 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly and structural transformations of high-nuclearity palladium-rich polyoxometalates†
Jamie M.
Cameron‡
,
Jing
Gao‡
,
De-Liang
Long
* and
Leroy
Cronin
*
WestCHEM, School of Chemistry, The University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Deliang.Long@glasgow.ac.uk; Lee.Cronin@glasgow.ac.uk; Web: http://www.croninlab.com Fax: (+44) (0)141-330-4888; Tel: (+44) (0)141-330-6650
First published on 17th January 2014
Abstract
A one-pot strategy exploiting the structure directing effects of SeIV and TeIV heteroatoms has yielded the highest nuclearity noble metal containing polyoxometalates to date; including the palladium-rich selenotungstate isomers K28[H12Pd10Se10W52O206]·65H2O (1) and K26[H14Pd10Se10W52O206]·68H2O (2), and the nanoscale tellurotungstate cluster Na40[Pd6Te19W42O190]·76H2O (3). These reaction systems exhibit remarkable structural flexibility and point to a new route towards the synthesis of complex heterometallic species, in which multiple lacunary polyoxometalate ‘building blocks’ have been assembled to trap a transient oxopalladate species suspected to play a role in the assembly of several common polyoxopalladates. Mass spectrometry has been applied to explore and compare the solution stability of compounds 1–3, demonstrating the markedly different properties of the SeIV and TeIV templated systems. Electrochemical analysis of 1 has been provided and is dominated by Pd redox processes, with reduction of the cluster resulting in electrodeposition of Pd metal and observation of the subsequent formation of PdO species, concurrent with previously reported oxopalladate containing species.
Introduction
Polyoxometalates (POMs) are an important class of polynuclear anionic metal–oxo clusters commonly formed via the assembly of small oxometalate units (traditionally W, Mo or V) into large and often remarkably complex architectures, frequently by careful control of the reaction parameters under one-pot conditions.1 The explosion in the number of publications concerning POMs over the last decade has, however, largely arisen as a result of the suitability of pre-formed lacunary POM clusters – in which vacancies in the cluster shell can be occupied by additional metal ions or may support the formation of larger metal moieties – to act as discrete ‘secondary building units’ in the formation of increasingly diverse higher-nuclearity mixed-metal clusters, commonly referred to as ‘transition metal substituted POMs’ (TMSPs).2 The degree of control over both structure and functionality that this promises has seen TMSPs explored at considerable length for use across a wide spectrum of fields ranging from catalysis to medical and material sciences to name but three.3Central to the assembly and function of these diverse heteropolyoxometalate (HPOM) ‘building blocks’ are the heteroatom species which template their formation.4 Recent years have seen a vast array of different elements from almost every group of the periodic table employed in this way,5 in which the electronic configuration of the heteroatom can have a profound effect on the self-assembly of the POM cluster, sometimes imparting specific function or properties to the oxometalate framework in the process.6 Notably, lone-pair containing heteroatoms (such as AsIII, SeIV or TeIVetc.) have been intensively exploited recently as a means by which lacunary clusters can be generated without the need for hydrolysis of a pre-existing parent clusters, allowing for the preparation of unique, often high nuclearity POM architectures7 and for further expansion of the class of TMSP materials.8
In particular, the incorporation of catalytically active noble metals (primarily Ru and Pd) within discrete POM units has attracted increasing attention as a means by which new functional and solution-stable catalytic materials might be discovered.9,10 Hill, Kortz and others have pioneered the study of well-defined lacunary POMs which form a range of novel noble metal containing species,11 two comprehensive reviews of which have been published recently and summarise these efforts.12 It is also important to note that some PdII species may comprise a fascinating subset of POM chemistry and primarily along with Kortz and coworkers,13 our own group has demonstrated how palladium can form a range of remarkable oxopalladate clusters including the giant, nanoscale {Pd84} wheel.14 These clusters are especially interesting in that they follow a quite different set of structural principles to more conventional POMs and in fact, the guiding building block concepts for this class of compounds are far less well-defined than those of polyoxotungstates. To date, however, the synthesis and characterisation of both noble metal containing POMs and polyoxopalladates remains relatively underexplored, particularly in comparison to the rich array of reported 1st row transition metal containing species.
Herein, we present three new, nanoscale, palladium-rich POM clusters synthesised via a facile one-pot approach which exploits the structure-directing effects of the group XVI heteroatoms, SeIV and TeIV.
Results and discussion
Selenite templated species
Crystallographic analysis of 1 reveals that it crystallises in a triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and exhibits two distinct pairs of lacunary tungstate units linked by three corner-sharing {WO6} octahedra to form two equivalent {Se3W26} units. These superlacunary selenotungstate species are bridged by two identical {Pd5Se2} moieties to form an overall architecture with nominal C2h symmetry (Fig. 1). The identical {Se3W26} units are related to each other in the structure of 1 by a 180° rotation around the vertical axis of the molecule (as depicted in Fig. 1) and as a result, 1 can effectively be described as being in a trans-conformation about the central cavity with two pairs of cluster ‘building blocks’ located on trans positions (note the colour differences). The two component selenotungstate ‘building blocks’ can be classified as either the tri-lacunary B-α-{SeW9O33} derivative of the W12-Keggin parent structure or the tetra-lacunary {Se2W14O52} derivative of the W18-‘Trojan Horse’ structure6b,15 which, to the best of our knowledge, is the first time that this species has been observed in the solid state. Of particular interest are the unusual {Pd5Se2} cores which are each supported by one {SeW9} and one {Se2W14} fragment from opposing {Se3W26} units and are themselves directly related via a 180° rotation along the horizontal axis of the molecule. All five PdII ions within each {Pd5Se2} unit exist in a square planar geometry coordinated by four oxygen atoms with an average Pd–O bond length of 1.999(3) Å. The coordination of those four palladium atoms which are bound directly to the lacunary POM fragments conforms largely to that observed previously in the literature for comparable systems,11c,e in which the PdII ion either forms a bond to oxo-groups on neighbouring {W3} triads in the case of {SeW9}, or similarly, to both the cap and belt regions of the {Se2W14} unit (Fig. 1c). The fifth PdII ion in each unit sits roughly perpendicular to the aforementioned palladium atoms, linking them via two shared μ3-oxo bridges, whilst two pyramidal SeIV ions cap the remaining available oxo groups, effectively stabilising the overall {Pd5Se2} unit.
