination resistance, which are related to the more effective binding of porphyrin dyad 5a due to the presence of two carboxylic acid anchoring groups in its molecular structure.
 | ||
| Scheme 1 | ||
Results and discussion
Synthesis and characterization
As shown in Scheme 1, porphyrin dyads 5a and 5b consist of two meso aryl-substituted zinc-metallated porphyrin units Zn[Porph1] and Zn[Porph2] (where Zn[Porph1] is 5-(4-aminophenyl)-10,15,20-triphenylporphyrin zinc and Zn[Porph2] is 5-(4-carboxyphenyl)-15-(4-aminophenyl)-10,20-bis(2,4,6-trimethylphenyl)-porphyrin zinc) which are covalently bridged, through aryl-amino groups at their peripheries, by a 1,3,5-triazine group. In dyad 5a the third position of the triazine ring is functionalized by a glycine moiety, while in 5b by an N-substituted piperidine moiety. Both dyads contain two potential anchoring groups for attachment onto the TiO2 surface of DSSC electrodes: the former with two carboxylic acid groups, while the latter with one carboxylic acid and one N-substituted piperidine group.The synthesis of dyads 5a and 5b was achieved via stepwise amination reactions of cyanuric chloride, the precursor of the 1,3,5-triazine group. The reactivity of cyanuric chloride is based on the temperature-dependent stepwise substitution of its three chlorine atoms through simple, one-pot reaction procedures that allow the sequential attachment of different nucleophiles, providing access to the synthesis of a variety of macrocycles,21 dendrimers22 and multiporphyrin arrays.23,24 We recently reported the syntheses of symmetrical and unsymmetrical porphyrin arrays dyads and triads linked by 1,3,5-triazine.20,25
The initial step for the syntheses of both dyads 5a and 5b involves the reaction of cyanuric chloride with H2[Porph] in the presence of the base DIPEA at 0 °C in THF (Scheme 2). The reaction was monitored by TLC, indicating the disappearance of the reactants and the formation of the mono-porphyrin-triazine adduct 1. The latter further reacted at room temperature with H2[TPP-NH2] affording the di-porphyrin-triazine adduct 2. The third chlorine atom of cyanuric chloride was substituted by a glycine–methyl ester moiety in a one-pot reaction at 65 °C resulting in the di-porphyrin-triazine-glycine methyl ester adduct 3a, as confirmed by 1H NMR spectroscopy and MALDI-TOF spectrometry. It is worth mentioning that in the 1H NMR spectrum of 3a, the signals of the aromatic H's ortho to the amino groups of H2[TPP-NH2], after attachment to the triazine ring, are downfield displaced compared to those of free H2[TPP-NH2]. In addition, its UV-vis absorption spectrum is characteristic of free-base porphyrins with a Soret band and four Q-bands. Reaction of 3a with excess of Zn(CH3COO)2·2H2O in MeOH–CH2Cl2 yielded the metallated di-porphyrin-triazine-glycine methyl ester adduct 4a, while basic hydrolysis of the methyl ester group of 4a resulted in the porphyrin dyad 5a, in almost quantitative yield, as indicated by 1H NMR, MALDI-TOF spectrometry, UV-vis absorption spectroscopy, and elemental analysis. The most noticeable features in the 1H NMR spectrum of 5a are the absence of the signal attributed to the methyl-ester H's after hydrolysis reaction (with respect to the 1H NMR spectrum of 4a), and the absence of the pyrrolic H's signals after metallation reaction (with respect to the 1H NMR spectrum of 3a). The latter is consistent with the UV-vis absorption spectrum of 5a that shows only two Q-bands, which is characteristic of Zn-metallated porphyrins.
 | ||
| Scheme 2 | ||
Porphyrin dyad 5b was synthesized in a similar manner, following the above-mentioned sequence of reactions. The piperidine group was introduced in a substitution reaction of the third chlorine atom of cyanuric chloride yielding the di-porphyrin-triazine-piperidine adduct 3b. Metallation reaction, followed by hydrolysis reaction, resulted in the formation of the desired porphyrin dyad 5b. The identity and purity of 5b were confirmed by 1H and 13C NMR spectroscopy, MALDI-TOF spectrometry, UV-vis absorption spectroscopy, and elemental analysis.
Photophysical studies
The UV-vis absorption spectra of porphyrin dyads 5a and 5b in solution (0.3 mM in CHCl3–EtOH = 1/1) are shown in Fig. 1a and 1b, respectively (black color lines). The spectra of both dyads exhibit typical porphyrin absorption bands, with intense Soret bands in the 400–450 nm range and two moderate Q bands in the 530–640 nm range and no other additional features. As is suggested by the theoretical calculation results (in the following sections), there is no intramolecular communication between the two porphyrin units attached to the triazine ring in the ground state of the dyads. The shoulders that appear in the low energy side of the Soret bands are probably due to very weak intermolecular exciton coupling between porphyrin units of different dyads in their excited states26 or lowering of molecular symmetry. | ||
| Fig. 1 Normalized UV-vis absorption spectra of (a) 5a and (b) 5b in solution (black color line) and after adsorbed onto TiO2 films (red color line). | ||
