
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally cleavable imine base/isocyanate adducts and oligomers suitable as initiators for radical homo- and copolymerization†
I.
Polenz
*ab,
A.
Laue
b,
T.
Uhrin
b,
T.
Rüffer
c,
H.
Lang
c,
F. G.
Schmidt
d and
S.
Spange
*b
aDr. Ingmar Polenz, Max-Planck Institute for Dynamics and Self-Organization Droplets, Membranes and Interfaces, D-37077 Goettingen, Germany. E-mail: ingmar.polenz@ds.mpg.de
bProf. Dr Stefan Spange, Dipl.-Chem. Andreas Laue and Dipl.-Chem. Tamás Uhrin, Chemnitz University of Technology, Polymer Chemistry Laboratory, D-09107 Chemnitz, Germany
cProf. Dr Heinrich Lang and Dr. Tobias Rüffer, Chemnitz University of Technology, Inorganic Chemistry Laboratory, D-09107 Chemnitz, Germany
dDr Friedrich Georg Schmidt, Evonik Industries AG, D-45772 Marl, Germany
First published on 2nd September 2014
Abstract
The addition of isocyanates to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds of imines gave triazinedione heterocycle structures; their thermal properties are reported. Mono-isocyanates were used to form 2
N double bonds of imines gave triazinedione heterocycle structures; their thermal properties are reported. Mono-isocyanates were used to form 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts with the imine bases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and 2-tert-butyl-1,1,3,3-tetramethylguanidine (tBuTMG). A 2:1 stoichiometry of the adducts was proven by NMR and IR spectroscopy and single crystal X-ray diffraction; certain cleavage temperatures (70 and 160 °C) were measured. Thermal analysis (TG-MS) of adducts indicated the release of free isocyanate during adduct cleavage. Furthermore, a new class of step-growth oligomers (MN = 750–7000 g mol−1) composed of multi-functional isocyanates and these imine bases was introduced. Their systematic spectroscopic and thermal analysis was shown, revealing the similarity in their chemical properties to the 2:1 adducts. Radical homo- and copolymerization of acrylates was initiated by the meta-stable adducts and oligomers of this work; the generation of novel telomeric block-copolymer architectures composed of polyacrylate and oligourea building blocks was demonstrated.
:1 adducts with the imine bases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and 2-tert-butyl-1,1,3,3-tetramethylguanidine (tBuTMG). A 2:1 stoichiometry of the adducts was proven by NMR and IR spectroscopy and single crystal X-ray diffraction; certain cleavage temperatures (70 and 160 °C) were measured. Thermal analysis (TG-MS) of adducts indicated the release of free isocyanate during adduct cleavage. Furthermore, a new class of step-growth oligomers (MN = 750–7000 g mol−1) composed of multi-functional isocyanates and these imine bases was introduced. Their systematic spectroscopic and thermal analysis was shown, revealing the similarity in their chemical properties to the 2:1 adducts. Radical homo- and copolymerization of acrylates was initiated by the meta-stable adducts and oligomers of this work; the generation of novel telomeric block-copolymer architectures composed of polyacrylate and oligourea building blocks was demonstrated.
Introduction
Since their discovery in 1848 by Wurtz, isocyanates have played an important role in industry.1 The synthesis of polyurethanes by the reaction of a polyester diol with a diisocyanate, discovered by Bayer and co-workers in 1937,2 made diisocyanates one of the major chemicals produced in the world.3,4 The main advantage of the use of isocyanates in industry is their high reactivity even at low temperatures or in highly viscous systems, ensuring a rapid conversion and an efficient mode of operation. One major drawback is the inherent toxicity of isocyantes.3 An approach to solve this problem is to temporarily transfer the isocyanate into meta-stable adducts, which are formed by reversible, isocyanate-based covalent bonds. The most common blocking agents bear hydrogen active nucleophile functionalities of the type X–H, where X can be oxygen, sulfur, nitrogen or carbon. The reagents have a moderate acidity in the pKa range of 10 to 20 and facilely undergo addition reactions with the electrophile isocyanate group at low temperatures, as depicted in Scheme 1.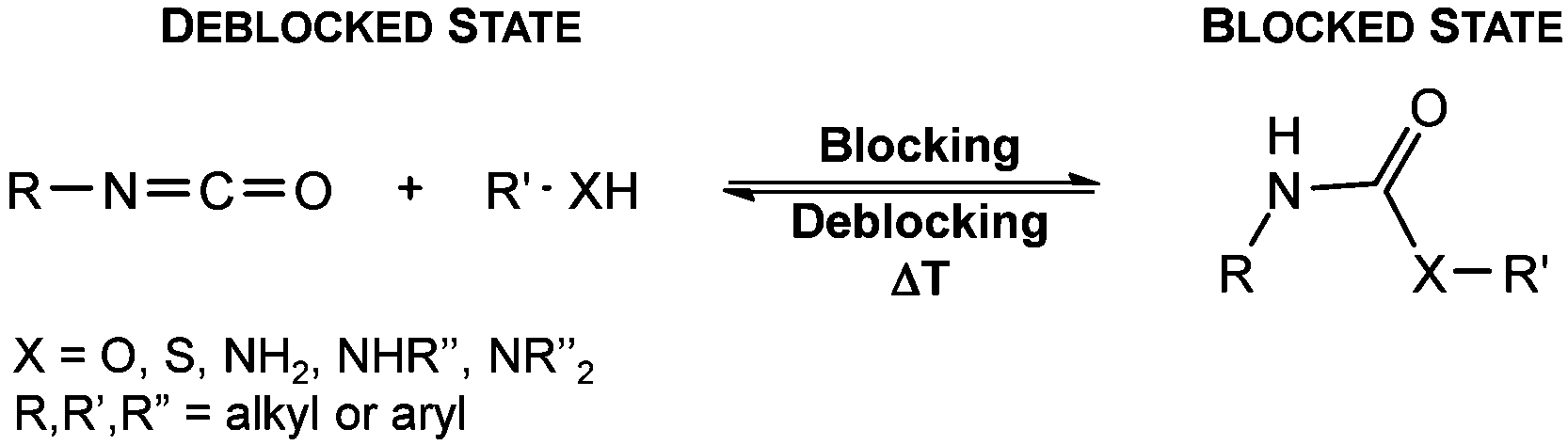 | ||
| Scheme 1 Principle of established isocyanate blocking reactions with acidic X–H compounds. | ||
At lower temperatures, isocyanates are in a blocked state. Therefore, the reactivity is decreased and hence the toxicity is remarkably reduced.3 Free isocyanates can be regenerated out of these adducts at elevated temperatures. The reversible blocking of isocyanates plays an important role in polyurethane engineering processes, which was reported extensively.3,5–8 Among others, malonic esters, oximes, phenols, lactams and secondary aniline derivatives have been studied in detail.9–14 Furthermore, the formation of urethdiones, dimers of isocyanate groups, is another way to produce blocked isocyanates. The advantage of those systems is that no additional blocking agent is required. However, this method is applicable solely to aromatic isocyanates. In addition, specific catalysts such as toxic phosphines are necessary for their formation and, due to the reversibility of the reaction, residues of highly reactive NCO-groups are still found in the reaction mixture.15 A recent contribution16 to the field revealed a new method for introducing an isocyanate group, blocked by 3,5-pyrrazole, into polymer architectures. However, post-modification of the isocyanate group with amines and alcohols was demonstrated to occur at 130 °C. Thus, up to now it is highly desirable in industry and academic research to find methods for the selective control of the isocyanate reactivity for reasonable and safe handling of this versatile group.