and exhibits two distinct pairs of lacunary tungstate units linked by three corner-sharing {WO6} octahedra to form two equivalent {Se3W26} units. These superlacunary selenotungstate species are bridged by two identical {Pd5Se2} moieties to form an overall architecture with nominal C2h symmetry (Fig. 1). The identical {Se3W26} units are related to each other in the structure of 1 by a 180° rotation around the vertical axis of the molecule (as depicted in Fig. 1) and as a result, 1 can effectively be described as being in a trans-conformation about the central cavity with two pairs of cluster ‘building blocks’ located on trans positions (note the colour differences). The two component selenotungstate ‘building blocks’ can be classified as either the tri-lacunary B-α-{SeW9O33} derivative of the W12-Keggin parent structure or the tetra-lacunary {Se2W14O52} derivative of the W18-‘Trojan Horse’ structure6b,15 which, to the best of our knowledge, is the first time that this species has been observed in the solid state. Of particular interest are the unusual {Pd5Se2} cores which are each supported by one {SeW9} and one {Se2W14} fragment from opposing {Se3W26} units and are themselves directly related via a 180° rotation along the horizontal axis of the molecule. All five PdII ions within each {Pd5Se2} unit exist in a square planar geometry coordinated by four oxygen atoms with an average Pd–O bond length of 1.999(3) Å. The coordination of those four palladium atoms which are bound directly to the lacunary POM fragments conforms largely to that observed previously in the literature for comparable systems,11c,e in which the PdII ion either forms a bond to oxo-groups on neighbouring {W3} triads in the case of {SeW9}, or similarly, to both the cap and belt regions of the {Se2W14} unit (Fig. 1c). The fifth PdII ion in each unit sits roughly perpendicular to the aforementioned palladium atoms, linking them via two shared μ3-oxo bridges, whilst two pyramidal SeIV ions cap the remaining available oxo groups, effectively stabilising the overall {Pd5Se2} unit.
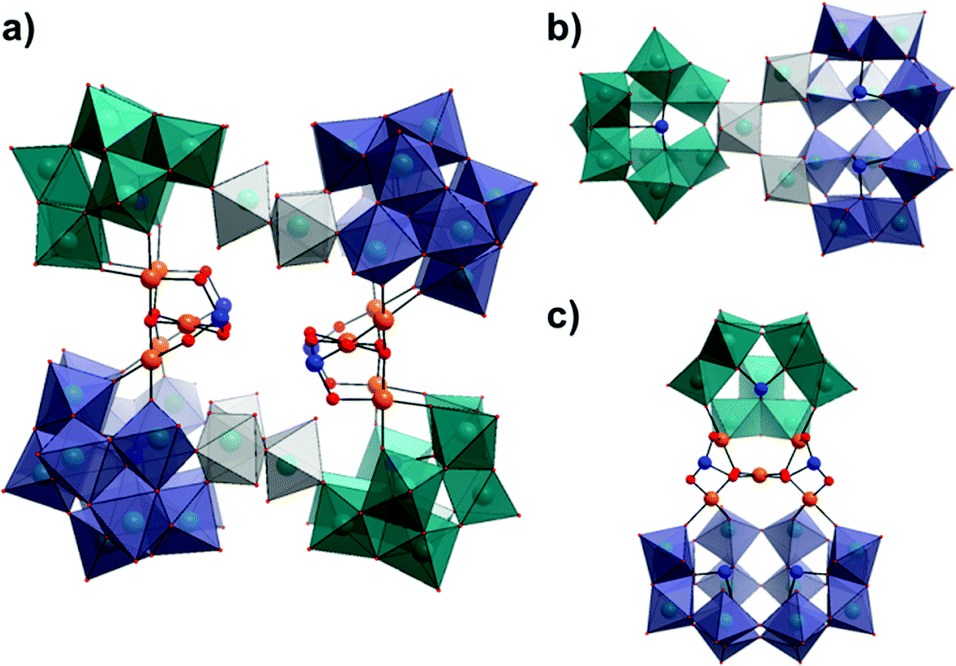 | ||
| Fig. 1 Combined polyhedral/ball and stick representation of the structure of 1 showing; (a) front view of the intact cluster [H12Pd10Se10W52O206]28−, (b) top-down view of one {Se3W26} half-unit and, (c) side-on view of the ({SeW9}{Pd5Se2}{Se2W14}) half-unit with the (WO6) linkers removed for clarity. (Colour code: {SeW9} units: teal polyhedra, {Se2W14} units: purple polyhedra, WO6 linkers: grey polyhedra, Pd = orange, Se = blue, O = red. Cations and water molecules have been omitted for clarity.) | ||
Furthermore, whilst we believe that this is the first reported example of this type of {Pd5Se2} moiety, it is particularly interesting to note that it is also possible to consider this structural unit as a key building block in the related field of polyoxopalladate chemistry. The previously reported family of palladate cages with the general formula [MPd12O8(XOy)8]n−,13a,c–f and the related series of Pd-‘nanostars’,13b,14a,16 of general formula [Pd17−xO10(XOy)10]n− (X = P, As or Se), may both be viewed as assembling via the fusion of either four or five {Pd5X2} units respectively, in which the capping heteroatoms form the vertices of the double diamond/star-like topology found in these species (Fig. 2). Comparatively, the {Pd5Se2} unit in 1 shows noticeable distortion as a result of the unequal binding sites provided by the {SeW9} and {Se2W14} units and exhibits a marked structural asymmetry in comparison to the analogous repeating structural units found in {Pd12−17} which possess a notional C2v symmetry. This observation seems especially relevant from a mechanistic point of view, providing both a second perspective on the possible mechanisms of polyoxopalladate cluster growth and posing an intriguing ‘chicken-and-egg’ question in terms of which structural units (i.e. palladate or tungstate) play the most important role in the assembly of 1.