The UV-vis absorption spectra of porphyrin dyads 5a and 5b adsorbed on TiO2 films (10 μm thickness) are presented in Fig. 1a and 1b, respectively (red color lines). Both spectra exhibit the usual porphyrin Soret and Q bands, but these are broader and slightly shifted compared to the corresponding solution spectra. In general, when porphyrins are adsorbed onto the TiO2 surface, they form either H- or J-aggregates. Soret bands that are broader and blue-shifted relative to the bands in solution characterize the former, while for the latter the Soret bands are sharp and red-shifted. The broad and red-shifted bands of porphyrin dyads 5a and 5b may be attributed to intermolecular interactions that result in the formation of J-type aggregates onto the TiO2 surface upon adsorption.27 Furthermore, the broader, red shifted and higher intensity Q bands suggest a strong electronic coupling between the dyads and the TiO2 surface28 and give an indication of enhanced light harvesting capacity to longer wavelengths of the solar spectrum of the two dyads. By using the onset absorption edge λonset of the Q bands and the expression
| Eoptg = 1240/λonset |
| Compound | Absorption λmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/nm (ε/mM−1 cm−1) a/nm (ε/mM−1 cm−1) |
Emission λmaxa (nm) |
E
optgb (eV) |
|---|---|---|---|
| a Measured at 298 K. b Calculated by onset absorption edge λonset of the Q bands and the expression Eoptg = 1240/λonset. | |||
| 5a | 425 (667.3), 557 (28.2), 597 (11.6) | 608, 656 | 1.91 |
| 5b | 425 (666.7), 557 (27.8), 597 (11.4) | 608, 657 | 1.94 |
In Fig. 2a and 2b are depicted the steady-state fluorescence spectra of 5a and 5b in CH2Cl2 solutions and adsorbed onto thin TiO2 films. Excitation of the porphyrin dyads in solution at the Soret band (420 nm) results in photoluminescence, with two peaks of unequal intensities at 608 nm and 656 nm (black color lines). However, when the dyads adsorbed onto thin TiO2 films are excited at the same wavelength, the fluorescence intensities for both compounds are significantly reduced, providing significant quenching (red color lines). The fluorescence quenching can be attributed to a photoinduced electron transfer process from the electron donating zinc-metallated porphyrin units of the dyads to the TiO2 surface.29
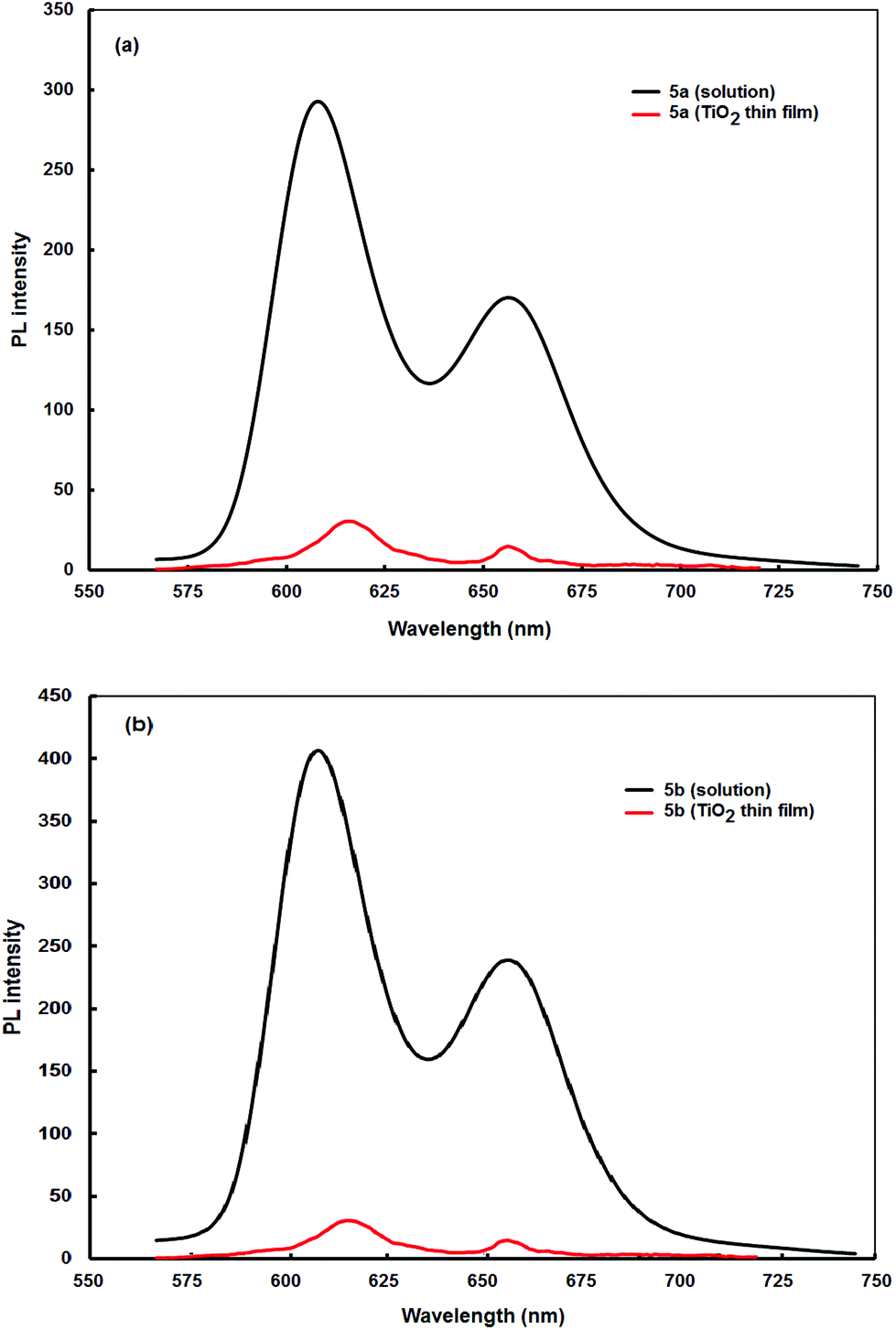 | ||
| Fig. 2 Isoabsorbing fluorescence spectra of (a) 5a and (b) 5b in solution (black color lines) and after adsorbed onto TiO2 films (red color line). | ||
Electrochemical studies
The electrochemical properties of porphyrin dyads 5a and 5b were investigated by cyclic voltammetry measurements. Their cyclic voltammograms in THF are depicted in Fig. 3, while the relevant electrochemical data are listed in Table 2. Both dyads exhibit two oxidation waves at E1ox = +1.04 V (quasi-reversible) and E2ox = +1.26 V for 5a, and E1ox = +1.29 V (quasi-reversible) and E2ox = +1.63 V vs. NHE for 5b. Furthermore, they exhibit one reduction process at Ered = −1.08 V (quasi-reversible) for 5a and Ered = −1.09 V (quasi-reversible) vs. NHE for 5b. The values for the redox potentials of these processes were determined more accurately by means of square-wave voltammetry measurements (Fig. S18†). The redox processes of both dyads are due to the usual stepwise oxidations by two electrons and reductions by one electron of the π-ring system of the porphyrin units. These data suggest that there is negligible electronic interaction between the two porphyrin units in the ground states of both dyads 5a and 5b.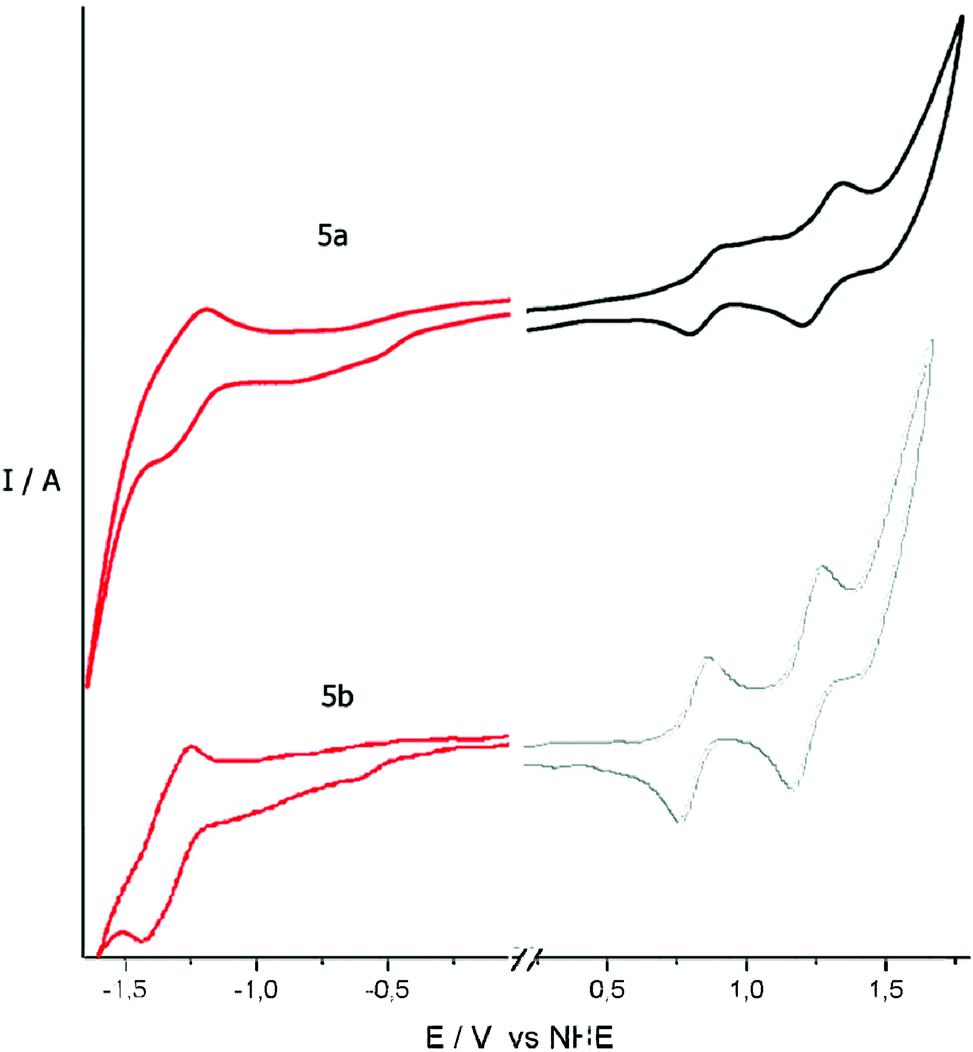 | ||
| Fig. 3 Cyclic voltammograms of 5a and 5b in THF vs. NHE. The ferrocene/ferrocenium (Fc/Fc+) redox couple wave is observed at 0.85 V and 0.86 V vs. NHE, for 5a and 5b, respectively. | ||
| Compound |
E
oxa (V) |
E
reda (V) |
HOMOb (eV) | LUMOb (eV) | E elecg (eV) | ΔGinjc (eV) |
ΔGregc (eV) |
|---|---|---|---|---|---|---|---|
| a Observed first oxidation and reduction potentials (vs. NHE). b HOMO and LUMO energy levels and band gaps calculated according to the relationships EHOMO = −(Eox + 4.4) eV and ELUMO = −(Ered + 4.4) eV, measured with reference to the redox potential −4.4 eV. c ΔGinj for electron injection from the LUMO of the dyads to the TiO2 CB (−0.5 V vs. NHE) and ΔGreg for electron transfer from I− to the ZnPor˙+ moiety regenerates the dyad in the neutral state and affords I3−, i.e., reductive quenching of the radical cation (0.4 V vs. NHE). | |||||||
| 5a | 1.04 | −1.08 | −5.44 | −3.32 | 2.12 | −0.58 | −0.64 |
| 5b | 1.29 | −1.09 | −5.69 | −3.31 | 2.38 | −0.59 | −0.89 |
Generally, the redox potentials of a sensitizer are related to the energetics of essential processes of DSSCs. The first oxidation potential E1ox and first reduction potential E1red of the sensitizer correspond to its highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies, respectively. An efficient sensitizer should have HOMO and LUMO energy levels that enable efficient electron and regeneration processes. Particularly, for efficient electron injection of the photosensitizer (after its photoexcitation) into the TiO2 conduction band (CB), the LUMO energy level of the sensitizer (E1red) should be higher than the TiO2 CB edge (−0.5 vs. NHE). Furthermore, in order to ensure efficient regeneration of the sensitizer cation by electron transfer from I− of the electrolyte (after the electron injection process), the HOMO energy level of the sensitizer (E1ox) should be lower than the redox potential of electrolyte I−/I3− couple (+0.4 V vs. NHE). As shown in Table 2, the E1red values of both dyes 5a and 5b are more negative than the TiO2 CB edge, which means that the electron injection from the photoexcited dyads into the TiO2 CB is favorable (ΔGinj < 0). Additionally, the E1ox values of both dyes 5a and 5b are more positive than the redox potential of the electrolyte couple I−/I3−, which indicates that there is sufficient driving force for regeneration of the oxidized sensitizer by electron transfer from I− of the electrolyte to afford I3− (ΔGreg < 0). Therefore, both electron injection and regeneration processes are thermodynamically favored for both dyads 5a and 5b.
