 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure related transport properties and cellular uptake of hyperbranched polyglycerol sulfates with hydrophobic cores†
Florian
Paulus
a,
Dirk
Steinhilber
a,
Pia
Welker
b,
Dorothea
Mangoldt
b,
Kai
Licha
b,
Harald
Depner
c,
Stephan
Sigrist
c and
Rainer
Haag
*a
aInstitut für Organische Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bmivenion GmbH, Robert-Koch-Platz 4, 10115, Berlin, Germany
cInstitut für Biologie, Freie Universität Berlin, Takustraße 6, 14195 Berlin, Germany
First published on 15th May 2014
Abstract
A set of six hydrophobically derivatized polymers based on polyglycerol sulfates have been investigated to determine the influence of scaffold architecture on the encapsulation properties of hydrophobic guests. Each of three block and statistical copolymers has been synthesized with phenyl, naphthyl, and biphenyl substituents in a one-pot procedure. The copolymers have been functionalized with sulfate groups in order to introduce an electrostatically repulsive surface that can stabilize the aggregated carriers. In addition, sulfates provide a highly active targeting moiety for inflammation and cellular uptake. UV measurements show a supramolecular encapsulation of the investigated guest molecules in the low μM range. The transport studies with pyrene and an indocarbocyanine dye further indicated a core–shell-type architecture which provides a distinct amphiphilicity as required for supramolecular guest complexation. The combination of a host functionality with an active sulfate targeting moiety has been used to investigate the structure related cellular transport properties.
Introduction
Perfectly branched dendrimers and hyperbranched polymers are often considered as scaffolds in polymer therapeutics.1–5 Dendritic macromolecules find broad applications as carrier systems in drug delivery and have been extensively studied for several decades.6–9 The application of macromolecular carrier systems utilizes several advantages, e.g., the enhanced permeation and retention (EPR) effect, protection of the active guest molecule from an auto-immune response, and prolonged blood circulation times.10,11 For the preparation of macromolecules which can serve as host molecules, two general strategies are utilized: (i) the organized polymeric micelles are stabilized by covalent intramolecular cross-linker,12–14 and (ii) the preparation of so-called unimolecular micelles, which resemble a micellar structure in one single molecule.15,16 The polymeric micelles mainly consist of a core–shell-like (CS) structure with a hydrophobic core and a hydrophilic shell with optional surface functionalization.A detailed study on the aggregation phenomena of amphiphiles and the influence of, e.g., temperature and concentration on the final micelles, determined basic principles for self-organization and spontaneous self-assembly.17 Based on this knowledge, a unimolecular, symmetrical, four-directional hydrocarbon cascade polymer was studied for its transport properties.18 The micellanoic acid derivatized spherical amphiphile with 36 carboxylic acid functionalities which were exclusively located on the surface showed good interactions with guest molecules, e.g., Phenol Blue (PB) or fluorescent chlortetracycline (CTC). The guest molecules were included within the lipophilic infrastructures of the spherical micellanoates. The carboxylic acid groups provided a good hydrophilicity of the host–gest-complex and sufficient water solubility. Other unimolecular, polyanionic micelles were synthesized via a multistep procedure using 3,5-dihydroxy benzyl alcohol building blocks and final modification of the surface functionalities to obtain carboxylates.19 The investigated architectures were able to solvate hydrophobic molecules, e.g., pyrene similar to micelles based on the well-known surfactant sodium dodecyl sulfate (SDS).
All depicted architectures emphasise the necessity for a high surface functionality to introduce to the shell with a polarity gradient different from the lipophilic core. Using hyperbranched polyglycerol (hPG), a biocompatible scaffold with similar properties which can be easily synthesized on a large, semi-industrial scale was introduced.8,20–25 Because of a spherical scaffold, high surface functionality, and a flexible polyether backbone, hPG has become a potent scaffold for various biomedical applications.26–28 In the present study we investigate the structure dependent transport properties of aromatically derivatized hyperbranched polyglycerol sulfates (hPGS). Peripheral sulfate groups were introduced due to their outstanding anti-inflammatory potential and as active targeting moieties in biomedical applications.29,30 To obtain the hydrophobically derivatized, amphiphilic architectures, three glycidol derivatives, namely, 2-(phenoxymethyl)oxirane (1), 2-(naphthalen-1-yloxymethyl)-oxirane (2) and 2-[(4-phenylphenoxy)methyl]oxirane (3), were copolymerized with glycidol. A set of six scaffolds, applying three block and three statistical copolymerizations, were synthesized. The molar monomer feed ratios were kept constant for the same hydrophobic comonomer to obtain a hydrophobic polymer part with Mn = 2000 g mol−1 and a hydrophilic hPG matrix with Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. The multifunctional polymeric scaffolds which can stabilize nanoparticles (NPs) with their amphiphilic structure and possess an active targeting moiety were investigated for the first time.
000 g mol−1. The multifunctional polymeric scaffolds which can stabilize nanoparticles (NPs) with their amphiphilic structure and possess an active targeting moiety were investigated for the first time.