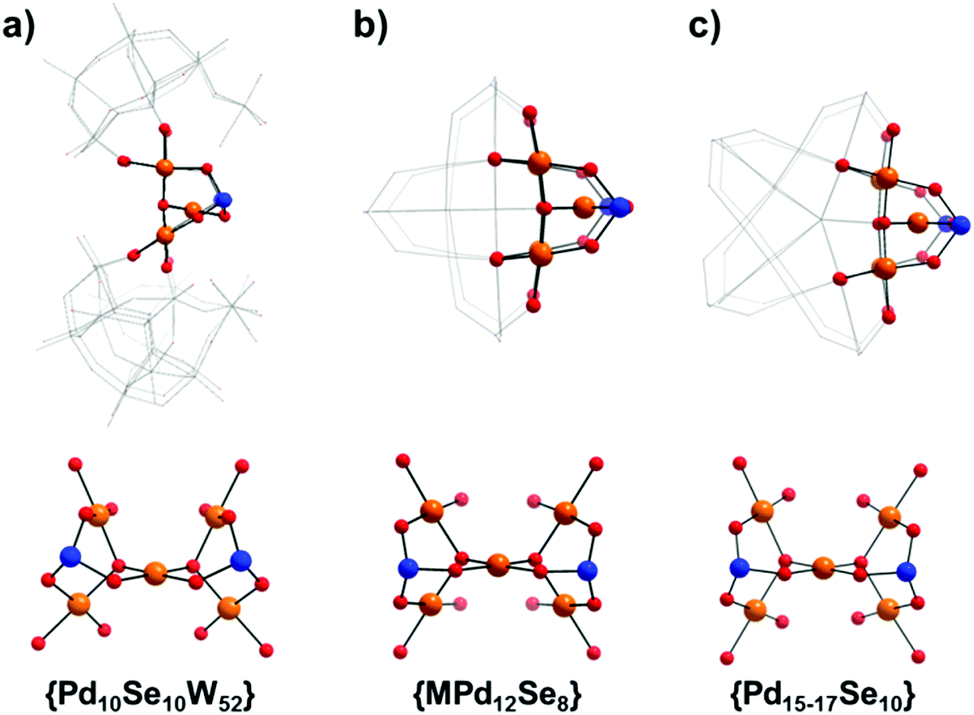 | ||
| Fig. 2 Comparison of the selenopalladate structural units common to both compounds 1 and 2 and the polyoxopalladate ‘cages’ and ‘nanostars’ of the general formulae shown.13f,g,16 The supporting POM framework/surrounding oxopalladate cluster associated with each {Pd5Se2} unit is shown in wireframe, providing an idea of the local environment of each unit. In the case of the polyoxopalladate clusters, it can clearly be seen how the {Pd5Se2} moiety can be considered as a repeating structural unit in which either; (b) four or (c) five selenopalladate groups are fused to form the overall structure in which the central PdII ion defines the four- or five-fold symmetry axes of the respective polyoxopalladate clusters. (Colour code: Pd = orange, Se = blue and O = red.) | ||
It should also be noted that the structure directing effect of the [SeO3]2− template is crucially important in this system, whereby through careful control of the reaction conditions we have been able to generate and combine multiple lacunary building blocks with additional heterometal atoms in situ. The lone pair on the template SeIV ions often precludes the formation of ‘closed’ POM species such as the Keggin or Wells–Dawson clusters, instead favouring the formation of multiple lacunary species over a wide range of conditions. These species can then be isolated (or ‘trapped’) either by addition of heterometals capable of occupying the vacancies in the POM framework or by allowing the tungstate lacuna to condense together into a larger cluster unit, occasionally involving the abstraction of additional tungstate ions present in solution.7,8 Whilst the concept of using lone pair containing heteroatoms in this way is not entirely new, we believe that this work is the first time that multiple lacunary building blocks derived from different parent species (i.e. Keggin- or Wells–Dawson-type clusters) have been combined into a single POM architecture.17 This is significant, both in that it may provide a pathway for the isolation of unusual heterometal species (such as the {Pd5Se2} core in 1) but also in that it indicates the hitherto unreported combinatorial nature of such systems, in which a series of inequivalent, metastable POM fragments are present and may be simultaneously accessed under the correct set of conditions.
On continued evaporation of the mother liquor from which the crystals of 1 were isolated, a second batch of product, K26[H14Pd10Se10W52O206]·68H2O (2), could be collected. Crystallographic analysis of compound 2 reveals that it is the structural cis-isomer of 1 and crystallises in a similar triclinic system with the space group P. In this species, the primary selenotungstate and {Pd5Se2} building blocks have been conserved but undergone a reorganisation, notionally through a 180° rotation of one complete {{SeW9}{Pd5Se2}{Se2W14}} unit about the clusters horizontal axis via cleaving of the {W3} tungstate linkers reducing the overall symmetry of the cluster to C2 (opposed to C2h) (Fig. 3). The {WO6} octahedra which form the {W3} linkers in 1 reorganise in compound 2 to form one corner-sharing {W2} bridge between the two {SeW9} fragments to form a {Se2W20} unit, and one corner-sharing {W4} unit which links the {Se2W14} units to yield a {Se4W32} species. In this way, it is conceivable to envisage the reorganisation of the entire cluster via breaking and reforming of just four W–oxo bonds, namely those shared between the W-centres in the {W3} linkers in 1 (shown in grey in Fig. 1 and 3). It seems possible therefore that the cis-isomer, 2, may be the thermodynamic product in this reaction system whilst the trans-isomer, 1, can be isolated first as the marginally less stable kinetic product. Whilst it may not be immediately apparent why this should be the case, it is possible to speculate that one explanation for the increased stability of 2 may be found in the respective configurations of both clusters and, specifically, the relationship of the two {Pd5Se2} cores to each other. The apparent 180° rotation of the {{SeW9}{Pd5Se2}{Se2W14}} half-unit in the formation of 2 leads the cis-isomer to adopt a ‘staggered’ configuration across the two halves of the molecule, whilst the two trans-related ‘half-units’ in 1 are eclipsed relative to another. Whilst at first glance, this seems to actually increase the steric constraints of the system (as evidenced by a slight reduction in the distances between the adjacent SeIV ions), this conformational change actually may subtly reduce the steric constraints within the central cavity of the cluster by reducing the number of chelating K+ ions needed to stabilise the capping [SeO3]2− units from two to one (Fig. S1†), thus providing a possible thermodynamic driving force for the isomerisation to occur.