Based on these electrochemical data, the HOMO–LUMO band gaps Eelecg of 5a and 5b were found to be 2.12 and 2.38 eV, respectively (Table 2). These are larger than the corresponding optical band gaps Eoptg estimated from the photophysical measurements, but this is a common feature of organic dyes of this type, which can be attributed to solvent effects.30
Theoretical calculations
Geometry optimizations on the 5a and 5b porphyrin dyads were performed within the framework of density functional theory (DFT), using the PBE, PBE0, and B3LYP functionals. The structures of 5a and 5b optimized using the B3LYP functional in the gas-phase are depicted in Fig. 4, while their optimized coordinates are provided in Table S1 in ESI.† Both molecules adopt “butterfly like” structures in which the triazine ring is nearly co-planar with the bridging, amino-substituted phenyl groups of the porphyrin units (which we collectively denote as the triazine framework), extending its π-conjugated system. The porphyrin plane dihedral angles with respect to the triazine framework in the range 63–68° for Zn[Porph1] and 67–72° for Zn[Porph2], for 5a, and 63–69° for Zn[Porph1] and 65–72° for Zn[Porph2] for 5b, depending on the level of theory. The non-planar orientation of the two porphyrin units, the relatively large inter-porphyrin distance, and the break of conjugation by the twisted phenyl rings diminish the electronic communication between neighboring π-electronic systems, which is reflected in the UV-vis absorption spectra, and electrochemical behavior of the dyads. | ||
| Fig. 4 Optimized structure of porphyrin dyads (a) 5a and (b) 5b. Carbon, nitrogen, hydrogen, oxygen and zinc atoms correspond to grey, blue, white, red and green spheres, respectively. | ||
Using the optimized structures of 5a and 5b, for each one of the PBE, PBE0, and B3LYP functionals, the corresponding optical gaps have been calculated, which are listed in Table 3, together with the HOMO and LUMO energies, the HOMO–LUMO gap, the optical gap Ecalc.opt as well as the main contributions to the first (allowed) excitation. A comparative study31 between hybrid functionals with and without long-range corrections for calculating the visible absorption spectra showed that the PBE0 functional computed excitation energies with the smallest average absolute deviation when linear calibrations to the values were not applied. Wong also reports32 on the good performance of the PBE0. We performed excited state calculations using all three functionals, which can additionally be used for comparison with the literature. The hybrid functionals overestimate only slightly the optical gaps for both structures by approximately 0.3 eV.
| HOMO (eV) | LUMO (eV) | HL (eV) | E calc.g (eV) | f | Assignment | μ (D) | |
|---|---|---|---|---|---|---|---|
| 5a | |||||||
| PBE | −4.78 | −3.01 | 1.77 | 1.77 (2.07) | 0.0005 (0.18) | H → L 99.7% | 4.67 |
| (H−1 → L 64.7%; H−3 → L+1 21.7%; | |||||||
| H → L+2 5.2%) | |||||||
| PBE0 | −5.42 | −2.41 | 3.00 | 2.33 | 0.051 | H → L+2 54.3%; H−1 → L+3 35.0% | 4.57 |
| H−1 → L 5.2%; H−2 → L+1 3.2% | |||||||
| B3LYP | −5.15 | −2.43 | 2.71 | 2.29 | 0.039 | H−1 → L 29.4%; H → L+2 27.1% | 4.51 |
| H−2 → L+1 20.2%; H−1 → L+3 18.3% | |||||||
| 5b | |||||||
| PBE | −4.74 | −2.99 | 1.75 | 1.75 (2.05) | 6.68 × 10−4 (0.17) | H → L 99.6% | 5.51 |
| (H−1 → L 61.4%; H−3 → L+1 17.4%; | |||||||
| H−2 → L 13.2%) | |||||||
| PBE0 | −5.39 | −2.40 | 2.99 | 2.33 | 5.81 × 10−2 | H → L+3 37.0%; H−2 → L+2 23.6% | 5.13 |
| H−1 → L 15.8%; H−3 → L+1 10.0% | |||||||
| H → L+2 7.6% | |||||||
| B3LYP | −5.18 | −2.40 | 2.78 | 2.29 | 4.10 × 10−2 | H−1 → L 36.4%; H−3 → L+1 24.7% | 5.17 |
| H → L+3 17.1%; H−2 → L+2 11.3% | |||||||
| H → L+2 5.7% | |||||||
In Table 3, the characteristics of the first allowed excitations are also provided. Only configurations that contribute more than 4% have been included. In the case of the PBE functional for both structures we additionally provide the values (given in parentheses) that correspond to the first excitation with significantly higher larger oscillator strength. For both structures we find a clear multi-configurational characteristic for the first excitation by all of the functionals. All of the functionals produce contributions from the same – near frontier – orbitals, however with some variation in the percentages. The contributions include transitions between the HOMO down to HOMO−3 and LUMO up to LUMO+3. The near HOMOs and near LUMOs are almost isoenergetic to the HOMO and LUMO with maximum energy differences about 0.22 and 0.04 eV for the near HOMOs and near LUMOs, respectively. The only noticeable effect on the optical gap from the substitution of the glycine moiety by piperidine is a significant increase in the contributions to the first excitation of transitions from the energetically (slightly) lower HOMO−3.
In contrast to the previous properties, a moderate increase in the dipole moment of the 5a dyad is noted upon substitution of the glycine by the piperidine moiety. The increase ranges from 12.2 to 18.0% with the lowest and highest increases computed using the PBE and PBE0 functionals respectively.