In this work, we report on the reversible adduct formation of mono- and bifunctional isocyanates with imine bases. Thermal properties and a detailed structure analysis of the products are provided. The adduct formation mechanism of mono-functional isocyanates with amidines was investigated earlier by Ulrich, Richter and co-workers.17–19 They reported that their strong exothermic reaction at room temperature results in the formation of 2:1 adducts bearing six-membered heterocyclic rings, as shown in Scheme 2a.4,20
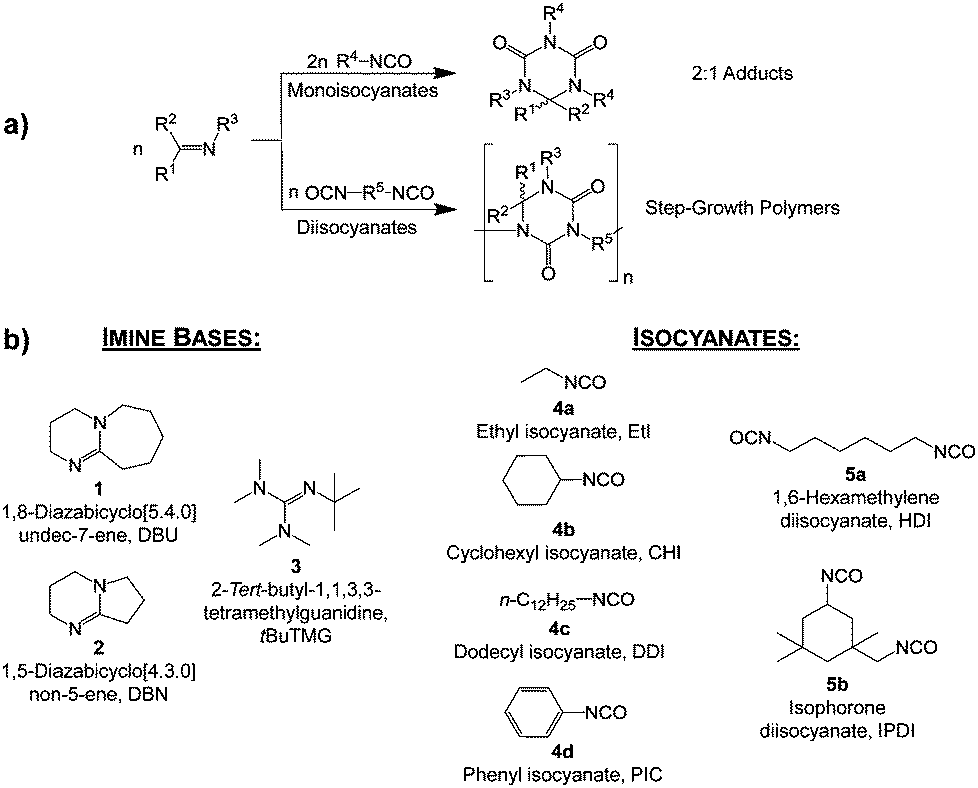 | ||
| Scheme 2 (a) Schematic view of the addition reactions of mono- and diisocyanates and imines yielding 2:1 adducts and step-growth polymers as well as (b) structures of imine bases and isocyanates used in this study. R1–R5 are alkyl groups. | ||
Recently, we have shown that isocyanates undergo radical formation when treated with imine bases and vinyl monomers.21,22 The subject of the present study is the investigation on obvious adduct formations of amidine and guanidine derivatives with isocyanates as special cases. The specific novelty of this work is that we employed a non-acidic blocking strategy. Furthermore, the quasi-bi-functionality of the imine moiety suggests its potential as a component for step-growth polymerization with diisocyanates. Herein, we show that multiple reactions of diisocyanates and imine bases result in a polyaddition mechanism yielding meta-stable step-growth polymers with a cyclic repeating unit (see Scheme 2a). A systematic study of this new polymer class is provided.
The structures of reactants used in this work are given in Scheme 2b. Thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and mass spectrometry are used to clarify the cleavage process in detail. With the approach described in this work we generate blocked isocyanates that are stable at room temperature and release free isocyanate at slightly elevated temperatures in the range between 70 and 160 °C. The adduct cleavage temperature is mainly influenced by the isocyanate reactivity and steric hindrance; adducts and oligomers composed of aromatic or voluminous isocyanates have a lower cleavage temperature than aliphatic isocyanates.
The cleavage analysis results reveal that radical intermediates are formed. In this context, we investigated the potential of the adducts and oligomers as initiators for the radical homo- and copolymerization of acrylic vinyl monomers. We showed that during the cleavage of the appropriate imine base–isocyanate adducts and oligomers, radical species are formed that, in combination with acrylates, result in the formation of high molecular weight polyacrylates. When using oligomers for the acrylate polymerization, novel telomeric block-copolymers composed of polyacrylate and oligourea building blocks are formed that are characterized by gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) spectroscopy and differential scanning calorimetry (DSC). With our approach, a novel path for the reversible formation of blocked isocyanates is given that enables the fabrication of functional block copolymers and materials based on the isocyanate structure.
Results and discussion
The reaction of isocyanates with imines proceeds immediately by the formation of 2:1 adducts at room temperature, as indicated in the Experimental section. This addition is similar to most reactions of isocyanates with N-nucleophiles, an exothermic reaction.4,20 Hence, all syntheses are carried out at 0 °C to prevent both side product formation and the back reaction. The DBN and DBU adducts with alkyl and aryl mono isocyanates are obtained in high to almost quantitative yields (56–99%). The adduct formation mechanism was proposed earlier by Ulrich, Richter and co-workers;17–19 the suggested reaction path and analysis results of this work are given in Scheme 3.