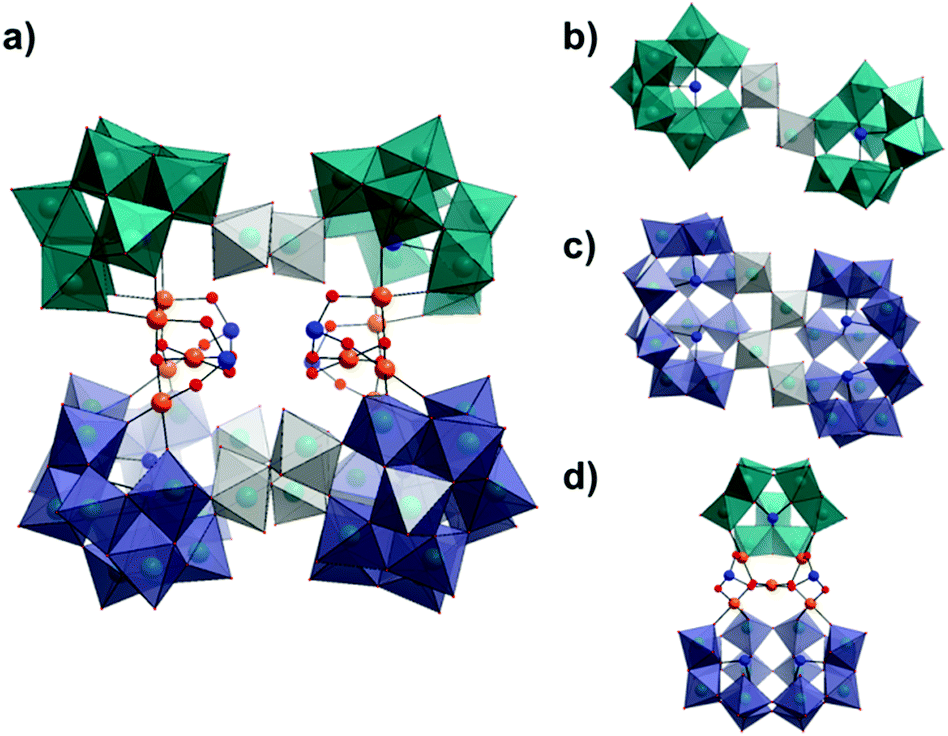 | ||
| Fig. 3 Combined polyhedral/ball and stick representation of the structure of 2 showing; (a) front view of the intact cluster [H14Pd10Se10W52O206]28−, (b) top-down view of the {Se2W20} unit, (c) top-down view of the {Se4W32} unit and, (d) side-on view of the identical ({SeW9}{Pd5Se2}{Se2W14}) half-unit with the (WO6) linkers removed for clarity. (Colour code: {SeW9} units: teal polyhedra, {Se2W14} units: purple polyhedra, WO6 linkers: grey polyhedra, Pd = orange, Se = blue, O = red. Cations and water molecules have been omitted for clarity.) | ||
High resolution negative-mode electrospray ionisation mass spectrometry (ESI-MS) was used to confirm the structure and solution stability of compounds 1 and 2 in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 solution of water and acetonitrile. Unsurprisingly, the spectra of both clusters are very similar in which the majority of peaks are close enough to effectively be considered as overlapping within the constraints of the experiment, whereby any small discrepancy is easily explained by the exchange of cations or, more frequently, associated solvate (Fig. 4). From these experiments it could be observed that both species retain their integrity in solution, with all of the major peaks identifiable in each spectrum corresponding to the intact POM cluster. Notably, the well-resolved major peaks in both spectra at ca. 2550 m/z correspond to the species [H17−xK5+x(H12Pd10Se10W52O206)·18H2O]6− and at higher m/z ranges, species corresponding to a dimeric unit of both 1 and 2 can be identified at ca. 3400 and 3900 m/z (see ESI† for detailed peak assignments). It should be noted that the high nuclearity and charge of these clusters makes direct assignment of the peak envelopes difficult, with most peak envelopes undergoing some degree of ‘broadening’ over a wider than expected m/z range, generally as a result of the loss of weakly associated solvent molecules or cation exchange within the spectrometer causing multiple closely related peaks to overlap into a single, enlarged envelope.18
:95 solution of water and acetonitrile. Unsurprisingly, the spectra of both clusters are very similar in which the majority of peaks are close enough to effectively be considered as overlapping within the constraints of the experiment, whereby any small discrepancy is easily explained by the exchange of cations or, more frequently, associated solvate (Fig. 4). From these experiments it could be observed that both species retain their integrity in solution, with all of the major peaks identifiable in each spectrum corresponding to the intact POM cluster. Notably, the well-resolved major peaks in both spectra at ca. 2550 m/z correspond to the species [H17−xK5+x(H12Pd10Se10W52O206)·18H2O]6− and at higher m/z ranges, species corresponding to a dimeric unit of both 1 and 2 can be identified at ca. 3400 and 3900 m/z (see ESI† for detailed peak assignments). It should be noted that the high nuclearity and charge of these clusters makes direct assignment of the peak envelopes difficult, with most peak envelopes undergoing some degree of ‘broadening’ over a wider than expected m/z range, generally as a result of the loss of weakly associated solvent molecules or cation exchange within the spectrometer causing multiple closely related peaks to overlap into a single, enlarged envelope.18
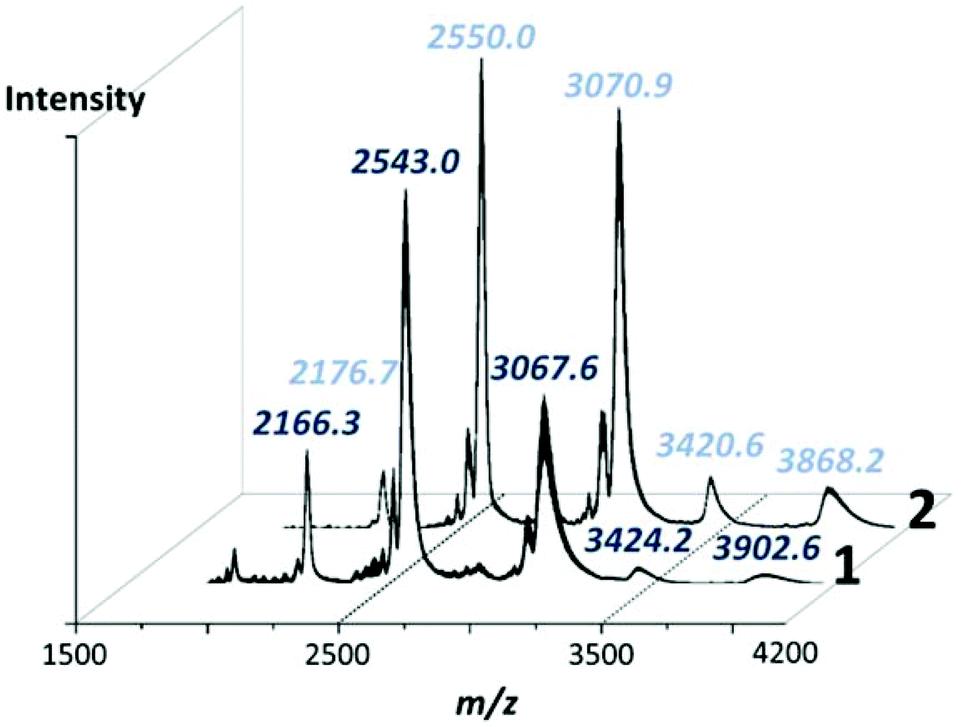 | ||
| Fig. 4 Negative mode mass spectra of 1 and 2 in the m/z range 1500 to 4200 showing the major peaks corresponding to the intact POM anions. Full assignment of the highlighted peaks is given in the ESI.† | ||
Tellurite templated species
Subsequent to the isolation of compounds 1 and 2 we were interested to see what effect substituting the SeIV heteroatoms for TeIV might have on this system. Following a similar one-pot approach we were able to isolate crystals of a quite different polyanion, Na40[Pd6Te19W42O190]·76H2O (3). Crystallographic analysis of compound 3 reveals that this cluster crystallises in a triclinic system with the space group P in which the arrangement of the POM cluster exhibits an approximate C3h symmetry (Fig. 5). In comparison to 1 and 2, 3 comprises only one type of lacunary tungstate unit, the rarely observed α-{TeW7O27}, six of which are linked together by pendant [TeO3]2− groups to support two identical {Pd3Te3O3} cores. Interestingly, the TeIV ions in compound 3 show three distinct coordination modes. The heteroatom templates within the {TeW7} units, which are analogous to species our group has reported previously,7d are remarkable in that they display an unusual highly distorted tetrahedral geometry rather than the more traditional pyramidal conformation as a result of an additional oxo bond shared with the pyramidal {TeO3} linkers which bridge the tungstate units. This is the first known example of a non-pyramidal, four-coordinate TeIV ion templating a POM unit. The three TeIV ions in each {Pd3Te3} core exist in a square pyramidal geometry in which the base of the pyramid forms a face linking two neighbouring {TeW7} units via two shared oxo-bonds whilst the apex of the pyramid is occupied by a μ3-oxygen shared with two PdII ions (Fig. S2†). Interestingly, the topology of the {Pd3Te3} core in 3 bears little similarity to that in 1 and 2 and appears to be unrelated to any fragments previously observed in polyoxopalladate chemistry, suggesting that the assembly of 3 may be more strongly influenced by the formation of the tungstate lacuna and related substituted species than by possible concurrent, or competing, assembly processes of oxopalladate species in solution.
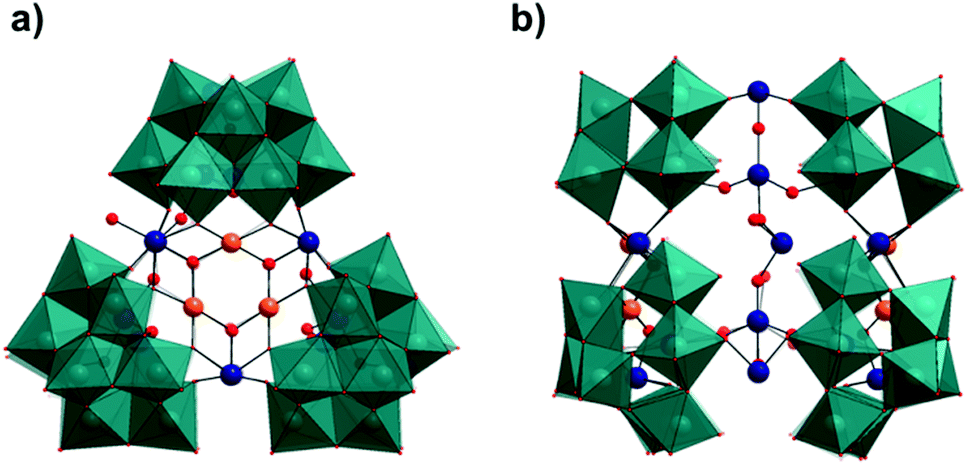 | ||
| Fig. 5 Combined polyhedral/ball and stick representation of the structure of 3 showing; (a) front on view of the intact cluster [Pd6Te19W42O190]40− highlighting the {Pd3Te3O3} core and, (b) side-on view of the intact cluster showing the varied linking modes of the pendant TeIV groups. (Colour code: WO6 = teal polyhedra, Pd = orange, Te = dark blue, O = red. Cations and water molecules have been omitted for clarity.) | ||
In the case of 3, negative mode ESI-MS yields a very different result than those recorded for clusters 1 and 2, hinting at the inherent instability of this species in solution. The mass spectrum of 3 appears to show heavy fragmentation of the parent species and in fact, no single peak can be identified that corresponds to the intact cluster, with the largest identifiable fragments occurring in a range between ca. 3–4 kDa (MW of 3 = 16.1 kDa) (Fig. S3, see Table S4 for selected peak assignments†). One probable explanation for this behaviour are the obvious structural differences between the two types of clusters, specifically in that the primary structural units in 1 and 2 are linked by W–O–W and Pd–O–Pd bonds whereas the primary linkers in 3 are based on the significantly more labile Te–O–Te and Te–O–W bonds. This observation is also commensurate with our previously reported work on macrocyclic tellurotungstate clusters containing a similar [TeW7O28]10− moiety, in which the macrocyclic units readily fragment into smaller species and could not be identified intact, further hinting at the inherent lability of such (TeOx) linked polyoxotungstate systems.7d Furthermore, the remarkable similarity in the fragmentation patterns of both 3 and the previously reported [Te8W28O112]24− species allow us to postulate that these two clusters may share a similar assembly pathway (Fig. S4†). Our experiences of synthesising 3 also generally support the conclusion that this cluster is inherently unstable, chiefly in that despite our best efforts, optimising the synthesis of this cluster proved challenging as there seems to exist only a narrow window of conditions in which 3 can be reliably isolated and, though our final reported conditions are entirely reproducible, the total yield of crystalline product from each batch remains low. Furthermore, our attempts to modify the synthesis and associated efforts to recrystallize 3 often led to different products; notably including a structurally related, lower nuclearity cluster, [Pd6Te3W28O108]14− (3′), but most commonly the previously reported and thermodynamically favourable [Pd3(TeW9O33)2]10− species8b (see ESI, Fig. S5,† for a more detailed discussion of these experiments). Taken together these observations suggest that 3 is likely to be an interesting, metastable product of this reaction system which, once removed from the mother liquor, can readily transform to form more thermodynamically favourable species in solution. Whilst a comprehensive exploration of this reaction system will not be provided here, we hope to report it in detail at a later date.