Results and discussion
Synthesis
The monomer to initiator ratio for all synthesized copolymers was adjusted to yield a hydrophilic PG matrix of Mn = 10000 g mol−1 with a hydrophobic contribution of Mn = 2000 g mol−1 to yield an overall targeted molecular weight of 12000 g mol−1. 1,3,5-Trihydroxybenzene (THB) was applied as an initiator for the block copolymerization to obtain a star-like hydrophobic core with an additional central benzene moiety (Scheme 1). Trimethylolpropane (TMP) was used as an initiator for the statistical copolymerization (Scheme 2). We assumed that the hydrophobicity of the core for the statistical copolymer was less important as the polymer architecture is less defined. Complete characterization of the synthesized block and statistical copolymers with 1H, 13C NMR spectra and GPC traces are presented in the ESI.†
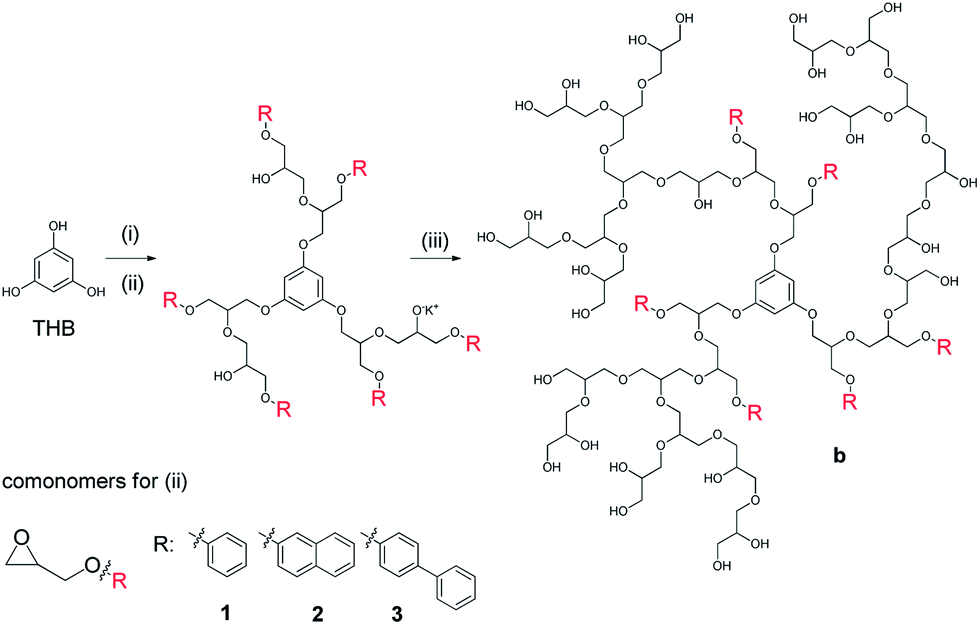 | ||
| Scheme 1 Synthesis of core–shell type, block copolymers b1 to b3 including steps: (i) KOtBu, NMP, 120 °C, 1 h; (ii) comonomer 1, 2, 3 in THF/NMP, 120 °C, addition over 18 h; (iii) glycidol in THF, 120 °C, addition over 18 h. Work up with H+ ion exchange resin. | ||
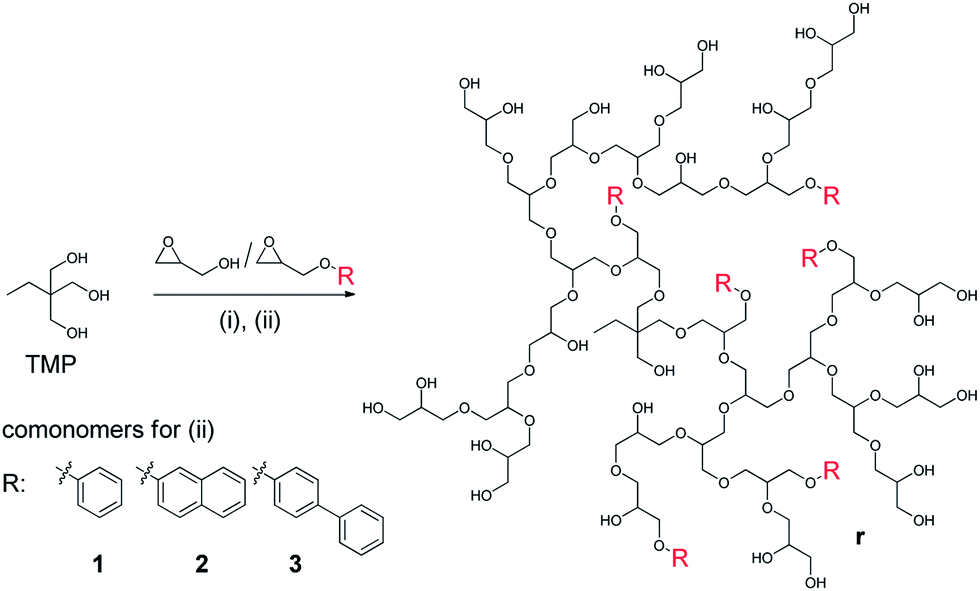 | ||
| Scheme 2 General mechanism for the synthesis of statistical, unsulfated copolymers as precursor for PGS r1 to r3 including steps: (i) KOtBu, NMP, 120 °C, 1 h, and (ii) glycidol/comonomer 1, 2, 3 mixture in THF/NMP, 120 °C, addition over 20 h. Work up with H+ ion exchange resin. | ||
Synthesis of block copolymers
The amphiphilic block copolymers were successfully synthesized by a one-pot, two-step procedure (see Scheme 1). After the synthesis of the hydrophobic core structure starting from the THB initiator, a PG shell was added to provide sufficient solubility in aqueous media. With the presented strategy, it was possible to obtain more defined core shell structures as the monomers were added stepwise.In comparison to post-synthetic modification protocols,32–34 where secondary hydroxyl groups were functionalized all over the scaffold, the hydrophobic moieties were exclusively located in the core in this new approach.