In Fig. 5a and 5b we have plotted the iso-surfaces (iso-value = 0.02) of the frontier molecular orbitals (FMOs) for porphyrin dyads 5a and 5b. All of the MOs that appear in the configuration assignments of Table 3 are shown, namely from HOMO down to HOMO−3 and LUMO up to LUMO+3 calculated using the B3LYP functional. All FMOs are highly localized on one (or the other) of the porphyrin units. One noticeable difference between the dyads is that in the case of 5b all of the contributing transitions to the first excitation are between orbitals localized on the same porphyrin of the dyad. However, in the case of 5a there are also significant contributions from transitions between orbitals localized on different porphyrins, specifically the H−2 → L+1 and H−1 → L+3 transitions. Since both have almost the same percentage no charge transfer is to be expected. To quantify the contributions of the moieties to the FMOs we have calculated the total and partial density of states (PDOS). The PDOS for the 5a and 5b dyads are shown in Fig. S1 (ESI†). We further partition the remaining atoms of the structure to the 2,6-diamine-1,3,5-triazine and the glycine or piperidine groups. In both structures the near frontier orbitals have 50% contributions from each of the porphyrin units, as expected by well separated localizations of the isosurfaces of Fig. 1 and 2 and the corresponding (similar) orbital energies. The triazine contributions are first noted at lower energies around −6.1 eV. The most noticeable difference is that the glycine moiety first contributions to states are at −7 eV and the piperidine at about −6.4 eV. However, the percentage of the piperidine contribution to these energetically lower states is much higher, about 19.3%, in comparison to 6% of glycine. Further contributions of piperidine are noted only at much lower energies of −9.2 eV and below. In both structures the second virtual state peak, which is about 0.7 eV higher than the LUMO peak and is well separated from other energetically either higher or lower states, is dominated by contributions from Zn[Porph2].
 | ||
| Fig. 5 Frontier and near frontier orbitals of the (a) 5a and (b) 5b porphyrin dyads. | ||
FTIR spectroscopy
Both porphyrin dyads 5a and 5b contain carboxylic acid groups which are known to be very efficient anchoring groups for attachment of dyes to the TiO2 surface of the DSSC electrodes. Additionally, the latter contains a piperidine binding site that could also act as an anchoring group to the metal oxide surface through its basic sp3 hybridized N atom. In order to get information about the binding, FTIR powder spectra were recorded in pure form and adsorbed on TiO2 films. The FTIR spectra of 5b and 5b/TiO2 films are shown in Fig. 6, while the corresponding spectra of 5a and 5a/TiO2 films are depicted in Fig. S19 (ESI†). | ||
| Fig. 6 FTIR spectra of (a) 5b (black color line) and (b) 5b adsorbed onto TiO2 film (red color line). | ||
The spectra of both porphyrin dyads 5a and 5b in pure form exhibit an absorption band at 1694 cm−1, which corresponds to the ν(COO) stretching vibration of the carboxylic acid group.33 In the corresponding spectra of the dyads after being adsorbed onto TiO2 surfaces, the band that corresponds to the ν(COO) stretching vibration shifts to lower frequencies at 1406 and 1524 cm−1, which correspond to the νsym (COO−) and νasymm (COO−) stretching vibrations, respectively. This is an indication that both dyads 5a and 5b are strongly bound to the TiO2 surface through their carboxylic acid groups. Particularly, in the case of dyad 5a, both types of carboxylic acid groups (on the phenyl group at the periphery and on the glycine group) are used for binding to the TiO2 surface.
Furthermore, in the IR spectrum of porphyrin dyad 5b in pure form (Fig. 6b, black color line), there are two absorption bands at 1626 and 3314 cm−1 (not shown in spectrum) which do not appear in the IR spectrum of 5a. These bands can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and N–H stretchings of the piperdine ring, respectively. In the corresponding IR spectrum of 5b/TiO2 film, these bands appear to be shifted. These observations suggest that in porphyrin dyad 5b, in addition to the carboxylic acid group, the piperidine group might also be involved in binding to the TiO2 surface through the nitrogen atom.
N and N–H stretchings of the piperdine ring, respectively. In the corresponding IR spectrum of 5b/TiO2 film, these bands appear to be shifted. These observations suggest that in porphyrin dyad 5b, in addition to the carboxylic acid group, the piperidine group might also be involved in binding to the TiO2 surface through the nitrogen atom.
Photovoltaic properties
The 5a and 5b based DSSCs (devices A and B, respectively) have been fabricated and their photovoltaic cell parameters, i.e. open circuit voltage (Voc), short circuit photocurrent (Jsc), fill factor (FF), and power conversion efficiency (PCE), are summarized in Table 4, while the corresponding current–voltage characteristics (J–V curves) are depicted in Fig. 7a. Device A, based on porphyrin dyad 5a, exhibits Voc = 0.68 V, Jsc = 10.78 mA cm−2, and FF = 0.72, resulting in an overall PCE value of 5.28%. In constrast, device B, based on dyad 5b, exhibits Voc = 0.62, Jsc = 8.56 mA cm−2 and FF = 0.66, giving a lower overall PCE value of 3.50%. In other words, the DSSC based on 5a, a porphyrin dyad with two carboxylic acid anchoring groups, results in higher Jsc and Voc, and PCE values than the DSSC based on 5b, a porphyrin dyad with one carboxylic acid and a piperidine anchoring group. | ||
| Fig. 7 (a) Current–voltage (J−V) characteristics, and (b) IPCE spectra of DSSCs based on 5a (device A, red color lines) and 5b (device B, black color lines). | ||
| DSSC sensitized by |
J
sca (mA cm−2) |
V
ocb (V) |
J
maxa (mA cm−2) |
V
maxb (V) |
FFc | PCEd (%) | Dye loading (10−7 mol cm−2) |
|---|---|---|---|---|---|---|---|
| a Short circuit current. b Open circuit voltage. c Fill factor. d Power conversion efficiency. | |||||||
| 5a (device A) | 10.78 | 0.68 | 8.24 | 0.64 | 0.72 | 5.28 | 3.45 |
| 5b (device B) | 8.56 | 0.62 | 5.93 | 0.59 | 0.63 | 3.50 | 2.12 |
One of the factors that are responsible for the higher PCE value of the 5a sensitized solar cell is its higher Jsc value. Since Jsc of a solar cell depends on its IPCE response, the difference between the Jsc values of the two solar cells is reflected on their IPCE responses. The IPCE spectra of the two cells, presented in Fig. 7b, closely resemble the absorption spectra of the dyes adsorbed on the TiO2 film, exhibiting characteristic peaks in the 400–470 nm, 540–580 nm, and 600–740 nm ranges. The higher IPCE response of the DSSC sensitized by 5a is consistent with its higher Jsc value.
In general, the IPCE of a DSSC is a combination of its light harvesting efficiency LHE (which depends on the absorption profile of the sensitizing dye and the amount of dye adsorbed on the TiO2 surface), the electron injection efficiency ηinj (which depends on the electron injection rate from the excited sensitizer into the TiO2 CB), and the charge collection efficiency ηcc (which depends on the electron diffusion rate in TiO2 towards the external circuit, and the recombination rate between the injected electrons in TiO2 and the redox electrolyte),8,34 and it is expressed as
| IPCE = LHE × ηinj × ηcc |
The amount of dye adsorbed (or dye loading) onto the TiO2 surface of a solar cell is of great importance, as it affects LHE and consequently the IPCE response.35 For the 5a and 5b sensitized solar cells, the dye loading values were calculated by immersing each one of the dye-sensitized TiO2 electrodes into a mixture of CH2Cl2 and NaOH(aq) solutions, and measuring the absorption spectra of de-adsorbed dyes. These were found to be 3.45 × 10−7 and 2.12 × 10−7 mol cm−2, respectively (Table 4). The higher dye loading value of the 5a based cell may be attributed to the more effective binding of dyad 5a on the TiO2 surface due to the presence of two carboxylic acid anchoring groups, while 5b, which contains one carboxylic acid anchoring group and a piperidine binding site, has a less effective binding capacity.
From the electrochemistry data of porphyrin dyads 5a and 5b presented in Table 2, it was calculated that ΔGinj for the 5a based DSSC is similar to that of the 5b based solar cell. This means that in both dyads there is the same driving force available for the electron injection from their excited states into the TiO2 CB. Moreover, ΔGreg was calculated to be larger for the DSSC based on 5b, contributing to a faster dye regeneration process relative to 5a.