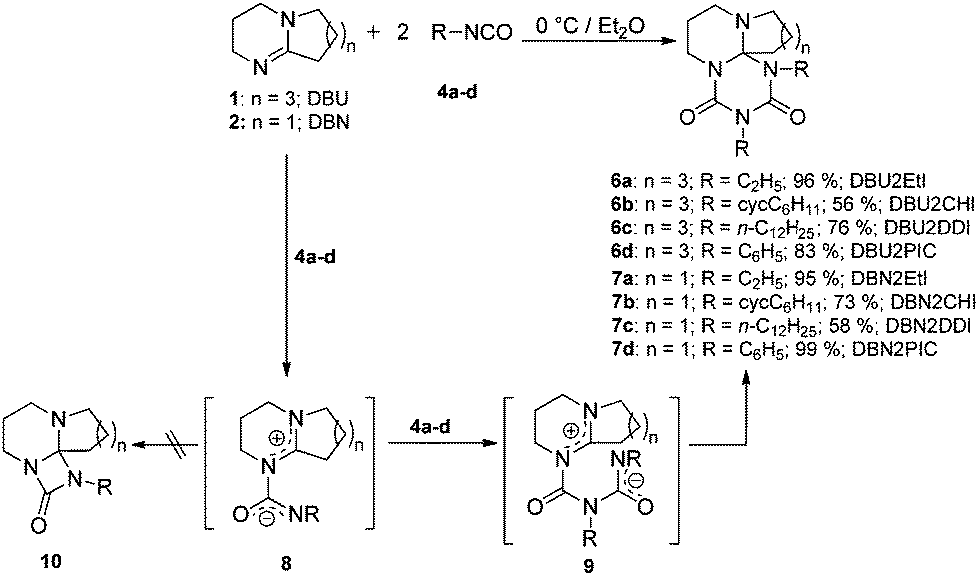 | ||
| Scheme 3 Schematic formation of the adducts 6 and 7 and the mechanism of stepwise addition of two mol isocyanate to one mol imine.17–19 | ||
Adduct formation proceeds via a two-step mechanism. In the initial step, one mole of isocyanate reacts with the nucleophilic imine function of the amidine to the zwitterionic intermediate 8. The rate of this reaction is mainly determined by the electron withdrawing character and the steric hindrance of the group R in 4 (see Scheme 2). The reaction continues by addition of another mole of isocyanate to the zwitterionic intermediate 8 to generate 9. The second isocyanate addition occurs faster than the first one. The formation of tricycles 10 is not observed; moreover, the addition of another mole of isocyanate yields tricycles 6 and 7. The structure of the so-formed six-membered rings of adducts 6 and 7 consists of a triazinedione heterocycle. In Fig. 1, the results of the X-ray single crystal structure of 6a is shown. The analysis clearly manifests the hypothesis of the formation of a tricyclic structure containing a triazinedione heterocycle. In particular, the quaternary carbon C10 (Fig. 1) is prominent for structures 6 and 7 that give characteristic 13C NMR shifts. The presence of tricyclic structures 6 and 7 is also proven by the signal of the quaternary C10 in the 13C NMR spectrum close to 90 ppm (CDCl3); more detailed information is included in the ESI.†
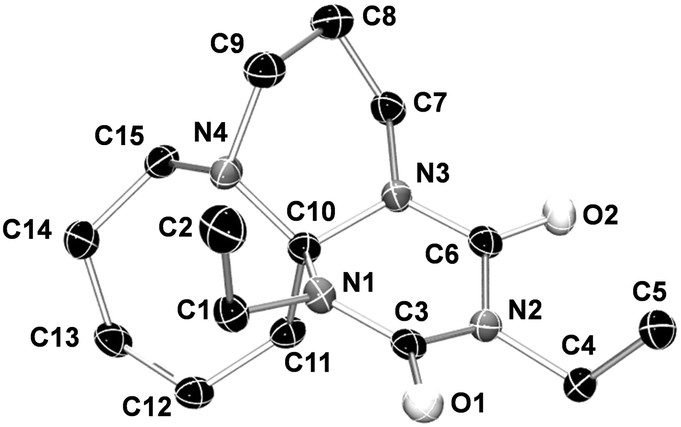 | ||
| Fig. 1 Results of the single-crystal X-ray diffraction analysis of 6a (97.3% ellipsoid probability). 6a is formed racemic, the S-enantiomer is depicted. A table of relevant bond lengths and angles is shown in the ESI.† | ||
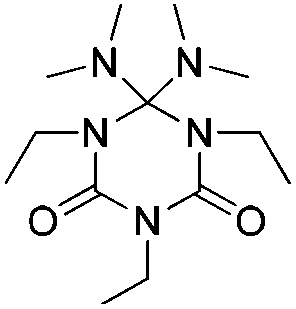
The adduct formation using tBuTMG, in conjunction with isocyanates is more complex than that of 6 and 7. Analysis results, single crystal structure of a model compound, as well as the mechanistic background of this reaction and the cleavage products, are provided in the ESI† (Fig. S2).
To elucidate the thermal profiles and the processes occurring at elevated temperatures of compounds 6 and 7, the DSC curve of 7a is shown in Fig. 2. When systematically heating the sample to 300 °C, the thermogram shows a sequence of two endothermic processes. The first peak (Tonset = 75.8 °C, Tpeak = 80.3 °C, Q = 0.27 J mol−1) indicates the melting of 7a, while at higher temperatures (Tonset = 161.8 °C, Tpeak = 194.5 °C, Q = 2.87 J mol−1), cleavage of 7a starts which is the opposite reaction to the exothermic 7a formation. Cleavage temperatures of 6 and 7 are in the range between 69 and 162 °C; the corresponding cleavage enthalpies are 0.02–2.87 J mol−1 and a function of the isocyanate type. Focusing solely on the isocyanate, the cleavage enthalpies and temperatures, and hence the stability, decreases for the following: R = C2H5 > cyc-C6H11 > n-C12H25 > C6H5. There are no significant adduct cleavage temperature differences when changing the imine base type (DBN or DBU).
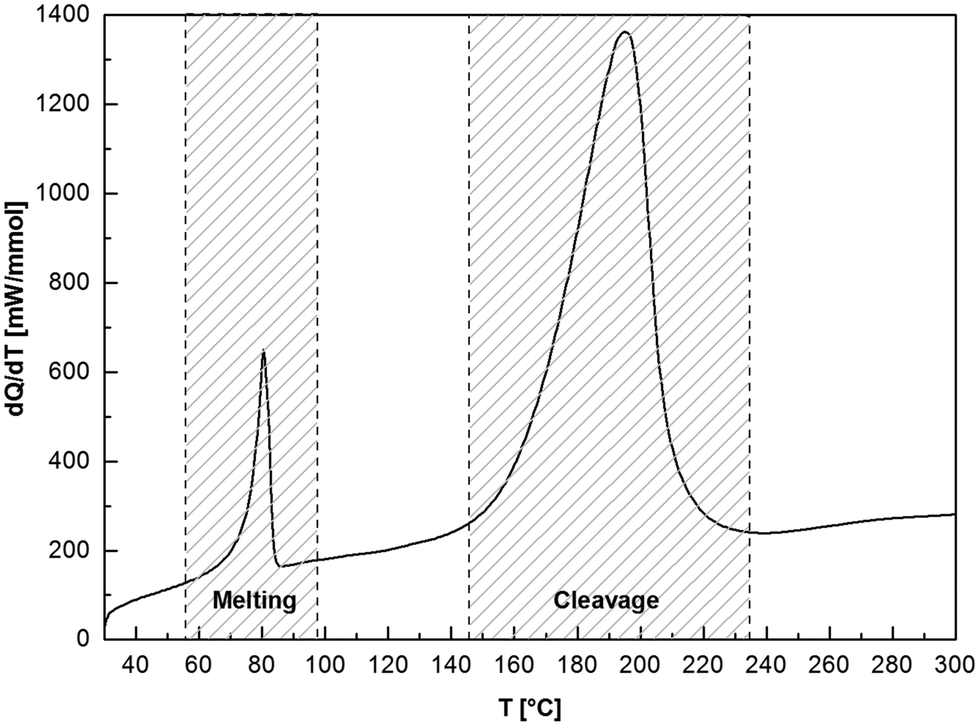 | ||
| Fig. 2 DSC-curve of 7a (DBN2EtI) at a heating rate of 10 K min−1. The melting occurs at Tonset = 75.8 °C, Tpeak = 80.3 °C, Q = 0.27 J mol−1 and cleavage is detected at Tonset = 161.8 °C, Tpeak = 194.5 °C, Q = 2.87 J mol−1. | ||
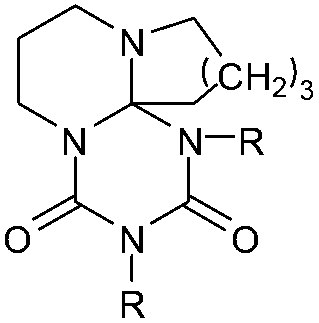
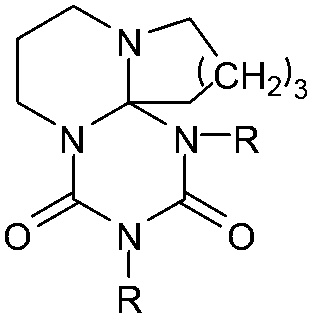
DSC curves of 6, 7 and 11 are measured to verify their thermal properties; analysis details of the adducts are given in Table 1. It was found that the adduct cleavage temperatures are much lower compared to established isocyanate blocking systems, such as blocked isocyanates based on ε-caprolactam, phenols or triazoles.5–8,11–13 Monomeric adducts are obtained by the reaction of DBU, DBN and tBuTMG with the monoisocyanates ethyl isocyanate 4a (EtI), cyclohexyl isocyanate 4b (CHI), dodecyl isocyanate 4c (DDI) and phenyl isocyanate 4d (PIC).