The amphiphilic structures formed large aggregates in buffered solution. Although the hydrophilic outer shell had about 5-fold the molecular mass of the hydrophobic core, it was not enough to prevent aggregation (see Table 1). Therefore we could not expect a unimolecular transport system. The main species present in the particle size distribution ranged between 113 and 160 nm for the OH-terminated block copolymers b1 to b3 before sulfation. The aggregation was not only present in DLS but also visible in the GPC measurements (see Table 1). Even the highly polar, organic solvent NMP could not prevent aggregation. Large aggregates with Mn > 100 kDa were detected by the RI-detector for all block copolymers. Although the main peak from the GPC measurements of all block copolymers was highly aggregated, a small side peak for the unimolecular, separated copolymers was detected. For all block copolymers the Mn values (referenced to PEG standard) were well below the targeted molecular weights except copolymer b1. The measured dispersities with 2.8 (b1) and 3.6 (b2) were high compared to values generally obtained from anionic ring opening polymerization. The dispersity for copolymer b3 was within the expected range.
| Entry | Initiator [mmol] | Comonomer [mmol] | Glycidol [mmol] | I r,target [%] | M n [kDa] (PDI) | M n,Aggr. [kDa] (PDI) | I r,measured [%] | DSc [%] | d [nm] ± SD | Copolymer |
|---|---|---|---|---|---|---|---|---|---|---|
| a Molecular weight of amphiphilic core structure determined by GPC in organic solvent NMP relative to PEG standard. “n.d.” indicates “not detectable”. b Comonomer incorporation was determined from 1H NMR integrals according to eqn (1)–(3) provided in the ESI. c DS was determined by elemental analysis with respect to the sulphur content of the polymer samples.29–31 d Hydrodynamic diameters of the sulfated copolymers measured after intensity; small particles included as secondary species in brackets (occurrence < 2.5%). | ||||||||||
| b1 | 5.00 | 66.6 | 718 | 9.3 | 4.3 (2.78) | 126 (1.91) | 2.6 | 99 | 88 ± 3 | PPhGE-block-hPGS (b1S) |
| b2 | 2.13 | 21.2 | 287 | 7.4 | 1.6 (3.55) | 130 (1.75) | 5.3 | 84 | 143 ± 8 (21 ± 1) | PNpGE-block-hPGS (b2S) |
| b3 | 3.93 | 35.4 | 574 | 6.2 | 2.4 (1.23) | 105 (1.81) | 5.6 | 64 | 136 ± 5 (18 ± 1) | PbPhGE-block-hPGS (b3S) |
| r1 | 0.97 | 13.4 | 144 | 9.3 | 5.1 (1.75) | n.d. | 6.3 | 96 | 222 ± 10 (8 ± 1) | PPhGE-co-hPGS(r1S) |
| r2 | 0.95 | 10.6 | 143 | 7.4 | 5.6 (1.83) | n.d. | 5.7 | 91 | 222 ± 11 (10 ± 1) | PNpGE-co-hPGS(r2S) |
| r3 | 0.94 | 8.8 | 143 | 6.2 | 5.0 (1.87) | n.d. | 4.5 | 93 | 237 ± 8 (21 ± 2) | PbPhGE-co-hPGS (r3S) |
The NMR measurements proved the incorporation of the THB initiator into the polymer scaffold (Fig. S2, (1–3)†). The amount of incorporated comonomer into the hydrophobic core determined by NMR was below the targeted content for b1 and b2 (see Fig. S2†). Although the monomer was added slowly to the initiator, small amounts were consumed by side reactions, e.g., with moisture, and therefore not incorporated into the core structure.35 The reduced values, however, can also be explained by a strong shielding of the inner core protons as well as reduced signals from aggregation. Due to heavy aggregation at higher concentrations, the NMR characterization was performed under high dilution. From experiences in NMR characterization of polymers, the concentration was below an adequate NMR concentration.
Synthesis of statistical composite polymers
The statistical composite polymers were synthesized by simply feeding the batch reactor with a premixed monomer mixture of glycidol with monomer 1, 2, and 3.The same comonomer to glycidol ratio was applied as for the block copolymers b1 to b3 (Scheme 2). Trimethylolpropane (TMP) was used as initiator. The monomer feed was slowly added to the reaction vessel and copolymers r1 to r3 were obtained as brownish, turbid wax after work up. The molecular weights determined by GPC in NMP were in the same range but below the targeted molecular weights. No aggregate peaks were detected in the GPC measurement for the OH-terminated statistical copolymers before sulfation, but particle sizes were determined by DLS between 235 and 409 nm. These sized aggregates are usually filtered on the pre-column of the GPC equipment. The Mn values for all copolymers were below the targeted molecular weights with dispersities between 1.8 and 1.9 which are comparable to polyglycerol homopolymers with similar size.