In order to gain a better understanding of the relationship between the observed photovoltaic properties and the related charge transfer processes in devices A and B their electrochemical impedance spectra (EIS) were recorded using a forward bias of 0.65 V. In the Nyquist plot of EIS of the 5b based solar cell (Fig. 8a, black color line), there are three semicircles clearly seen. The semicircle in the left side of the spectrum (high frequency region) is associated with the charge transfer resistance at the platinum counter electrode–electrolyte interface, the larger semicircle in the middle frequency region is related to the charge recombination resistance at the TiO2/dye/electrolyte interface, while the semicircle on the left side of the spectrum is associated with the Nernst diffusion resistance of the redox couple in the electrolyte.36 However, in the Nyquist plot of the 5a based solar cell (Fig. 8a, red color line) there are only two semicircles present, since the semicircle due to Nernst diffusion within the electrolyte overlaps with the middle frequency large semicircle, which is an indication of fast electron transport and long lifetime in the TiO2 film. In addition, the semicircle radius in the middle frequency range is larger than that of the 5b based solar cell, indicating that the charge recombination is suppressed in the device sensitized by 5a.
 | ||
| Fig. 8 (a) Nyquist plots and (b) Bode phase plots of electrochemical impedance spectra of DSSCs based on 5a (device A, red color lines) and 5b (device B, black color lines). | ||
The middle frequency peaks in the Bode phase plots of the EIS of devices A and B (Fig. 8b) are related to the corresponding electron lifetimes (τe). The higher frequency peak for the 5b based DSSC is indicative of a shorter electron lifetime.37 More specifically, the electron lifetimes (τe) for the 5a and 5b sensitized devices were calculated according to the relation

The reason for the different photovoltaic performances of the DSSCs based on the porphyrin dyads 5a and 5b can be associated with the molecular structures of the corresponding sensitizers, and more specifically with the anchoring groups used for attachment to the TiO2 film. As was previously mentioned, porphyrin dyad 5a is attached to the TiO2 surface through two carboxylic acid groups. Hence, its binding can be described by the branched multi-porphyrin system bound to TiO2 through two carboxylic acid groups (red color lines) as shown in Scheme 3a. Dyad 5b is claimed to be bound to the TiO2 surface through a carboxylic acid and a piperidine group. Considering that the donor atom on the piperidine group of 5b is an N-substituted nitrogen atom (directly linked to the triazine ring) and that the position of the nitrogen lone pair is very sterically hindered to be exposed to the acid sites of the TiO2 surface (as revealed by the geometry optimized structure of 5b), its binding onto the TiO2 surface through the piperidine group is not easy to happen. However, taking into account that the TiO2 film surface is not ideally flat, but very rough with pores and cavities of very large dimensions, adsorption of 5b through piperidine attachment is possible to happen, as is also suggested by FTIR spectroscopy. Consequently, the attachment of porphyrin dyad 5b onto the TiO2 surface could be described either by the linear multi-porphyrin system bound through one carboxylic acid group in Scheme 3b(i) or by a branched multi-porphyrin system bound through one carboxylic acid (red color line) and a piperidine group (blue color line), as shown in Scheme 3b(ii).
 | ||
| Scheme 3 | ||
One of the factors that affect the efficiency of multi-porphyrin systems in DSSCs is their capability to inject electrons efficiently into the TiO2 CB, which is related to the type of anchoring group. In general, carboxylic acids as anchoring groups result in very efficient DSSCs, compared to other anchoring groups (e.g. phosphonic acid groups, nitrogen donor atom groups).38,39 This has been ascribed to the fact that carboxylic acids ensure efficient adsorption of sensitizers onto metal oxide surfaces and support an excellent electronic coupling between the levels of the excited sensitizer and those of the metal oxide (by means of a variety of binding modes40–42). The presence of two effectively bound carboxylic acid groups in 5a, in contrast to only one carboxylic acid group and a less effectively bound piperidine group in 5b, results in the formation of a more compact layer of dye 5a on the TiO2 film (which is evident by the increased dye loading), and a cooperative effect in the electron transfer towards the TiO2 surface, leading to a faster and more efficient electron injection process (which is manifested by a longer electron lifetime and higher charge recombination resistance).
Energy related factors, associated with the transfer of the absorbed solar energy from the porphyrin units into the TiO2 CB, might also be responsible for the different efficiencies of the solar cells based on 5a and 5b. Each transfer between the porphyrin units (before electron injection) is associated with a minimum energy requirement ΔEi (i = 1, 2). Assuming that the triazine group mediates effectively intra-molecular energy and electron transfer between the porphyrin units, and that dyad 5b could be better approximated by the linear multi-porphyrin system described in Scheme 3b(i), the electron injection into the TiO2 CB in the multi-porphyrin system in Scheme 3a would be less energetically demanding than in Scheme 3b(i), and more efficient, due to the presence of two effectively bound carboxylic acid groups, leading to a better overall photovoltaic performance.
Conclusions
By utilizing the stepwise and temperature dependent reactivity of cyanuric chloride, two new porphyrin dyads have been synthesized, which are bridged by a triazine group through their peripheral aryl-amino groups. One terminal carboxyphenyl group functionalizes both dyads, while 5a contains a carboxylic acid of a glycine moiety attached to the triazine group and 5b contains a piperidine group. The study of photophysical and electrochemical properties of dyads suggests that there is no significant intramolecular electronic communication between the porphyrin units in their ground states. Dyads 5a and 5b have been used as sensitizers for the fabrication of DSSCs, which resulted in PCE values of 5.28 and 3.50%, respectively. The higher PCE value of the 5a sensitized solar cell is attributed to its higher short circuit current (Jsc), open circuit voltage (Voc), and fill factor (FF) values, as well as to a larger dye loading. These could be due to more effective binding of 5a to the TiO2 surface of the electrode, ascribed to the presence of two carboxylic acid anchoring groups (compared to one carboxylic acid anchoring group and one piperidine binding site of 5b). As a result, a longer electron lifetime (τe) and larger recombination resistance are observed for the former solar cell. Further improvement of the PCE values of the DSSCs reported herein may be achieved by using a modified photoanode, and we are currently working in this direction.Experimental
General methods, materials, and techniques
All manipulations were carried out using standard Schlenk techniques under nitrogen atmosphere. 2,4,6-Trichloro-1,3,5-triazine (cyanuric chloride), diisopropylethylamine (DIPEA), glycine methyl ester hydrochloride, Zn(CH3COO)2·2H2O, Na2SO4, KOH and other chemicals and solvents were purchased from usual commercial sources and used as received, unless otherwise stated. Tetrahydrofuran (THF) was freshly distilled from Na/benzophenone. 5-(4-Aminophenyl)-10,15,20-triphenyl-porphyrin (H2[TPPNH2]) and 5-(4-carbomethoxyphenyl)-15-(4-aminophenyl)-10,20-bis(2,4,6-trimethylphenyl)-porphyrin (H2[Porph]) were prepared according to literature procedures.25,43Metallation. To a CH2Cl2 solution (12 mL) of 3a (40 mg, 0.026 mmol) a saturated solution (4 mL) of Zn(CH3COO)2·2H2O in MeOH (500 mg per 20 mL) was added and the reaction mixture was stirred at room temperature overnight. The organic phase was washed with H2O (3 × 10 mL), dried with Na2SO4 and the solvent was removed under reduced pressure. The crude product 4a was purified by column chromatography (silica gel, CH2Cl2–EtOH 4%), resulting in 39 mg of a purple solid (yield: 90%). 1H NMR (300 MHz, CDCl3): δ (ppm) 8.98 (m, 11H), 8.79 (m, 5H), 8.37 (d, J = 8.1 Hz, 2H), 8.29 (d, J = 8.1 Hz, 2H), 8.18 (s br, 9H), 7.94 (d, J = 7.8 Hz, 4H), 7.70 (m, 10H), 7.22 (s, 4H), 4.16 (d, J = 5.1 Hz, 2H), 4.06 (s, 3H), 3.70 (s, 3H), 2.58 (s, 6H), 1.78 (s, 12H). HRMS (MALDI-TOF): m/z calcd for C102H78N14O4Zn2: 1690.4757 [M + 2H]+, found 1690.4746. UV-vis (CH2Cl2), λ/nm (ε × 10−3 M−1 cm−1): 422 (771.2), 550 (33.4), 591 (11.0). Anal. calcd for C102H76N14O4Zn2: C, 72.38; H, 4.53; N, 11.59. Found: C, 72.44; H, 4.62; N, 11.41.