| Adduct | R= | T cleavage [°C] | Q cleavage [J mol–1] | |
|---|---|---|---|---|
| a More details on the formation mechanism of 11 are included in the ESI. | ||||
|
6a (DBU2EtI) | C2H5 | 154 | 1.219 |
| 6b (DBU2CHI) | cyc-C6H11 | 115 | 0.580 | |
| 6b (DBU2DDI) | C12H25 | 76 | 0.035 | |
| 6b (DBU2PIC) | C6H5 | 69 | 0.126 | |
|
7a (DBN2EtI) | C2H5 | 162 | 2.866 |
| 7b (DBN2CHI) | cyc-C6H11 | 125 | 0.623 | |
| 7c (DBN2DDI) | C12H25 | 101 | 0.241 | |
| 7d (DBN2PIC) | C6H5 | 73 | 0.019 | |
|
11 (TMG3EtI)a | C2H5 | 176 | 1.586 |
We recorded ESI-MS and TG-MS spectra to further study separate fragments from the cleavage. The ESI-MS spectrum of 7a is shown in Fig. 3 and a reasonable fragmentation mechanism is given in Fig. 3a. Fragments at m/z = 196 ([DBU–EtI]H+) and m/z = 125 ([DBU]H+) are clearly detected. This indicates a stepwise elimination of two moles of isocyanate from 7a. The intermediate 12 is assumed to have a radical character. The release of free isocyanate from 7a at elevated temperatures is also confirmed by TG-MS measurements; spectra are in the SI. In the TG-MS analysis of 7a, EtI 4a is detected at 192.5 °C. The free isocyanate 4a can be trapped by nucleophilic substances such as methanol or n-butylamine, quantitatively.23,24 When using diisocyanates instead of monoisocyanates, the reaction proceeds as a polyaddition reaction to form oligomeric structures with the triazinedione repeating unit as shown in Scheme 4. Oligomers 13–15 were obtained after washing and drying as foamy white solids that are soluble in polar organic solvents such as chloroform and methanol. Number average molecular weights of 13–15 are in a range between 750 and 7000 g mol−1 (poly dispersity = MW/MN = 1.35–1.75, Table 2). The degree of polymerization of the products is in a range between 2 and 21, so we consider the products to be oligomers. Moreover, oligomers of tBuTMG and isophorone diisocyanate (IPDI) are not obtained; a drastic steric hindrance of both monomers prevents any reaction. The presence of the cyclic triazinedione repeating units in oligomers 13–15 is facilely identified by 13C NMR spectroscopy on the quaternary C-atom that is formed during the cyclization reaction. The 13C NMR spectrum shows the characteristic quaternary C-atom of the oligomers resonating at 92 (13, 14) and 103 ppm (15) (Fig. 1). Furthermore, a broad signal at 121 ppm in the 13C NMR spectra of 13–15 reveals the presence of isocyanate end groups. This hypothesis is also confirmed by IR spectroscopy. Vibrational spectra of oligomers 13–15 show a broad band at 2170 cm−1 that can be assigned to the asymmetric stretching mode of the NCO group, νas. Further analysis results of oligomers 13–15 are summarized in the ESI.† A more detailed synthetic procedure as well as further chemical and physical properties of those oligomers obtainable from amidines and diisocyanates will be reported in a separate work.
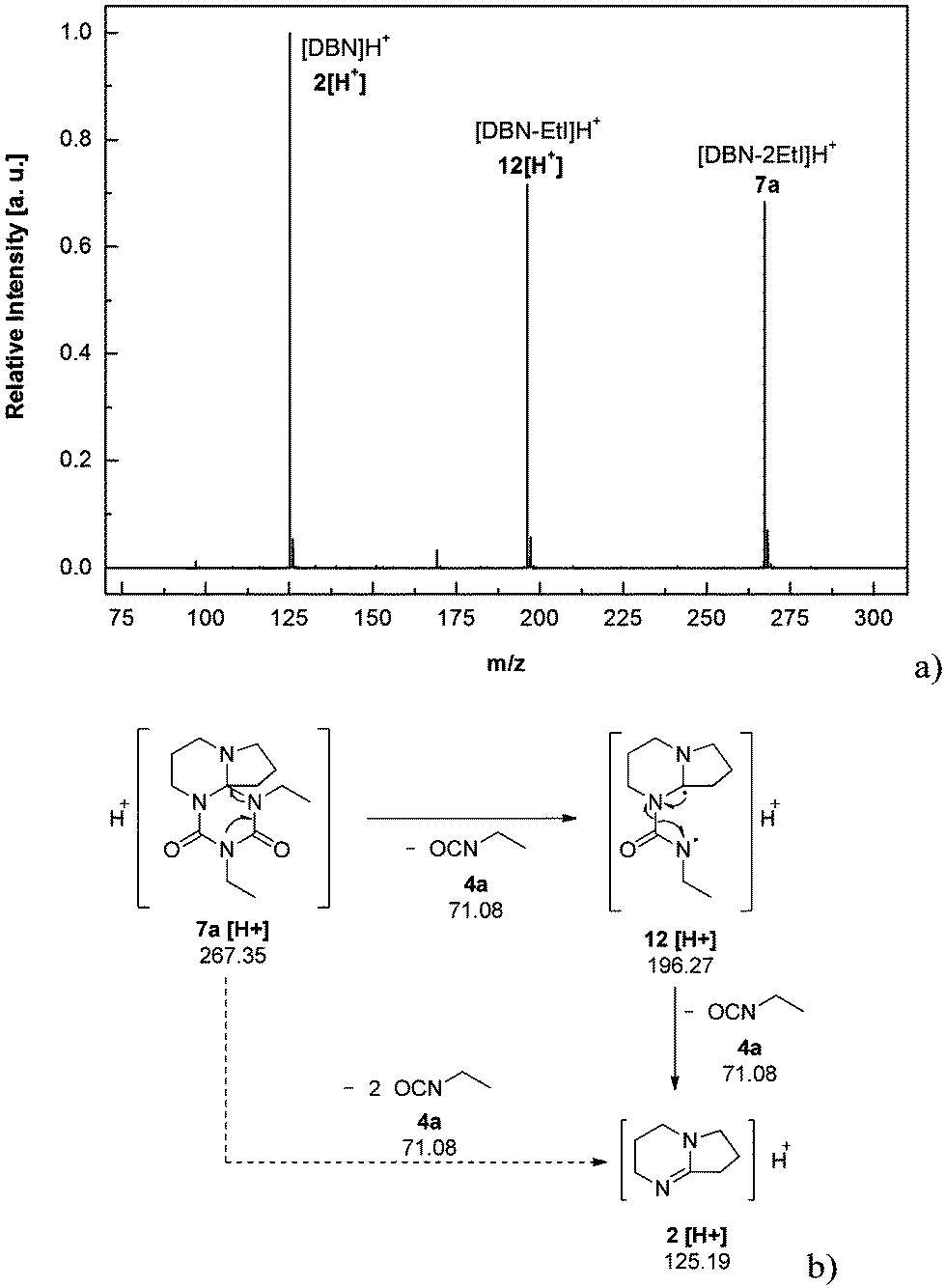 | ||
| Fig. 3 ESI-MS-spectrum of DBN2EtI 7a (a) and presumed fragmentation mechanism of 7a (b). Fragments in (a) correspond to species: m/z = 125.11 [DBN]H+, m/z = 196.15 [DBN–EtI]H+ and m/z = 267.21 [DBN2EtI]H+. The excitation energy is 8500 V. | ||
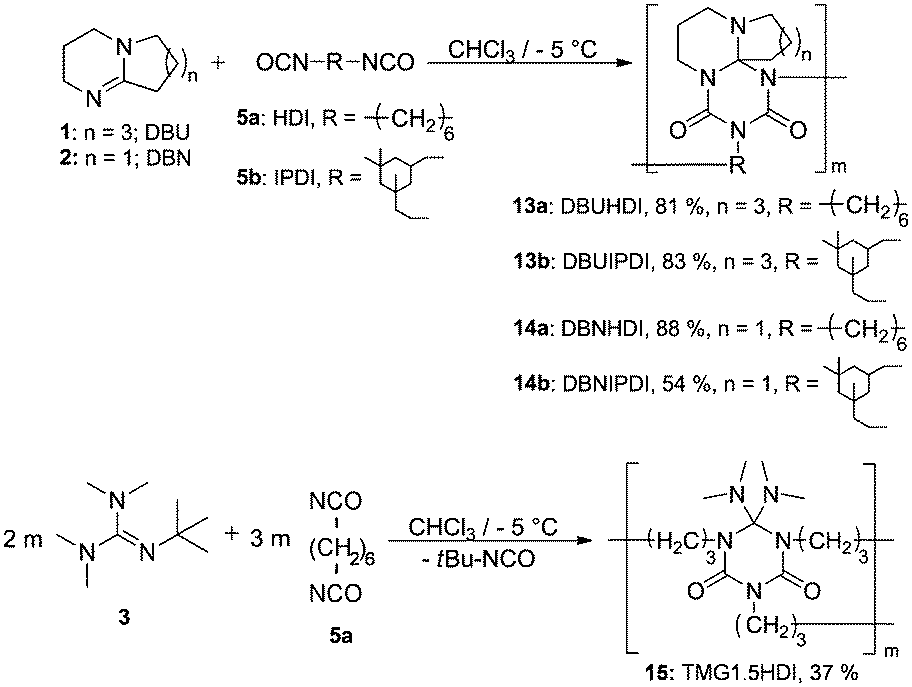 | ||
| Scheme 4 Schematic view of the oligomer synthesis DBUHDI 13a, DBUIPDI 13b, DBNHDI 14a, DBNIPDI 14b and TMG1.5HDI 15. | ||
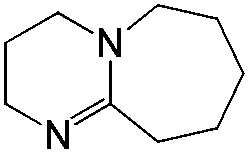
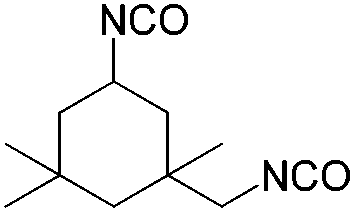
| Amine | Isocyanate | Oligomer | M N [g mol–1] | M W/MN |
|---|---|---|---|---|
|

|
13a | 5000 | 1.71 |
|
13b | 1300 | 1.35 | |
|
14b | 2400 | 1.60 | |

|
14a | 6400 | 1.48 | |
|

|
15 | 780 | 1.30 |