The resonance signals from the initiator protons as well as the reduced comonomer signals may be explained with the broad molecular weight distribution resulting in a large number of different copolymer species. Although DMSO was used for the NMR measurements, strong aggregation was observed which consequently reduces the quality of the NMR resonance signals.
The hydrophobic comonomers were incorporated into the PG matrix similarly to the block copolymers but were again below the targeted values, especially for r1. Assuming comparable reactivity to the other monomers and a statistical incorporation into the scaffold, the resonance signal was probably reduced by shielding and aggregation phenomena similar to the block copolymers. This effect was more pronounced because of the aggregate size determined by DLS. To determine the reactivity of each glycidol derivative applied here, the reaction kinetics need to be investigated in more detail. From this investigation it would be possible to determine the exact position of the hydrophobic cores within the polyglycerol matrix.
Polyglycerol sulfate derivatives with hydrophobic cores
All the investigated copolymer species were sulfated to introduce an electrostatic repulsion/stabilization, including a targeting and solubilization function. The strategy was necessary because preliminary encapsulation experiments with pyrene indicated heavy aggregation during loading which caused the complexes to precipitate even at low concentrations. Therefore, all copolymer species were sulfated using an excess of sulfamic acid (NH3SO3) in NMP as presented in Scheme 3.36 | ||
| Scheme 3 Sulfation of hydroxyl terminated polymer architectures: (i) NH3SO3, NMP, 90 °C, 3 d; (ii) neutralization with NaOH to pH 10–12. | ||
The lower reactivity than in other SO3 complexes was compensated by increasing both reaction temperature and time.36,37 The degrees of sulfation (DS) are presented in Table 1 (data from combustion analysis available in Table S1†). Although the sulfation of all copolymers was conducted according to the same protocol, the DS for polymers b2S and b3S decreased. This reduction may be a result from aggregation during the reaction. Although all samples were sonicated prior to heating and extensively stirred throughout the reaction, aggregation could not be prevented during functionalization. Therefore, a certain number of hydroxyl groups were not sulfated. Its amphiphilic character and the accessibility of the sulfate groups is in line with the discussed assumption.
The NMR characterization revealed an immense shielding of the core protons for the block copolymers b1S to b3S (Fig. S4, 1–3†). This might also have been a result from aggregation as determined from DLS (Fig. 1). A small deviation from the baseline indicated the presence of the aromatic moieties, however. The spectra for the statistical copolymers r1S to r3S (Fig. S4, 4–6†) proved the shielding and aggregation theory, and the homogeneously incorporated hydrophobic moieties showed adequate resonance signals.
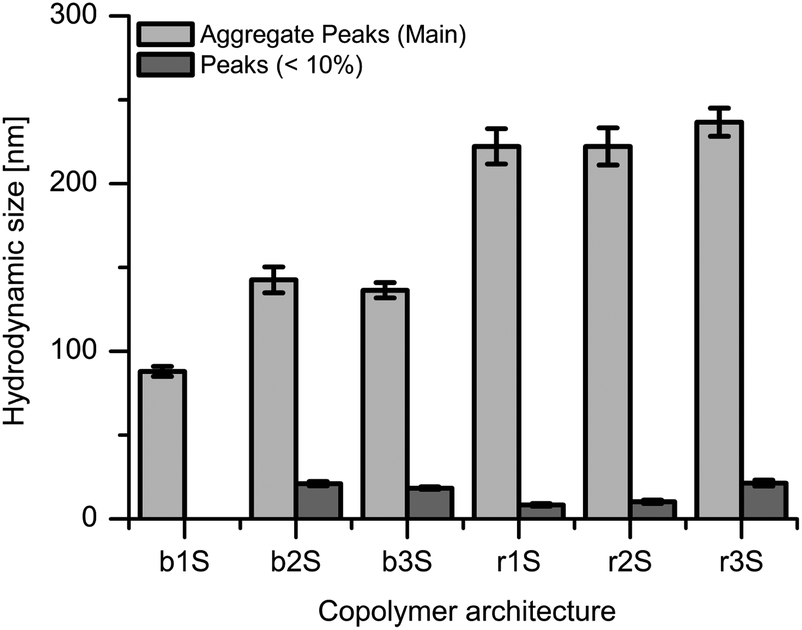 | ||
| Fig. 1 Hydrodynamic particle size by intensity of sulfated copolymers with 1 mg mL−1 in 150 mM PBS. | ||
The sulfated copolymers were investigated by DLS to determine the particle sizes under buffered conditions prior to dye encapsulation (see Fig. 1). All architectures except b1S showed two distinct particle sizes. However, the occurrence of the peaks indicated that the polymers were mainly aggregated. The PGS copolymers showed similar hydrodynamic sizes depending on the nature of the architecture, which were either block or statistical copolymers. The block copolymers were around 139 ± 3 nm and the statistical copolymers around 227 ± 7 nm in size. Copolymer b1S had a significant deviation from the related structures which could be explained by insufficient amphiphilicity. In general, the introduction of the anionic surface charge by sulfation reduced the particle sizes compared to the OH-terminated copolymers.