Electrochemistry
Cyclic voltammetry (CV) was also employed as a means to explore the electrochemical behaviour of these clusters. Due to the instability of 3 and the difficulty in ensuring that a single species is being measured in solution, we cannot accurately report the electrochemical behaviour of this compound here. CV of compounds 1 and 2 was performed in 0.2 M NaSO4 solution and, as expected, no significant difference was found between the two species in which the electrochemical behaviour was dominated by irreversible Pd-centred redox processes (Fig. S6†). Scanning of the potential in the negative direction leads to a single broad irreversible cathodic peak centred at −0.45 V corresponding to the reduction of the PdII centres in 1 to Pd0 and an electrodeposition process on the working electrode. Notably, this occurs at a slightly lower potential than is normally reported in the literature11g,13c,d and whilst it is possible that trapping the {Pd5Se2} species within the tungstate framework does have an overall effect on its electrochemical behaviour, it has been noted in multiple previous studies that electrochemical reduction and oxidation of Pd-species is a particularly complex process and it is therefore difficult to draw conclusions from this study alone.19 As the scan proceeds in the positive direction, two oxidation waves can be observed at +0.60 V and +0.88 V respectively. These correspond to the re-oxidation of Pd0 and the formation of resulting oxides, which tallies well with that which has previously been reported in related systems. This is further confirmed by the appearance of a second cathodic wave at +0.21 V which is characteristic of the reduction of PdO.Conclusions
In conclusion, we have presented three new Pd-containing polyoxotungstate species which are the highest nuclearity noble-metal containing polyanions to have been isolated thus far. A one-pot reaction strategy exploiting the structure directing effects of SeIV template heteroatoms has allowed us to trap a {Pd5Se2} unit within a robust selenotungstate species, providing an interesting mechanistic insight into the formation of the related class of polyoxopalladate materials and combining, for the first time in a single POM architecture, two distinct lacunary ‘building blocks’ from different parent anions. Further experiments using the coordinatively flexible TeIV heteroatom template and linker have allowed us to isolate a metastable tellurotungstate hexamer containing two unique {Pd3Te3O3} moieties and allowing us to draw insights into the assembly of both this species and a previously reported, related class of tellurotungstate macrocycles assembled from the same unusual {TeW7} units. We believe that this approach could be an important new means to isolate a new series of complex transition metal substituted polyoxometalate species, in which the assembly processes are not governed by stable, well-defined, pre-synthesised lacunary building blocks but by the dynamic, combinatorial solution chemistry of lone-pair containing heteropolyoxometalates synthesised via a bottom-up approach under one-pot conditions.Experimental section
Materials and physical measurements
All reagents were purchased commercially and used without further purification. Elemental analyses for Pd and W were conducted on an Agilent 7700x ICP-MS and alkali metal content was determined on a Sherwood Scientific M410 INDUSTRIAL flame photometer. Thermogravimetric analysis was conducted using a TA Instruments Q500 Thermogravimetric Analyser at a heating rate of 10 °C min−1 under air. FT-IR spectra were collected on a Shimadzu FTIR 8400 s spectrometer fitted with a golden gate ATR attachment. Electrospray ionisation mass spectrometry (ESI-MS) was performed on a Bruker micrOTOF-Q spectrometer (full details of MS measurements available in ESI†). Cyclic voltammetry was performed on a CH Instruments 600D potentiostat using a standard three electrode set-up, with a 3 mm diameter glassy carbon working electrode and Pt wire counter electrode, with all potentials quoted against an Ag/AgCl reference electrode. All measurements were performed at room temperature in thoroughly de-aerated solutions under a positive pressure of dry Ar/N2.X-ray crystallography
Single crystals of compounds 1 and 2 were mounted in oil on a glass capillary and datasets were collected on an Oxford Diffraction Gemini A Ultra diffractometer (with ATLAS CCD detector), whilst 3 was mounted in oil on a flexible mount loop and data was collected on a Bruker Apex II diffractometer (with Quazar CCD detector), equipped with a graphite monochromator (λ(MoKα) = 0.71073 Å). All measurements were conducted at 150(2) K. Absorption effects for datasets 1 and 2 were corrected via an analytical method using a multifaceted crystal model,20 whilst corrections were applied to compound 3via an empirical method21 and data reductions performed using the CrysAlisPro or Apex2 software respectively. All structures were solved by direct methods and structural refinement was carried out using SHELX-97 by means of a full matrix least squares on F2 method,22 performed within the WinGX software suite.23 Selected details of the data collection and structural refinement of compounds 1–3 can be found within the ESI (Table S1†) and full details are available in the corresponding CIF files. Crystallographic data (excluding structure factors) have been deposited (ICSD 426772–426774).Acknowledgements
We thank the EPSRC funding (grants EP/H024107/1; EP/I033459/1; EP/J015156/1), EU FP7 Micreagents (318671), the Chinese Scholarship Council, WestCHEM and the University of Glasgow for supporting this work. LC also thanks the Royal-Society Wolfson Foundation for a Merit Award. The authors would also like to thank Dr Jun Yan (Zhongnan University) for useful discussions and preliminary help.Notes and references
- (a) M. T. Pope and A. Müller, Angew. Chem., Int. Ed., 1991, 30, 34–48 CrossRef; (b) J. Livage, Coord. Chem. Rev., 1998, 178–180(Part 2), 999–1018 CrossRef CAS; (c) C. P. Pradeep, D.-L. Long and L. Cronin, Dalton Trans., 2010, 39, 9443–9457 RSC; (d) A. Müller and P. Gouzerh, Chem. Soc. Rev., 2012, 41, 7431–7463 RSC.