The thermal properties of compounds 13–15 are similar to their monomeric adducts. Fig. 4 shows the cyclic DSC analysis of 14a. When heating 14a initially, a glass transition process, occurring at 63 °C (Q = 4.50 J g−1), was recorded. The reversibility of this process was proven by testing the solubility of the oligomer. A sample heated to 70 °C for 2 h is still completely soluble in chloroform. This glass transition is followed by an endothermic process at 150 °C (Q = 64.95 J g−1) that can be assigned to the engaging cleavage. Systematically, after heating a sample to 185 °C for a period of 3 h, the foamy oligomer becomes insoluble in chloroform. After heating the sample above the cleavage, an exothermic process is detected (Tonset ∼ 200 °C, Q = −7.03 J g−1) that can be assigned to an irreversible transformation and cross-linkage of 14a. An insoluble brownish rubber-like product was obtained after the thermal treatment indicating an inter- and intramolecular cross-linkage. Free isocyanate functions cannot be detected in the product, which was proven by IR spectroscopy. After the second thermal treatment, no cleavage and cross-linking processes are detected; only a glass transition (84 °C) of the tempered oligomer was recorded. In Fig. S3 in the ESI† liquid high resolution 13C NMR and solid state 13C CP MAS NMR spectra of tempered 14a at 70, 185 and 210 °C are presented, supporting the observations described above. In addition, IR studies on the cleavage of adduct 7d are shown in Fig. S5.† It is notable that products containing an aromatic or sterically hindered isocyanate show the lowest cleavage temperature, which is caused by the increased structural stress in these compounds. In Table 3 all adduct and oligomer cleavage temperatures presented in this work are summarized.
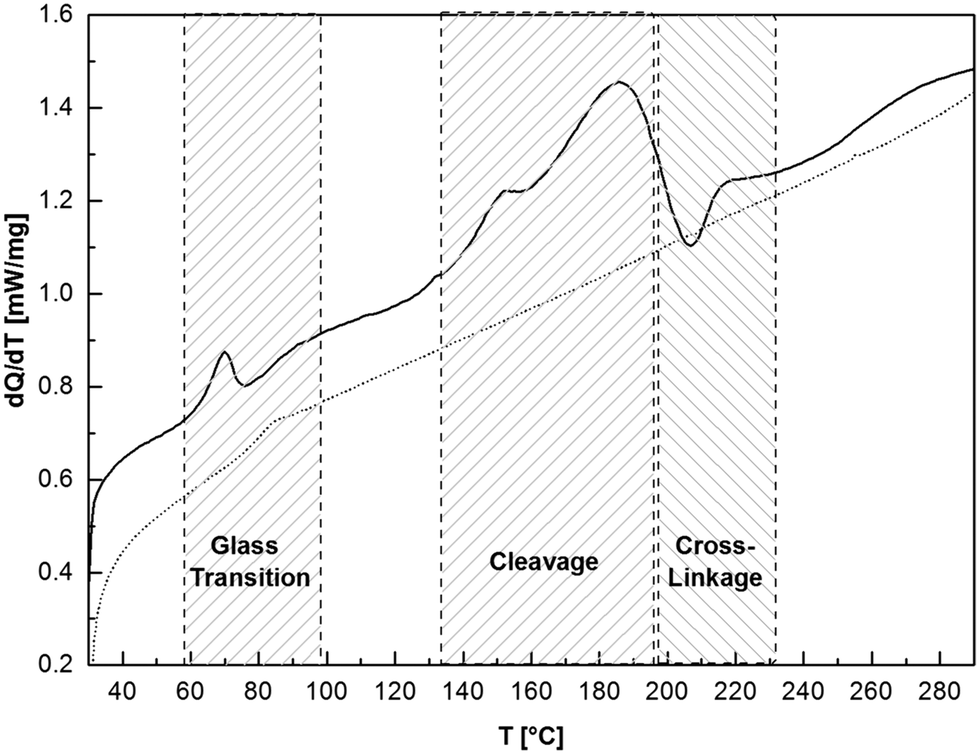 | ||
| Fig. 4 Cyclic DSC curves of DBNHDI 14a (1st –, 2nd …) at a heating rate of 10 K min−1. At the first thermal treatment the glass-transition occurs at Tonset = 62.7 °C, Tpeak = 70.0 °C, Q = 4.50 J g−1, cleavage at Tonset = 150.1 °C, Tpeak = 184.8 °C, Q = 64.95 J g−1 and a cross-linking process is detected at Tonset = 199.7 °C, Tpeak = 207.0 °C, Q = −7.03 J g−1. A glass transition is detected at the second heating at Tpeak = 84 °C. | ||
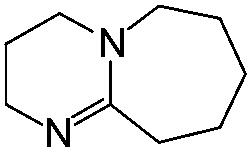
| Imine | ||||
|---|---|---|---|---|
|
|
|
|
||
| Isocyanate | DBU | DBN | tBuTMG | |
| PIC |
|
69 | 72 | — |
| DDI | n-C12H25–NCO | 76 | 73 | — |
| CHI |
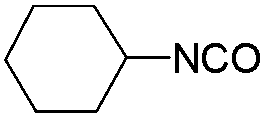
|
115 | 125 | — |
| EtI |

|
154 | 162 | 176 |
| IPDI |
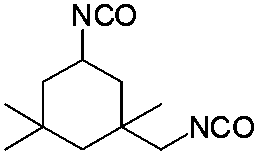
|
116 | 137 | — |
| HDI |
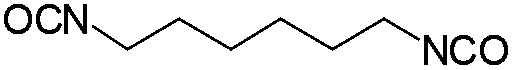
|
130 | 106 | 152 |
One basic question still remains – what is the nature of the fragments at the adduct and oligomer cleavage? For the re-cyclization mechanism of thermal treatment, we assumed the formation of radical species (Fig. 3). To prove our assumption the adducts are thermally cleaved in the presence of acrylic monomers. Remarkably, the monomeric and oligomeric imine base–isocyanate adducts initiate a radical polymerization of methyl methacrylate (MMA) and other acrylic monomers simply by thermal treatment, resulting in high molecular weight polymers. To evidently show the radical character of the propagating species, copolymerization experiments were carried out using co-monomers MMA and methacrylonitrile (MAN). We determined the copolymerization parameters, rMMA and rMAN, by standard methods26,27 and compared the results with literature known r-values of anionic and radical copolymerization of these co-monomers. Table 4 shows the experimental results of the copolymerization using DBU2EtI 6a as the initiator at 80 °C and results from literature, according to radical and anionic copolymerization experiments. These results clearly confirm that the propagating species of this polymerization is of a radical nature.