Encapsulation of guest molecules
The encapsulation properties of the sulfated copolymers were investigated using pyrene (4) and indocarbocyanine dye (5, ICC, Cy3) as presented in Fig. 2. Both dyes were encapsulated using the previously described film method.34,38 The ICC dye (5) was investigated because of an applicable emission pattern with λ > 600 nm, which had been already exploited in several cellular uptake studies with ICC labeled polyglycerol sulfates due to its tissue permeating properties.39–42 In the conducted assays, however, the ICC was covalently attached to the PGS scaffold which included numerous functionalization steps. We wanted to investigate a potential use in therapeutic applications, with the supramolecular carriers synthesized in this study. Two of the investigated architectures showed transport capacities (TC) in the low μM range. Fig. 3 presents the TC for b2S and b3S measured by UV absorbance based on extinction coefficients of ε = 45300 cm−1 M −1 for 4 and ε = 132130 cm−1 M −1 for 5.43,44
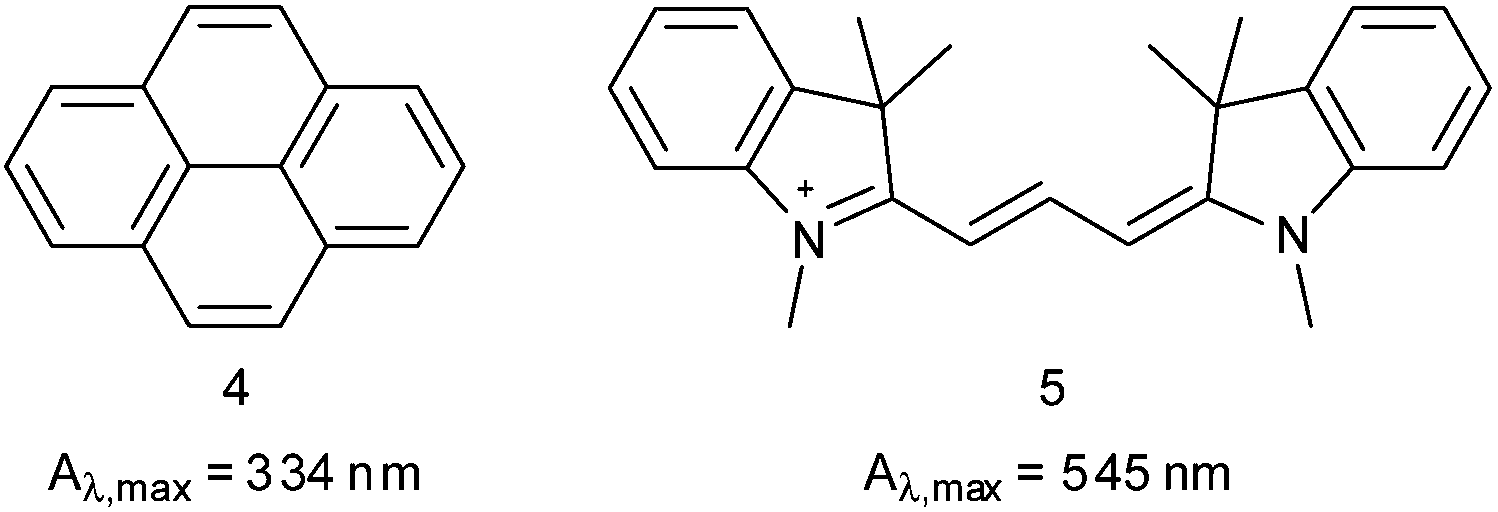 | ||
| Fig. 2 Structures of pyrene (4) and ICC dye (5) used for encapsulation studies. | ||
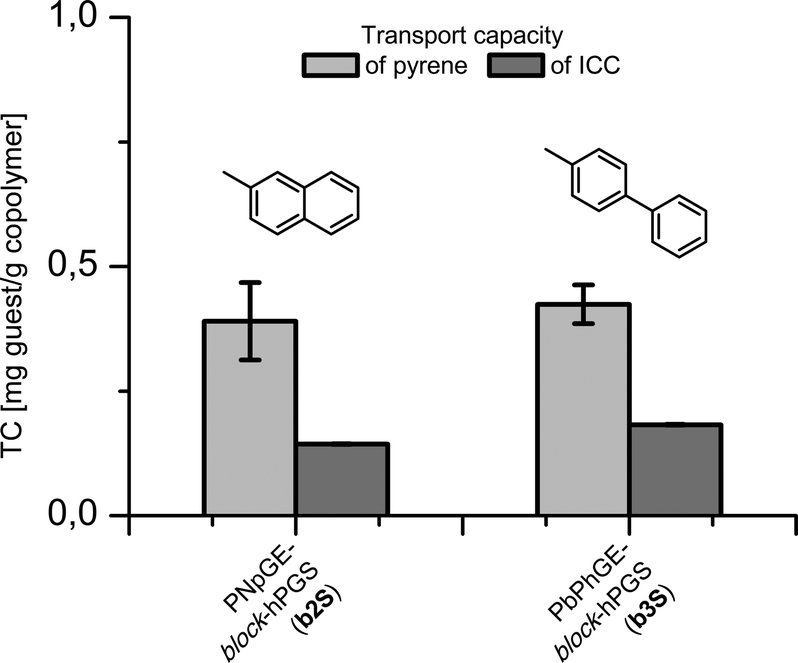 | ||
| Fig. 3 TC of 4 and 5 in sulfated block copolymers b2S and b3S determined by UV measurement in MeOH based on ε = 45300 cm−1 M−1 for pyrene and ε = 132130 cm−1 M−1 for the ICC dye. | ||
Both TCs of b3S with 0.424 and 0.182 mg g−1 copolymer, and of b2S with 0.390 and 0.144 mg g−1 (4 and 5), are low compared to previously investigated architectures, but high enough for the cellular uptake studies.33,38,45 Regarding the hydrodynamic sizes before and after dye encapsulation, both conjugates showed completely different loading structure relationships (see Table 2). The biphenyl derivatized structure b3S had similar particle sizes to those in the unloaded architecture. The amphiphilicity induced by hydrophobic core seems to successfully stabilize the supramolecular aggregates.