Ester hydrolysis. To a THF solution (22 mL) of 4a (0.030 g, 0.018 mmol), 6 mL MeOH, 7.5 mL H2O and KOH (0.450 g, 0.008 mol) were added. After stirring the reaction mixture at room temperature overnight, the organic solvents were removed under reduced pressure and then a solution of HCl (0.5 M) was added dropwise, until pH ∼ 6. The precipitate was filtered, washed with water, extracted with CH2Cl2 and purified by column chromatography (silica gel, CH2Cl2–EtOH 10%). The product 5a was isolated as a purple solid. Yield: 0.026 g (87%). 1H NMR (300 MHz, CDCl3–MeOD: 1/0.01): δ (ppm) 8.90 (d, J = 4.5 Hz, 3H), 8.79 (m, 8H), 8.67 (s br, 2H), 8.58 (s br, 4H), 8.24 (m, 3H), 8.15 (m, 15H), 7.62 (m, 10H), 7.23 (s, 4H), 7.11 (s br, 3H), 6.52 (s, 1H), 3.68 (s br, 2H), 2.50 (s, 6H), 1.67 (s, 12H); HRMS (MALDI-TOF): m/z calcd for C100H74N14O4Zn2: 1662.4444 [M + 2H]+, found 1662.4450 UV-vis (THF), λ/nm (ε × 10−3 M−1 cm−1): 425 (667.3), 557 (28.2), 597 (11.6). Anal. calcd for C100H72N14O4Zn2: C, 76.85; H, 4.45; N, 11.36. Found: C, 76.77; H, 4.56; N, 11.42.
Metallation. To a CH2Cl2 solution (12 mL) of 3b (40 mg, 0.026 mmol), a saturated solution (4 mL) of Zn(CH3COO)2·2H2O in MeOH (500 mg per 20 mL) was added and the reaction mixture was stirred at room temperature overnight. The organic phase was washed with H2O (3 × 10 mL), dried with Na2SO4 and the solvent was removed under reduced pressure. The crude product 4b was purified by column chromatography (silica gel, CH2Cl2–EtOH 4%) resulting in 35 mg of a purple solid (yield: 80%). 1H NMR (300 MHz, CDCl3): δ (ppm) 9.07 (d, J = 4.5 Hz, 2H), 9.01 (d, J = 4.8 Hz, 2H), 8.93 (s, 7H), 8.79 (m, 5H), 8.40 (d, J = 8.1 Hz, 2H), 8.30 (d, J = 7.8 Hz, 2H), 8.21 (m, 11H), 8.04 (m, 4H), 7.73 (m, 11H), 7.24 (s, 2H), 7.09 (s, 2H), 4.08 (s, 3H), 3.94 (s br, 10H), 2.60 (s, 6H), 1.79 (s, 12H). 13C NMR (75 MHz, CDCl3): δ (ppm) 167.6, 165.2, 164.7, 156.3, 150.6, 150.4, 149.7, 143.0, 139.4, 139.1, 137.6, 137.2, 135.1, 134.6, 132.8, 132.1, 131.2, 129.4, 127.8, 127.6, 126.7, 121.2, 119.6, 118.1, 44.9, 26.1, 25.0, 21.7. HRMS (MALDI-TOF): m/z calcd for C104H82N14O2Zn2: 1686.5172 [M + 2H]+, found 1686.5164. UV-vis (CH2Cl2), λ/nm (ε × 10−3 M−1 cm−1): 422 (769.9), 550 (30.8), 591 (11.1). Anal. calcd for C104H80N14O2Zn2: C, 73.97; H, 4.78; N, 11.61. Found: C, 73.77; H, 4.86; N, 11.57.
Ester hydrolysis. To a THF solution (22 mL) of 4b (0.030 g, 0.018 mmol), 6 mL MeOH, 7.5 mL H2O and KOH (0.450 g, 0.008 mol) were added. After stirring the reaction mixture at room temperature overnight, the organic solvents were removed under reduced pressure and then a solution of HCl (0.5 M) was added dropwise, until pH ∼ 6. The precipitate was filtered, washed with water, extracted with CH2Cl2 and purified by column chromatography (silica gel, CH2Cl2–EtOH 5%). The product 5b was isolated as a purple solid. Yield: 0.025 g (83%). 1H NMR (300 MHz, CDCl3–MeOD: 1/0.01): δ (ppm) 8.77 (m, 16H), 8.20 (m, 14H), 8.01 (s, 4H), 7.64 (s, 11H), 7.16 (s, 4H), 6.90 (s, 2H), 6.49 (s, 3H), 3.90 (s br, 10H), 2.53 (s, 6H), 1.73 (s, 12H); 13C NMR (75 MHz, CDCl3–MeOD: 1/0.01): δ (ppm) 164.5, 149.6, 149.5, 149.2, 149.0, 148.8, 148.7, 145.7, 142.7, 140.3, 139.1, 138.9, 138.6, 138.4, 136.8, 135.7, 134.4, 134.2, 132.6, 132.3, 131.9, 131.5, 130.3, 130.0, 127.5, 126.6, 124.9, 124.4, 123.9, 120.2, 120.1, 119.9, 118.0, 65.9, 34.4, 34.0, 30.4, 29.2, 25.7, 25.5, 24.3, 24.1, 21.4, 21.3, 21.0. HRMS (MALDI-TOF): m/z calcd for C103H80N14O2Zn2 1672.5015 [M + 2H]+, found 1672.5023. UV-vis (THF), λ/nm (ε × 10−3 M−1 cm−1): 425 (666.7), 557 (27.8), 597 (11.4). Anal. calcd for C103H78N14O2Zn2: C, 73.88; H, 4.69; N, 11.71. Found: C, 73.76; H, 4.76; N, 11.64.