Furthermore, imine base/isocyanate adducts and oligomers were used for a radical polymerization of MMA. Polymerization conditions and MMA conversions XMMA as well as GPC analysis results of PMMA of selected MMA-polymerizations using adducts 6a,b,d, 7 and 11, as well as oligomers 13 and 15 are shown in Table 5.
| Adduct (Ad) | [MMA]/M | [Adduct]/M | t/h | X MMA/% | M N/g mol–1 | M W/MN |
|---|---|---|---|---|---|---|
| 6a | 3.53 | 0.49 | 6 | 13 | 368,400 | 1.66 |
| 6b | 3.52 | 0.48 | 18 | 8 | 33,000 | 1.35 |
| 6d | 3.52 | 0.46 | 2 | 74 | 36,700 | 1.72 |
| 13a | 9.09 | 0.16 | 2 | 13 | 354,800 | 1.85 |
| 13b | 5.22 | 0.08 | 4 | 21 | 112,200 | 2.38 |
| 7a | 3.59 | 0.49 | 6 | 15 | 264,300 | 1.41 |
| 7b | 3.52 | 0.48 | 18 | 25 | 33,100 | 2.26 |
| 7c | 3.52 | 0.48 | 18 | 7 | 82,400 | 1.62 |
| 7d | 3.52 | 0.48 | 2 | 82 | 23,500 | 1.89 |
| 11 | 3.51 | 0.52 | 6 | 55 | 112,500 | 1.91 |
| 15 | 9.98 | 0.06 | 1 | 28 | 180,100 | 2.26 |
The higher the stability of adducts and oligomers 6a,b,d, 7, 11 and 13, 15, the higher the average molecular weight of PMMA is, which is consistent with the corresponding initiator concentration. As an example, for the least stable adducts, DBU2PIC 6d and DBN2PIC 7d, the MMA polymerization proceeds rapidly until all of the initiator is consumed. Polymerization of MMA using 6d and 7d is possible even at moderate temperatures (45–70 °C). Consequently, low molecular weight PMMA is isolated (MN = 23.500–36.700 g mol−1). In comparison, the more stable adducts with higher cleavage temperatures give high molecular weight PMMA (112.000–368.400 g mol−1) and the non-cleaved adduct is isolated after polymerization. However, in the case of polymerization with 6b, whose cleavage temperature (115 °C) is below that of 6a (154 °C), the polymer yield is significantly lower. The authors assume that the stability of the radicals formed at the cleavage of 6b (secondary alkyl isocyanate) is increased disfavouring an initiation. We are able to reuse stable adducts for polymerization after their purification. Adducts and oligomers are isolated from the reaction mixture by precipitating the product PMMA in methanol. The initiators are completely soluble in this solvent.
Interestingly, when polymerizing MMA using oligomers 13 and 14, even after purification of the product, still 13 and 14 is being detected in the 1H, 13C NMR and GPC. Product NMR spectra and GPC graphs resulting from the MMA polymerization using DBUHDI 13a are depicted in Fig. 5. No starting material signal is found in the GPC of PMMA-b-DBUHDI, indicating the presence of only one product species. The polymerization product is – like PMMA – insoluble in methanol. In contrast, DBUHDI 13a is well soluble in methanol. Even after an extensive purification of the methanol insoluble product by multiple precipitation and washing in methanol, signals of 13a were still detected. Obviously, the oligomers are covalently bonded to the PMMA chain and a novel class of block-copolymers containing both a polyacrylate and an oligourea backbone is generated. The AB-block structure of PMMA-b-DBUHDI is confirmed by the NMR spectra that have only one broad resonance of the OCH3 ester group of PMMA. Taking into account quantitative elementary analysis and quantitative 1H NMR analysis of the product, the PMMA:DBUHDI ratio is 52:1. Thus, short oligomer blocks of DBUHDI are covalently bonded to long PMMA chains. Hence, the structure more resembles that of a telomeric block-copolymer.