| Carrier | d ± SDa [nm] | Pyrene (4) | ICC (5) | ||
|---|---|---|---|---|---|
| TC [mgguest/gpolymer] | d loaded [nm] | TC [mgguest/gpolymer] | d loaded [nm] | ||
| a Hydrodynamic diameters of the aggregated copolymers measured after intensity; small particles included as secondary species in brackets (occurrence < 2.5%). Standard deviations are included with ± SD. Transport capacities are presented in mg (guest) per g polymer. | |||||
| b2S | 143 ± 8 (21 ± 1) | 0.390 | 133 ± 3 (22 ± 2) | 0.144 | 187 ± 6 |
| b3S | 136 ± 5 (18 ± 1) | 0.424 | 144 ± 3 (24 ± 2) | 0.182 | 131 ± 3 (23 ± 2) |
The naphthyl derivatized architecture showed a similar particle size to the unloaded structure upon loading with pyrene, but the hydrodynamic diameter increased by roughly 25% upon loading with ICC dye. In this case, neither the electrostatic repulsion nor the amphiphilic core shell structure could stabilize the aggregates. In both polymer dye complexes, the surface charge prevented precipitation. Although the degree of sulfation (DS) decreased for b2S and b3S, the lower DS combined with sufficient amphiphilic properties was able to stabilize the aggregates without a guest molecule. The achieved loading is mainly affected by the overall low hydrophobic content within the polymer architecture (1:5). A distinct structure activity relationship was determined, as both block copolymers contained a bi-aromatic moiety b2S and b3S, and were able to carry a guest molecule.
No dye encapsulation was detected for the copolymers r1S to r3S. Although the same comonomer incorporation as for the block copolymers was achieved, the statistical distribution over the whole scaffold obviously reduced the hydrophobicity below the required threshold. From a previous study, we know that hydrophobic core functionality is important for encapsulation properties and can influence the maximum loading capacity.34 Here, a higher core functionalization, similar to the synthesized statistical composite polymers r1S to r3S, resulted in a decrease of loaded guest molecule, whereas lower functionalization showed higher capacities. In general, the hydrophobic ratio is crucial for the encapsulation of guest molecules as a distinct amphiphilicity has to be introduced to the PG scaffold.
Cellular uptake of host–guest complexes
The dye loaded block copolymers b2S and b3S were investigated for their cellular uptake properties into the QGP-1 pancreatic carcinoma cell line, A549 human lung carcinoma epithelial cell line and A2780 ovarian carcinoma cell line. Previous studies showed an enhanced cellular uptake of dye labeled dPGS.41,42,46 The cellular uptake was compared after 4 h and 24 h by utilizing fluorescence activated cell sorting (FACS) according to a standardized protocol.40 The uptake into highly proliferating A2780 and A549 cells increased with incubation time and was similar for the cross-comparison of the normalized values for both nanocarriers b2S and b3S (see Fig. 4, Table S2†). The amount of dye which was endocytosed into the cell was approximately 6-fold for A2780 cells after 4 h and 3-fold after 24 h compared to the free dye. The cellular uptake into A549 was even more pronounced and about 6- to 7-fold after 4 and 24 h, respectively. The uptake into QGP-1 cells showed an oppositional development (Table S3, Fig. S5†). The normalized fluorescence intensity decreased from 4 to 24 h incubation time. The reduced fluorescence signals can be explained by apoptosis,47 which was induced either by the ICC dye or the nanocarriers when incubated with this special, slow-growing cell line,48 compared to the cellular uptake into A2780 and A549 cells.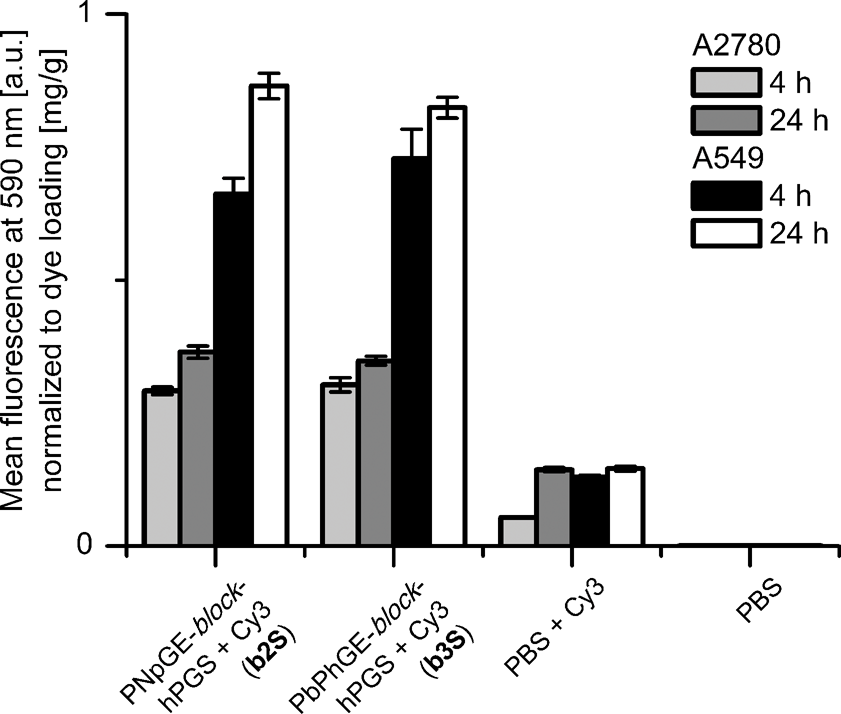 | ||
| Fig. 4 Mean fluorescence at 590 nm (Cy3 dye detection) is displayed for A2780 (4 h: light gray, 24 h: dark gray columns) and A549 (4 h: black, 24 h: white columns) cells incubated with copolymers b2S and b3S, free ICC dye, and PBS control. | ||
However, to determine the inherent cytotoxicity of the compounds further studies have to be conducted. The results from the uptake into QGP-1 cells may be deceptive, with regard to the occurring apoptosis, as dead cells could not be detected. From the results obtained by FACS analysis we conclude that the supramolecular PGS-dye complexes b2S and b3S endocytosed significantly better than the free dye. This is a first indication for the transport potential of the synthesized carriers.