- (a) D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC; (b) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS PubMed; (c) D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- (a) Special edition: C. L. Hill (guest ed.), Chem. Rev., 1998, 98, 1–390; (b) J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed; (c) Special edition: U. Kortz (guest ed.), Eur. J. Inorg. Chem., 2009, 2009, 5055–5276; (d) Special edition: L. Cronin and A. Müller (guest ed.), Chem. Soc. Rev., 2012, 41, 7325–7648; (e) Special edition: D.-L. Long and L. Cronin (guest ed.), Dalton Trans., 2012, 41, 9799–10106; (f) C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC.
- (a) S. G. Mitchell, P. I. Molina, S. Khanra, H. N. Miras, A. Prescimone, G. J. T. Cooper, R. S. Winter, E. K. Brechin, D.-L. Long, R. J. Cogdell and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 9154–9157 CrossRef CAS PubMed; (b) L. Vilà-Nadal, S. G. Mitchell, A. Rodriguez-Fortea, H. N. Miras, L. Cronin and J. M. Poblet, Phys. Chem. Chem. Phys., 2011, 13, 20136–20145 RSC; (c) H. N. Miras, M. Sorus, J. Hawkett, D. O. Sells, E. J. L. McInnes and L. Cronin, J. Am. Chem. Soc., 2012, 134, 6980–6983 CrossRef CAS PubMed; (d) J. M. Cameron, G. N. Newton, C. Busche, D.-L. Long, H. Oshio and L. Cronin, Chem. Commun., 2013, 49, 3395–3397 RSC.
- (a) M. Ishaque Khan, S. Cevik and R. Hayashi, Dalton Trans., 1999, 1651–1654 RSC; (b) I. A. Weinstock, J. J. Cowan, E. M. G. Barbuzzi, H. Zeng and C. L. Hill, J. Am. Chem. Soc., 1999, 121, 4608–4617 CrossRef CAS; (c) S. Uchida, M. Hashimoto and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 2814–2817 CrossRef CAS; (d) D.-L. Long, P. Kögerler, A. D. C. Parenty, J. Fielden and L. Cronin, Angew. Chem., Int. Ed., 2006, 45, 4798–4803 CrossRef CAS PubMed; (e) N. Leclerc-Laronze, J. Marrot, G. Hervé, R. Thouvenot and E. Cadot, Chem.–Eur. J., 2007, 13, 7234–7245 CrossRef CAS PubMed; (f) D.-L. Long, Y.-F. Song, E. F. Wilson, P. Kögerler, S.-X. Guo, A. M. Bond, J. S. J. Hargreaves and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4384–4387 CrossRef CAS PubMed; (g) B. S. Bassil and U. Kortz, Dalton Trans., 2011, 40, 9649–9661 RSC.
- (a) D.-L. Long, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2004, 43, 1817–1820 CrossRef CAS PubMed; (b) D.-L. Long, H. Abbas, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2005, 44, 3415–3419 CrossRef CAS PubMed; (c) C. Baffert, J. F. Boas, A. M. Bond, P. Kögerler, D.-L. Long, J. R. Pilbrow and L. Cronin, Chem.–Eur. J., 2006, 12, 8472–8483 CrossRef CAS PubMed; (d) C. Fleming, D.-L. Long, N. McMillan, J. Johnston, N. Bovet, V. Dhanak, N. Gadegaard, P. Kögerler, L. Cronin and M. Kadodwala, Nat. Nanotechnol., 2008, 3, 289–233 CrossRef PubMed; (e) J. Thiel, C. Ritchie, C. Streb, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2009, 131, 4180–4181 CrossRef CAS PubMed; (f) L. Vilà-Nadal, K. Peuntinger, C. Busche, J. Yan, D. Lüders, D.-L. Long, J. M. Poblet, D. M. Guldi and L. Cronin, Angew. Chem., Int. Ed., 2013, 52, 9695–9699 CrossRef PubMed.
- (a) K. Wassermann, M. H. Dickman and M. T. Pope, Angew. Chem., Int. Ed., 1997, 36, 1445–1448 CrossRef CAS; (b) J. Yan, J. Gao, D.-L. Long, H. N. Miras and L. Cronin, J. Am. Chem. Soc., 2010, 132, 11410–11411 CrossRef CAS PubMed; (c) J. Yan, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 4117–4120 CrossRef CAS PubMed; (d) J. Gao, J. Yan, S. G. Mitchell, H. N. Miras, A. G. Boulay, D.-L. Long and L. Cronin, Chem. Sci., 2011, 2, 1502–1508 RSC; (e) M. N. Corella-Ochoa, H. N. Miras, A. Kidd, D.-L. Long and L. Cronin, Chem. Commun., 2011, 47, 8799–8801 RSC.
- (a) A. C. Stowe, S. Nellutla, N. S. Dalal and U. Kortz, Eur. J. Inorg. Chem., 2004, 2004, 3792–3797 CrossRef; (b) J. Gao, J. Yan, S. Beeg, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2012, 51, 3373–3376 CrossRef CAS PubMed; (c) J. Gao, J. Yan, S. Beeg, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2012, 135, 1796–1805 CrossRef PubMed; (d) W.-C. Chen, H.-L. Li, X.-L. Wang, K.-Z. Shao, Z.-M. Su and E.-B. Wang, Chem.–Eur. J., 2013, 19, 11007–11015 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- (a) T. M. Anderson, W. A. Neiwert, M. L. Kirk, P. M. B. Piccoli, A. J. Schultz, T. F. Koetzle, D. G. Musaev, K. Morokuma, R. Cao and C. L. Hill, Science, 2004, 306, 2074–2077 CrossRef CAS PubMed; (b) R. Neumann and A. M. Khenkin, Inorg. Chem., 1995, 34, 5753–5760 CrossRef CAS; (c) R. Neumann and M. Dahan, Nature, 1997, 388, 353–355 CrossRef CAS PubMed; (d) C. Dablemont, A. Proust, R. Thouvenot, C. Afonso, F. Fournier and J.-C. Tabet, Inorg. Chem., 2004, 43, 3514–3520 CrossRef CAS PubMed; (e) T. Kurata, A. Uehara, Y. Hayashi and K. Isobe, Inorg. Chem., 2005, 44, 2524–2530 CrossRef CAS PubMed; (f) Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed; (g) A. Sartorel, M. Carraro, G. Scorrano, R. D. Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed; (h) A. E. Kuznetsov, Y. V. Geletii, C. L. Hill, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2009, 131, 6844–6854 CrossRef CAS PubMed; (i) S. Angus-Dunne, R. C. Burns, D. C. Craig and G. A. Lawrance, Z. Anorg. Allg. Chem., 2010, 636, 727–734 CrossRef CAS; (j) T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425 CrossRef CAS PubMed.