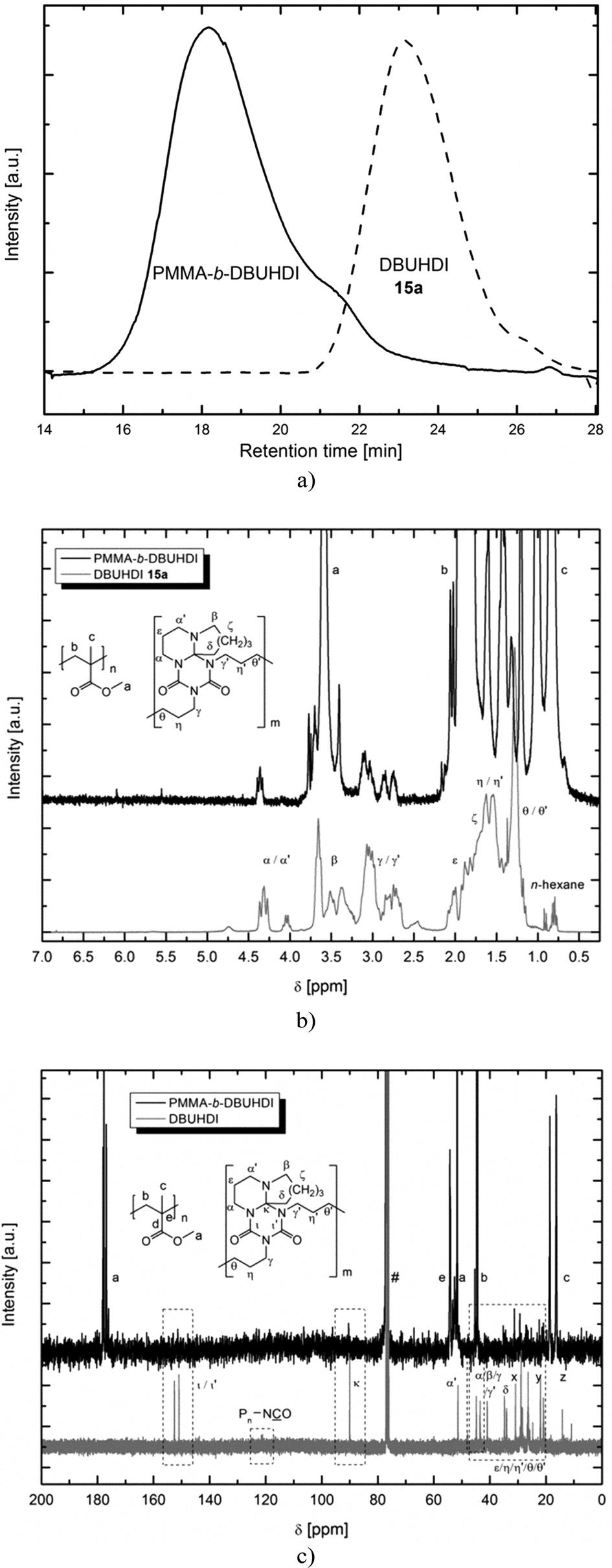 | ||
| Fig. 5 (a) GPC, (b) 1H- and (c) 13C NMR analysis of 13a (–) and block-copolymer PMMA-b-DBUHDI (–). Signals of DBUHDI in the 13C NMR spectrum of PMMA-b-DBUHDI are highlighted by boxes. The number average molecular weight MN of 13a (---) is 5.000 g mol−1 (PDI = 1.71) and that of PMMA-b-DBUHDI (–) is 354.800 g mol−1 (PDI = 1.85). | ||
The short oligomer block is still intact in its reactivity. We record endothermic and exothermic peaks of DBUHDI in the telomeric block copolymers PMMA-b-DBUHDI and PMA-b-DBUHDI in the cyclic DSC, as shown in Fig. 6. The characteristic DSC signals of DBUHDI are slightly shifted to increased temperatures in the block copolymers (Fig. 6). After the thermal treatment of the block-copolymers, insoluble rubber-like (PMA-b-DBUHDI) and robust glassy (PMA-b-DBUHDI) polymers are obtained. Multiple cross-linking of the block-copolymer sequence caused by the radical degradation of the DBUHDI-block occurred. This process is irreversible and influences the properties of the polyacrylate block: no further peaks of DBUHDI are detected in PMA-b-DBUHDI at the second heating process. In addition, the glass transition temperature of PMA is shifted to higher temperatures (20 °C), indicating an increased rigidification of the polyacrylate backbone. These experiments reveal the novel block-copolymers to be promising candidates for the development of new types of programmable composite coatings and resins based on the acrylate and polyurea structure increment.
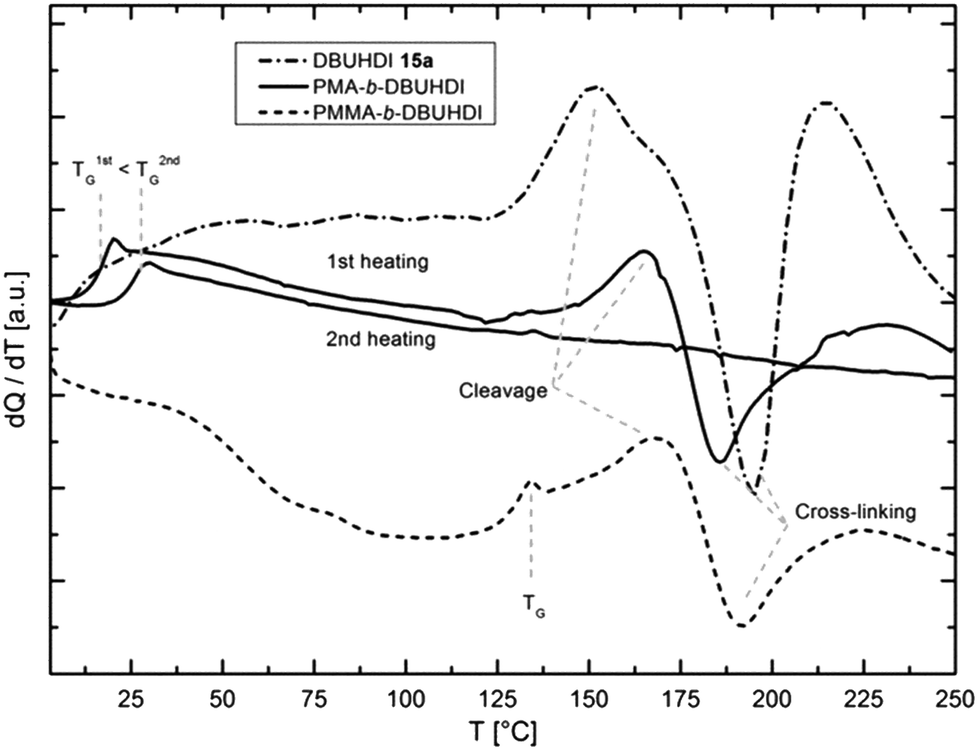 | ||
| Fig. 6 Cyclic DSC curves of DBUHDI (-.-), PMA-b-DBUHDI (1st –, 2nd –) and PMMA-b-DBUHDI (---) at a heating rate of 10 K min−1. DBUHDI: cleavage: Tonset = 130 °C, Q = 39.4 J g−1; cross-linkage: Tonset = 180.0 °C, Q = −38.5 J g−1; 2nd endothermic peak: Tonset = 203.6 °C, Q = 38.2 J g−1. Block copolymer 1st thermal treatment: PMMA-b-DBUHDI: glass transition Tonset = 127 °C, endothermic peak Tonset = 155 °C, exothermic peak Tonset = 175 °C, endothermic peak Tonset = 201 °C; PMA-b-DBUHDI: glass transition Tonset = 13 °C, endothermic peak Tonset = 144 °C, exothermic peak Tonset = 168 °C, endothermic peak Tonset = 200 °C. Block copolymer 2nd heating PMA-b-DBUHDI: glass transition Tonset = 20 °C. | ||
Conclusions
We have shown that the reaction of isocyanates and imines yields metastable thermally cleavable 2:1 adducts with a heterocyclic triazinedione structure. Their stability is mainly a function of the isocyanate nature, which was proven by DSC measurements. The cleavage of the adducts occurs at low temperatures between 70 and 160 °C. Thermal treatment of the adducts leads to a stepwise re-cyclization process with the release of free isocyanate which is shown by ESI-MS studies. Thus, the use of the adducts as latent isocyanate sources is indicated. When converting diisocyanates with the imine bases of this work, a new class of metastable oligomers with six-membered triazinedione rings as repeating units was formed. The number average molecular weights MN of these step-growth oligomers are in a range between 750 and 7000 g mol−1. The oligomers bear reactive NCO-end groups and their cleavage temperatures are similar to those of the monomeric adducts. After the thermal treatment of the oligomers at elevated temperatures, insoluble rubber-like products are formed that result from the stepwise fragmentation of the instable tricyclic structures, followed by intra- and intermolecular rearrangements and cross-linkage. Those adducts and oligomers were effectively used as initiators for radical polymerizations of acrylate monomers. Telomeric polyacrylate-oligourea block-copolymer structures were formed when using oligomers as initiators for the acrylate polymerization. These reactive, well-defined block-copolymers are a novel polymer class with a direct covalent bonding of polyacrylate chains to meta-stable oligourea based cyclic triazinedione structures, which open doors for approaches for the synthesis of programmable hybrid coatings and resins based on these polymer classes.