Further characterization of the cellular uptake and transport properties was performed by confocal laser scanning microscopy (CFM) after incubation times of 4 and 24 h into all three cell lines. The merged pictures from the uptake into QGP-1 cells (Fig. 5) indicated an increased and controlled uptake into the perinuclear region of the living cells after 4 h. Because of the mentioned reduced fluorescence signals after 24 h obtained by FACS analysis (Fig. S5†), no CFM pictures were recorded. In comparison, the free dye did not endocytose into QGP-1 cells. The CFM pictures from the uptake into A2780 cells (Fig. S6†) showed a controlled uptake of the polymer–dye complex. Compared to the free ICC dye which was randomly distributed over the whole cell nuclei, the complex accumulated in the perinuclear region. The CFM pictures of the A549 cells present a similar uptake and transport behaviour (Fig. S7†). However, hardly any uptake of the free dye was visible.
 | ||
| Fig. 5 Confocal laser scanning fluorescence microscopy images from QGP-1 cells after incubation with (a) b2S + ICC dye, (b) b3S + ICC dye, (c) free ICC dye, and (d) control PBS for 4 h. The merged pictures (a–d) show the ICC dye in red, stained nuclei in blue (DAPI), and the actin cytoskeleton (Alexa Fluor 488 Phalloidin label) in green; (e–h) present ICC dye channel for (e) b2S + ICC dye, (f) b3S + ICC dye, (g) free ICC dye, and (h) control PBS visualized as intensity grey values. | ||
The present study confirmed the enhanced uptake of PGS into the perinuclear region of the cell. In the previous studies, however, the ICC dye was covalently attached to the PG scaffold and therefore could not be liberated.41,42
Experimental section
Materials
All chemicals were purchased from Acros Organics and used as received if not otherwise indicated. Glycidol was dried over CaH2 and distilled under reduced pressure. The purified monomer was stored at 4 °C under inert atmosphere and only used up to two months. All solvents were purchased from Sigma-Aldrich and used without further purification. Dry tetrahydrofuran (THF) was obtained from a solvent purification system. Dry N-methyl-2-pyrrolidone (NMP) and potassium tert-butoxide (KOtBu) were used and stored under AcroSeal® conditions and used as received. Monomers 2 and 3 were synthesized according to published procedures.49,50 Indocarbocyanine dye (ICC) was obtained from mivenion GmbH.Block copolymerization
The batch reactor was extensively dried at 140 °C under reduced pressure. After cooling to 65 °C, 1,3,5-Trihydroxybenzene (THB), KOtBu (0.10 mol-eq. from initiator; 1 M in THF), and dry NMP were added under inert conditions. The temperature was increased to 120 °C whereas butanol was distilled and the initiator homogeneously dissolved. The glycidol derivative 1, 2 or 3 (see Table 1) was mixed with THF/NMP, and slowly added to the reactor over a period of 18 h using a precision dosing pump. Subsequently, a solution of glycidol in THF was added to the reactor over 18 h and additionally stirred for another 2 h. After cooling to ambient temperature, the polymer mixture was diluted with methanol and cation exchange resin (Dowex® Monosphere® 650C UPW, Supelco, Bellefonte, USA) was added. After stirring for 3 d, the turbid solution was filtered and remaining solvent was evaporated under reduced pressure. Purification of a partial batch amount was performed by dialysis in saturated aqueous NaCl solution. 1H, 13C NMR spectra and GPC traces are available in Fig. S1–S3.†Statistical copolymerization
The batch reactor was extensively dried as described. Trimethylolpropane (TMP) was melted at 65 °C under reduced pressure. KOtBu (0.15 mol-eq. from initiator; 1 M in THF) and NMP were added under inert conditions and the temperature was increased to 120 °C. A defined monomer mixture of glycidol with 1, 2 or 3 (see Table 1) in THF was prepared and slowly added to the reactor over a period of 20 h, whereas the THF was immediately distilled from the reaction mixture. After complete monomer addition and stirring for additional 2 h, the polymer mixture was diluted with methanol and cation exchange resin was added. After stirring for 3 d, the polymer solution was filtered and solvent was evaporated under reduced pressure. Purification of a partial batch amount was performed by dialysis in water for 3 d. 1H, 13C NMR spectra and GPC traces are available in Fig. S1–S3.†Sulfation of polymer architectures