NMR spectra
1H and 13C NMR spectra were recorded on a Bruker DPX-300 MHz and a Bruker DPX-75 MHz spectrometer, respectively, as solutions in deuterated solvents by using the solvent peak as the internal standard.Mass spectra
High-resolution mass spectra were recorded on a Bruker UltrafleXtreme MALDI-TOF/TOF spectrometer.FTIR spectra
FTIR were recorded on a Perkin Elmer 16PC FTIR spectrometer.Photophysical measurements
UV-vis absorption spectra were measured on a Shimadzu UV-1700 spectrophotometer using 10 mm path-length cuvettes. Emission spectra were measured on a JASCO FP-6500 fluorescence spectrophotometer equipped with a red sensitive WRE-343 photomultiplier tube (wavelength range: 200–850 nm).Electrochemistry measurements
Cyclic and square wave voltammetry experiments were carried out at room temperature using an AutoLab PGSTAT20 potentiostat and appropriate routines available in the operating software (GPES, version 4.9). Measurements were carried out in freshly distilled and deoxygenated THF, with a scan rate of 100 mV s−1, with a solute concentration of 1.0 mM in the presence of tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte. A three-electrode cell setup was used with a platinum working electrode, a saturated calomel (SCE) reference electrode, and a platinum wire as a counter electrode. The system was calibrated by ferrocene.Computational methods
Theoretical calculations were performed within the framework of density functional theory (DFT). Initial gas-phase geometry optimizations were performed employing the gradient corrected functional PBE44 of Perdew, Burke and Ernzerhof in combination with the TZVP basis set,45 which is of triple-ζ quality. For each structure several rotamers were used as starting geometries. To increase the computational efficiency (without loss in accuracy) using the PBE functional, the resolution of the identity method46 was used for the treatment of the two-electron integrals. The resulting structures were further optimized using both the hybrid exchange-correlation functional PBE047 (without adjustable parameters) of Perdew, Burke and Ernzerhof, as well as the hybrid functional of Becke, Lee, Yang and Parr, B3LYP,48 using the same basis set. Tight convergence criteria were placed for the SCF energy (up to 10−7 Eh) and the one-electron density (rms of the density matrix up to 10−8) as well as for the norm of the Cartesian gradient (residual forces both average and maximum smaller than 1.5 × 10−5 a.u.) and residual displacements (both average and maximum smaller than 6 × 10−5 a.u.). Time-dependent density functional theory (TD-DFT) was used for excited state calculations, using the aforementioned functionals and basis set on the corresponding ground state structures. In each case the optical gap was calculated. The first round of geometry optimizations was performed using the Turbomole package,49 while all of the follow up calculations were performed using the Gaussian package.50Preparation of TiO2 electrodes and DSCCs
The DSSCs were fabricated with electrodes based on fluorine-doped tin oxide (FTO) glass substrates, which were pre-cleaned by sonication in decon 90, distilled water, iso-propanol (i-PrOH), ethanol (EtOH), and dried in air. The working electrodes were prepared by firstly forming a blocking layer of 0.2 M titanium di-isopropoxide bis(acetylacetonate) in i-PrOH by spray pyrolysis on a pre-cleaned FTO coated glass substrate. This was followed by deposition of a nano-crystalline layer of TiO2 by the doctor blade technique, using a dye sol TiO2 paste (DSL 18NR-T). The TiO2 coated FTO electrodes were heated at 500 °C for 30 min. After cooling to room temperature, they were immersed into a 0.02 M TiCl4 aqueous solution for 20 minutes, washed with distilled water and ethanol, and annealed again at 500 °C for 20 min. The thickness of the TiO2 electrodes was measured using a thin film thickness measurement system (Nano calc XR Ocean Optics, Germany) and they were found to be in the range of 10–12 μm. Finally, they were immersed into the corresponding solutions of 5a and 5b (5 × 10−4 M in CHCl3–EtOH = 1/1) for 12 h and washed with THF to give the dye-sensitized TiO2 working electrodes. The counter electrode was prepared by spin coating of a H2PtCl4 solution (0.002 g in 1 mL i-PrOH) onto a pre-cleaned FTO coated glass substrate and then heating at 450 °C for 15 min in air. The sensitized working electrode was assembled with the Pt coated FTO electrode into a sandwich type cell and sealed with the hot-melt polymer surlyn. To complete the DSSC fabrication, the electrolyte solution containing LiI (0.05 M), I2 (0.03 M), 1 methyl-3-n-propylimidazolium iodide (0.6 M) and tert-butylpyridine (0.5 M in a mixture of acetonitrile and valeronitrile, 85:15 volume ratio) was introduced into the space between the two electrodes through a drilled hole in the platinum coated FTO by vacuum backfilling.
Photovoltaic measurements
The current–voltage (J–V) characteristics of the DSSCs under simulated air mass 1.5 global, and illumination intensity of 100 mW cm−2 were measured using a computer controlled Keithley source meter and illuminated using the solar simulator TS space system class AAA. Incident photon to current efficiency (IPCE) spectra were measured using a Bentham IPCE system (TMc 300 monochromator computer controlled). Electrochemical impedance spectra (EIS) in the dark were recorded using a CHN electrochemical workstation. They were measured by applying a dc bias equivalent to the open circuit voltage of DSSC in the frequency range 0.1 to 100 kHz.Acknowledgements
Financial support from the European Commission (FP7-REGPOT-2008-1, Project BIOSOLENUTI No. 229927) is greatly acknowledged. This research has also been co-financed by the European Union (European Social Fund) and Greek national funds (Heraklitos II) through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework Research Funding Program (THALIS-UOA-MIS 377252). We are also thankful to Petra J. Cameron, Department of Chemistry, University of Bath, Bath, BA2 7AY, United Kingdom, for allowing fabrication and characterization of the DSSCs. Finally COST Action I, CM1202 is also acknowledged.References
- (a) N. Robertson, Angew. Chem., Int. Ed., 2008, 47, 1012–1014 CrossRef CAS PubMed; (b) R. Eisenberg and D. G. Nocera, Inorg. Chem., 2005, 44, 6799 CrossRef CAS PubMed; (c) N. Oreskes, Science, 2004, 306, 1686 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- (a) A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed; (b) K. L. Wu, S. T. Ho, C. C. Chou, Y. C. Chang, H.-A. Pan, Y. Chi and P. T. Chou, Angew. Chem., Int. Ed., 2012, 51, 5642 CrossRef CAS PubMed.
- (a) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed; (b) M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613 CrossRef CAS PubMed; (c) J. Xu, H. Wu, X. Jia and D. C. Zou, Chem. Commun., 2012, 48, 7793 RSC.
- K. Kalayansundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347 CrossRef.
- (a) L. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057 RSC; (b) F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720 CrossRef CAS PubMed; (c) Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Cao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CrossRef CAS.
- (a) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed; (b) S. P. Singh, M. S. Roy, K. R. J. Thomas, S. Balaiah, K. Bhanuprakash and G. D. Sharma, J. Phys. Chem. C, 2012, 116, 5941 CrossRef CAS; (c) S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291 CrossRef CAS; (d) B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220 CrossRef CAS PubMed; (e) C. Qin, W.-Y. Wong and L. Han, Chem.–Asian J., 2013, 8, 1706 CrossRef CAS PubMed; (f) S. Chang, H. Wang, Y. Hua, Q. Li, X. Xiao, W.-K. Wong, W. Y. Wong, X. Zhu and T. Chen, J. Mater. Chem. A, 2013, 1, 11553 RSC; (g) Y. Hua, S. Chang, D. Huang, X. Zhou, X. Zhu, J. Zhao, T. Chen, W.-Y. Wong and W.-K. Wong, Chem. Mater., 2013, 25, 2146 CrossRef CAS.
- (a) L. Li and E. W. G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC; (b) H. Zhan, S. Lamare, A. Ng, T. Kenny, H. Guernon, W.-K. Chan, A. B. Djurišić, P. D. Harvey and W.-Y. Wong, Macromolecules, 2011, 44, 5155 CrossRef CAS; (c) W.-K. Wong, X. Zhua and W.-Y. Wong, Coord. Chem. Rev., 2007, 251, 2386 CrossRef CAS PubMed; (d) X. Zhu, W.-K. Wong, W.-Y. Wong and X. Yang, Eur. J. Inorg. Chem., 2011, 30, 4651 CrossRef.
- (a) H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809 CrossRef CAS PubMed; (b) I. Radivojevic, A. Varotto, C. Farley and C. M. Drain, Energy Environ. Sci., 2010, 3, 1897 RSC; (c) M. V. Martínez-Díaz, G. de la Torrea and T. Torres, Chem. Commun., 2010, 46, 7090 RSC; (d) M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759 CrossRef CAS; (e) M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145 RSC; (f) H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032 RSC; (g) T. Bessho, S. M. Zakeeruddin, C. Y. Yeh, E. W. G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646 CrossRef CAS PubMed; (h) H. He, A. Gurung and L. Si, Chem. Commun., 2012, 48, 5910 RSC.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- P. J. Angiolillo, V. S.-Y. Lin, J. M. Vanderkooi and M. J. Therien, J. Am. Chem. Soc., 1995, 117, 12514 CrossRef CAS.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. C. Du, R. Katoh, A. Furube and D. L. Officer, J. Am. Chem. Soc., 2009, 131, 15621 CrossRef CAS PubMed.
- (a) J. K. Park, J. P. Chen, H. R. Lee, S. W. Park, H. Shinokubo, A. Osuka and D. Kim, J. Phys. Chem. C, 2009, 113, 21956 CrossRef CAS; (b) A. B. F. Martinson, T. W. Hamann, M. J. Pellin and J. T. Hupp, Chem.–Eur. J., 2008, 14, 4458 CrossRef CAS PubMed; (c) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed.