Experimental section
Materials
The isocyanates ethyl isocyanate (EtI, Aldrich), phenyl isocyanate (PIC, ABCR), cyclohexyl isocyanate (CHI, Merck) dodecyl isocyanate (DDI, ABCR), 1,6-hexamethylene diisocyanate (HDI) and isophorone diisocyanate (IPDI) (Evonik Industries AG) and imine bases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN, Aldrich), and 2-tert-butyl-1,1,3,3-tetramethylguanidine (tBuTMG) were used as received. Monomers methyl methacrylate (MMA) and methacrylonitrile (MAN) were purchased from Sigma Aldrich, distilled prior to use and stored under argon in the freezer (−20 °C). Diethyl ether (Et2O) and chloroform were purified by standard methods.25Measurements
NMR spectra were recorded with a Bruker Avance DRX 250 apparatus (1H: 250.13 MHz, 13C: 62.90 MHz) using CDCl3 as the solvent.Size exclusion chromatography (SEC) measurements were performed using a PL-GPC 50 plus from Polymer Laboratories equipped with a PL-AS RT auto sampler and a PC-RI detector. A PLgel MIXED-D column was used with THF as an eluent with a flow rate of 1 mL min−1 at 40 °C.
ATR-FT-MIR spectra were measured with a FTS-165 spectrometer from BioRad equipped with a golden gate device.
Elemental analysis was performed with a Vario El apparatus from Elementar Analysesysteme GmbH, Hanau.
The ESI mass spectra were recorded with a micrOTOF QII (ESI-Qq-TOF) from Bruker Daltonik. Excitation voltages range between 4500 and 8500 V.
Differential scanning calorimetry measurements were performed on a DSC 1 of Mettler Toledo. All measurements are done in alumina pans at a heating rate of 10 K min−1.
Single crystal X-ray diffraction was done with an Oxford Gemini diffractometer at 110 K applying Mo-Kα (0.71 Å) and Cu-Kα (1.54 Å) radiation. Structure resolution was done using direct methods by means of SHELXS-91.28 Structure refinement was performed with the software SHELX-97.29
TG-MS measurements were recorded with a Thermo-Microscales TG 209 Iris® coupled with a mass spectrometer MS 403C Aëlos® from Netzsch. While heating the sample a constant helium gas flow (30 ml min−1) is applied. As detector of the mass spectrometer a quadrupol-MS (QMA200) is used (measuring range m/z = 0–300).
General acrylate polymerization procedure using adducts 6, 7 and 11 and oligomers 12–14
A solution of 200 mg adduct or oligomer dissolved in 5.3 mL (25 mmol) of MMA was stirred at 80 °C for 3 h. After polymerization, the viscous solution was cooled to room temperature, diluted in 30 mL of chloroform and slowly dripped into 300 mL of ice-cooled methanol under stirring. The precipitate was centrifuged off, dissolved in 15 mL of chloroform and re-precipitated in methanol. After repeating this process three more times, the polymer was dried in a vacuum oven (5 mbar) at room temperature for 12 h. Polyacrylate or polyacrylate-oligourea block-copolymer was isolated as colorless powder in yields ranging between 7 and 82% (details see Table 5).Products were characterized by 1H/13C NMR, FT-IR, DSC and elemental analysis, partially by TG-MS and ESI-MS. Measurement results are concluded in the ESI.† Block co-polymerizations using MA were also performed according to this method; the mixture was stirred under reflux at an oil bath temperature of 100 °C.
Acknowledgements
We thank the Evonik Industries AG for founding of this work.Notes and references
- A. Wurtz and J. Liebig, Ann. Chem., 1849, 71, 326 CrossRef.
- O. Bayer, DE patent, DRP 728981, 1937 Search PubMed.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80 CrossRef CAS PubMed.
- H. Ulrich, Chemistry and Technology of Isocyanates, John Wiley & Sons Ltd, Chichester, England, 1996 Search PubMed.
- Z. Wicks, Prog. Org. Coat., 1975, 3, 73 CrossRef CAS.
- Z. Wicks, Prog. Org. Coat., 1981, 9, 3 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks Jr., Prog. Org. Coat., 1999, 36, 148 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks Jr., Prog. Org. Coat., 2001, 41, 1 CrossRef CAS.
- B. W. Kostyk and Z. W. Wicks Jr., J. Polym. Sci., 1979, 17, 2423 CAS.
- H. Kothandaraman and R. Thangavel, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2653 CrossRef CAS.
- A. Mühlebach, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 753 CrossRef.
- (a) S. Subramani, Y. J. Park, Y. S. Lee and J. H. Kim, Prog. Org. Coat., 2003, 48, 71 CrossRef CAS; (b) X. Tassel, D. Barbry and L. Tighzert, Eur. Polym. J., 2000, 36, 1745 CrossRef CAS.
- S. Subramani, I. W. Cheong and J. H. Kim, Prog. Org. Coat., 2004, 51, 329 CrossRef CAS PubMed.
- A. Sultan Nasar, S. Subramani and G. Radhakrishnan, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1815 CrossRef.
- (a) H. J. Laas, R. Halpaap and J. Pedain, J. Prakt. Chem., 1994, 336, 185 CrossRef CAS; (b) E. Querat, L. Tighzert, J. P. Pascault and K. Dušek, Angew. Makromol. Chem., 1996, 242, 1 CrossRef CAS.
- S. Bode, M. Enke, H. Göris, S. Hoeppener, R. Weberskirch, M. D. Hager and U. S. Schubert, Polym. Chem., 2014, 5, 2574 RSC.
- H. Ulrich, B. Tucker and A. A. R. Sayigh, Angew. Chem., Int. Ed. Engl., 1968, 7, 291 CrossRef CAS.
- R. Richter, Tetrahedron Lett., 1968, 48, 5037 CrossRef.
- R. Richter, Chem. Ber., 1968, 101, 3002 CrossRef CAS.
- S. Ozaki, Chem. Rev., 1972, 72, 457 CrossRef.
- I. Polenz, F. G. Schmidt and S. Spange, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1655 CrossRef.
- I. Polenz, F. G. Schmidt and S. Spange, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 687 CrossRef CAS.
- J. Alsarraf, F. Robert, H. Cramail and Y. Landais, Polym. Chem., 2013, 4, 904 RSC.
- J. Alsarraf, Y. A. Ammar, F. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249 CrossRef CAS.
- K. Schwetlick, Organikum, Wiley-VCH Verlag, Weinheim, 23 edn, 2009 Search PubMed.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer Handbook, Wiley-Interscience, New York, 1999 Search PubMed.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 2, 259 CrossRef.
- (a) A. Altomare, G. Cascarano, C. Giacovazzo and A. Gualardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXTL Version 5.1, An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Siemens Analytical X-Ray Instruments, Madison, WI, 1990 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1012671, 1012672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4py01002g |
| This journal is © The Royal Society of Chemistry 2014 |