- (a) L.-H. Bi, U. Kortz, B. Keita, L. Nadjo and H. Borrmann, Inorg. Chem., 2004, 43, 8367–8372 CrossRef CAS PubMed; (b) L.-H. Bi, M. Reicke, U. Kortz, B. Keita, L. Nadjo and R. J. Clark, Inorg. Chem., 2004, 43, 3915–3920 CrossRef CAS PubMed; (c) L.-H. Bi, U. Kortz, B. Keita, L. Nadjo and L. Daniels, Eur. J. Inorg. Chem., 2005, 2005, 3034–3041 CrossRef; (d) L.-H. Bi, M. H. Dickman and U. Kortz, CrystEngComm, 2009, 11, 965–966 RSC; (e) R. Villanneau, S. Renaudineau, P. Herson, K. Boubekeur, R. Thouvenot and A. Proust, Eur. J. Inorg. Chem., 2009, 2009, 479–488 CrossRef; (f) N. V. Izarova, A. Banerjee and U. Kortz, Inorg. Chem., 2011, 50, 10379–10386 CrossRef CAS PubMed; (g) S. Duval, J. Marrot, C. Simonnet-Jegat, I. M. Mbomekalle, M. Sokolov and E. Cadot, Dalton Trans., 2012, 41, 3174–3184 RSC.
- (a) P. Putaj and F. Lefebvre, Coord. Chem. Rev., 2011, 255, 1642–1685 CrossRef CAS PubMed; (b) N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492–9510 CrossRef CAS PubMed.
- (a) E. V. Chubarova, M. H. Dickman, B. Keita, L. Nadjo, F. Miserque, M. Mifsud, I. W. C. E. Arends and U. Kortz, Angew. Chem., Int. Ed., 2008, 47, 9542–9546 CrossRef CAS PubMed; (b) N. V. Izarova, R. N. Biboum, B. Keita, M. Mifsud, I. W. C. E. Arends, G. B. Jameson and U. Kortz, Dalton Trans., 2009, 9385–9387 RSC; (c) N. V. Izarova, M. H. Dickman, R. N. Biboum, B. Keita, L. Nadjo, V. Ramachandran, N. S. Dalal and U. Kortz, Inorg. Chem., 2009, 48, 7504–7506 CrossRef CAS PubMed; (d) M. Barsukova, N. V. Izarova, R. N. Biboum, B. Keita, L. Nadjo, V. Ramachandran, N. S. Dalal, N. S. Antonova, J. J. Carbó, J. M. Poblet and U. Kortz, Chem.–Eur. J., 2010, 16, 9076–9085 CrossRef CAS PubMed; (e) M. Barsukova-Stuckart, N. V. Izarova, R. Barrett, Z. Wang, J. van Tol, H. W. Kroto, N. S. Dalal, B. Keita, D. Heller and U. Kortz, Chem.–Eur. J., 2012, 18, 6167–6171 CrossRef CAS PubMed; (f) M. Barsukova-Stuckart, N. V. Izarova, R. A. Barrett, Z. Wang, J. van Tol, H. W. Kroto, N. S. Dalal, P. Jiménez-Lozano, J. J. Carbó, J. M. Poblet, M. S. von Gernler, T. Drewello, P. de Oliveira, B. Keita and U. Kortz, Inorg. Chem., 2012, 51, 13214–13228 CrossRef CAS PubMed; (g) Z.-G. Lin, B. Wang, J. Cao, B.-K. Chen, Y.-Z. Gao, Y.-N. Chi, C. Xu, X.-Q. Huang, R.-D. Han, S.-Y. Su and C.-W. Hu, Inorg. Chem., 2012, 51, 4435–4437 CrossRef CAS PubMed.
- (a) F. Xu, R. A. Scullion, J. Yan, H. N. Miras, C. Busche, A. Scandurra, B. Pignataro, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2011, 133, 4684–4686 CrossRef CAS PubMed; (b) F. Xu, H. N. Miras, R. A. Scullion, D.-L. Long, J. Thiel and L. Cronin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11609–11612 CrossRef CAS PubMed.
- R. Q. Cabrera, D.-L. Long, L. Cronin and P. F. McMillan, CrystEngComm, 2010, 12, 2568–2572 RSC.
- M. Delferro, C. Graiff, L. Elviri and G. Predieri, Dalton Trans., 2010, 39, 4479–4481 RSC.
- The combination of multiple inequivalent lacunary fragments within a single POM framework is relatively uncommon but it has been demonstrated in several previous instances, mostly within the structurally labile silicotungstate series of HPOMs (see ref. 4a,5e for example). Combining lacunary building blocks from different parent clusters has however proven to be a considerable challenge as a result of the differing range of pH under which Keggin and Wells–Dawson fragments are generally stable.
- This phenomena is common, particularly in high nuclearity POM clusters, and we have observed it before in related previous work (see ref. 8b,c for example). Whilst this makes accurate absolute peak assignments difficult, the relationship of the major peaks in each spectrum allows us to unambiguously identify them as sequentially charged anions containing the cluster of interest, particularly where the peak envelope is poorly resolved or especially broadened.
- (a) A. E. Bolzán and A. J. Arvia, J. Electroanal. Chem., 1992, 322, 247–265 CrossRef; (b) C.-C. Hu and T.-C. Wen, Electrochim. Acta, 1996, 41, 1505–1514 CrossRef CAS.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- R. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data and refinement parameters, ESI-MS experimental details and full assignments, discussion of structure, assembly and solution transformations of 3, cyclic voltammetry. ICSD 426772–426774. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00075c |
| ‡ These authors contributed equally towards the preparation of this manuscript. |
| This journal is © the Partner Organisations 2014 |