- Y. Liu, H. Lin, J. T. Dy, K. Tamaki, J. Nakazaki, D. Nakayama, S. Uchida, T. Kubo and H. Segawa, Chem. Commun., 2011, 47, 4010 RSC.
- (a) H. Zhong, E. Xu, D. Zeng, J. Du, J. Sun, S. Ren, B. Jiang and Q. Fang, Org. Lett., 2008, 10, 709 CrossRef CAS PubMed; (b) J. Pang, Y. Tao, S. Freiberg, X.-P. Yang, M. D'Iorio and S. Wang, J. Mater. Chem., 2002, 12, 206 RSC; (c) J.-W. Kang, D.-S. Lee, H.-D. Park, Y.-S. Park, J.-W. Kim, W.-I. Jeong, K.-M. Yoo, K. Go, S.-H. Kim and J.-J. Kim, J. Mater. Chem., 2007, 17, 3714 RSC; (d) A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- C. A. M. Afonso, N. M. T. Lourenc
![[o with combining cedilla]](https://www.rsc.org/images/entities/char_006f_0327.gif) and A. A. Rosatella, Molecules, 2006, 11, 81 CrossRef CAS PubMed.
and A. A. Rosatella, Molecules, 2006, 11, 81 CrossRef CAS PubMed. - (a) S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian and Q. J. Fang, Phys. Chem. B, 2010, 114, 10374 CrossRef CAS PubMed; (b) H. Zhong, H. Lai and Q. Fang, J. Phys. Chem. C, 2011, 115, 2423 CrossRef CAS.
- A. Luechai, J. Gasiorowski, A. Petsom, H. Neugebauer, N. S. Sariciftci and P. Thamyongkit, J. Mater. Chem., 2012, 22, 23030 RSC.
- J. Liu, K. Wang, X. Zhnag, C. Li and X. You, Tetrahedron, 2013, 69, 190 CrossRef CAS PubMed.
- G. E. Zervaki, M. S. Roy, M. K. Panda, P. A. Angaridis, E. Chrissos, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2013, 52, 9813 CrossRef CAS PubMed.
- D. W. P. M. Löwik and C. R. Lowe, Eur. J. Org. Chem., 2001, 2825 CrossRef.
- (a) M. B. Steffensen and E. Simanek, Org. Lett., 2003, 5, 2359 CrossRef CAS PubMed; (b) W. Zhang and E. E. Simanek, Org. Lett., 2000, 2, 843 CrossRef CAS PubMed.
- (a) T. Carofiglio, A. Varotto and U. Tonellato, J. Org. Chem., 2004, 69, 8121 CrossRef CAS PubMed; (b) K. Ichihara and Y. Naruta, Chem. Lett., 1995, 631 CrossRef CAS; (c) T. Carofiglio, E. Lubian, I. Menegazzo, G. Saielli and A. Varotto, J. Org. Chem., 2009, 74, 9034 CrossRef CAS PubMed.
- T. Lazarides, G. Charalambidis, A. Vuillamy, M. Reglier, E. Klontzas, G. Froudakis, S. Kuhri, D. M. Guldi and A. G. Coutsolelos, Inorg. Chem., 2011, 50, 8926 CrossRef CAS PubMed.
- G. E. Zervaki, E. Papastamatakis, P. A. Angaridis, M. Singh, R. Kurchania, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2014 DOI:10.1002/ejic.201301278.
- (a) D. Kim and A. Osuka, Acc. Chem. Res., 2004, 37, 735 CrossRef CAS PubMed; (b) N. Aratani, A. Osuka, Y. H. Kim, D. H. Jeong and D. Kim, Angew. Chem., Int. Ed., 2000, 39, 1458 CrossRef CAS; (c) T. K. Ahn, Z. S. Yoon, I.-W. Hwang, J. K. Lim, H. Rhee, T. Joo, E. Sim, S. K. Kim, N. Aratani, A. Osuka and D. Kim, J. Phys. Chem. B, 2005, 109, 11223 CrossRef CAS PubMed.
- (a) C.-F. Lo, L. Luo, E. W.-G. Diau, I. J. Chang and C.-Y. Lin, Chem. Commun., 2006, 1430–1432 RSC; (b) C. Y. Lee, C. She, N. C. Jeong and J. T. Hupp, Chem. Commun., 2010, 46, 6090 RSC.
- (a) J. A. Shelnutt, K. D. Straub, P. M. Rentzepis, M. Gouterman and E. R. Davidson, Biochemistry, 1984, 23, 3946–3954 CrossRef CAS; (b) H. He, A. Gurung, L. Si and A. G. Sykes, Chem. Commun., 2012, 48, 7619 RSC.
- E. M. Barea, R. Caballero, F. Fabregat-Santiago, P. de La Cruz, F. Langa and J. Bisquert, J. Chem. Phys. Chem., 2010, 11, 245 CrossRef CAS PubMed.
- H. Imahori, H. Iijima, H. Hayashi, Y. Toude, T. Umeyama, Y. Matano and S. Ito, ChemSusChem, 2011, 4, 797 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123 CrossRef CAS.
- B. M. Wong, J. Phys. Chem. C, 2009, 113, 21921 CAS.
- R. B. Ambre, G.-F. Chang, M. R. Zanwar, C.-F. Yao, E. W.-G. Diau and C.-H. Hung, Chem.–Asian J., 2013, 8, 2144 CrossRef CAS PubMed.
- K. E. Jasim, Dye Sensitized Solar Cells – Working Principles, Challenges and Opportunities, in Solar Cells – Dye-Sensitized Devices, ed. L. A. Kosyachenko, InTech, 2011, ISBN: 978-953-307-735-2, available from: http://www.intechopen.com/books/solar-cells-dye-sensitized-devices/dye-sensitized-solar-cellsworking-principles-challenges-and-opportunities Search PubMed.
- (a) H. Hayashi, K. Hosomizu and Y. Mantano, J. Phys. Chem. B, 2004, 108, 5018 CrossRef; (b) S. Hayashi, S. Eu and H. Imahori, Chem. Commun., 2007, 2069 Search PubMed.
- (a) Q. Wang, J. E. Moser and M. Grätzel, J. Phys. Chem. B, 2005, 109, 14945 CrossRef CAS PubMed; (b) F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117 CrossRef CAS PubMed; (c) D. B. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923 CrossRef CAS PubMed.
- (a) J. Bisquert, Phys. Chem. Chem. Phys., 2003, 5, 5360 RSC; (b) J. Bisquert, A. Zaban, M. Greenshtein and I. Mora-Sero, J. Am. Chem. Soc., 2004, 126, 13550 CrossRef CAS PubMed.
- (a) M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514 CrossRef CAS PubMed; (b) M. K. Nazeeruddin, R. Humphry-Baker, P. Liska and M. Grätzel, J. Phys. Chem. B, 2003, 107, 8981 CrossRef CAS.
- B. J. Brennan, M. J. Llansola Portole's, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605 RSC.
- P. Falaras, Sol. Energy Mater. Sol. Cells, 1998, 53, 163 CrossRef CAS.
- N. W. Duffy, K. D. Dobson, K. C. Gordon, B. H. Robinson and A. Mc-Quillan, J. Chem. Phys. Lett., 1997, 266, 451 CrossRef CAS.
- T. Ma, K. Inoue, K. Yao, H. Noma, T. Shuji, E. Abe, J. Yu, X. Wang and B. Zhang, J. Electroanal. Chem., 2002, 31, 537 Search PubMed.
- K. Ladomenou, T. Lazarides, M. K. Panda, G. Charalambidis, D. Daphnomili and A. G. Coutsolelos, Inorg. Chem., 2012, 51, 10548 CrossRef CAS PubMed.
- P. Perdew, J. K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- TURBOMOLE (version 5.6), Universität Karlsruhe, 2000 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 03 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, and total and partial density of states of structures 5a and 5b from DFT calculations, 1H NMR and 13C NMR spectra of compounds 3a, 4a, 5a, 3b, 4b, and 5b, and square-wave voltammograms of 5a and 5b. See DOI: 10.1039/c3qi00095h |
| This journal is © the Partner Organisations 2014 |