Sulfation of the polymer architectures was performed as follows: 120 mg of each copolymer was dissolved in NMP and 1.5 to 1.8 equivalents NH3SO3 per hydroxyl group were added. The samples were sonicated for 10 min and subsequently stirred at 90 °C for 3 d. After cooling to room temperature the polymer mixture was precipitated into diethyl ether and stirred for 30 min. The solvent was decanted and purification was performed by dialysis in saturated aqueous NaCl solution for 3 d with solvent exchange twice a day. 1H NMR spectra are available in Fig. S4.† The degree of sulfation (DS) was determined by combustion analysis (see Table 1 and S1†).Encapsulation of guest molecules
A stock solution of the guest molecules (4 and 5) with 5 mg mL−1 in THF or dichloromethane (DCM) was prepared. 200 μL of the stock solution was transferred into a small vial and the solvent was evaporated. In the meantime, a stock solution of each synthesized copolymer with 1 mg mL−1 in in Dulbecco's phosphate buffered saline (DPBS, 1×, Sigma-Aldrich, Germany) was prepared. After evaporation of the solvent, 3 mL of each polymer solution and 3 mL of pure PBS was added to the dried dyes. After excessive stirring for 24 h, the samples were filtered through 0.45 μm Minisart RC 15 syringe filters (Sartorius Stedim Biotech, Germany).Instrumentation
NMR spectra were recorded on a Joel ECX 400 MHz or a Bruker Avance 700 MHz spectrometer as indicated. Spectra were recorded in ppm and referenced to indicated, deuterated solvents. Molecular weight distributions were determined by means of GPC coupled to a refractive-index detector (RI) obtaining the complete distribution (Mn, Mp, Mw, dispersity). The unsulfated copolymers were measured in NMP with 0.05 M LiBr (1.5 mg mL−1) as mobile phase with a flow rate of 0.8 mL min−1 on a GPC (Thermo Separation Products, Thermo Scientific, Waltham, USA) consisting of a solvent delivery system with pump P-100 and an auto sampler AS-100. Two 30 cm columns (PPS: Polymer Standards Service GmbH, Germany; Gram 100 Å, 1000 Å with 7 μm particle size) were used to separate the samples. The columns were operated in a column oven at 70 °C. The calibration was performed using linear calibration standard (PSS GmbH, Germany). Aqueous samples (sulfated architectures) were measured under highly diluted conditions (10 mg mL−1) from a GPC consisting of an Agilent 1100 solvent delivery system with pump, manual injector, and an Agilent differential refractometer. Three 30 cm columns (PPS GmbH, Germany; Suprema 100 Å, 1000 Å, 3000 Å with 5 and 10 μm particle size) were used to separate aqueous polymer samples using water with 0.05% NaNO3 as the mobile phase at a flow rate of 1 mL min−1. The columns were operated at ambient temperature with the RI detector at 50 °C. The calibration was performed by using linear pullulan calibration standard (PPS GmbH, Germany). WinGPC Unity from PSS was used for data acquirement and interpretation. Particle size was determined by DLS in UV-transparent low-volume disposable polystyrene cuvettes on a Zetasizer Nano ZS (Malvern Instruments Ltd., UK) equipped with a 4 mW He–Ne laser (λ = 633 nm). The samples were measured at a concentration of 1 mg mL−1 in DPBS at pH 7.4. Prior to the measurement all samples were filtered through 0.45 μm Minisart RC 15 syringe filters. Each sample was equilibrated for 1 min at 25 °C and measured with 10 scans for 15 s (173° back-scattering). All stated values are the mean of at least 3 independent measurements and standard deviation is indicated with ± SD. UV/Vis spectra were recorded on a SCINCO S-3100 UV/Vis spectrometer equipped using UV Disposable Cuvettes 220–900 nm (BrandTech Scientific, Inc., Essex, USA). The transport capacities were determined as follows: 1 mL of aqueous polymer solution in PBS was freeze dried. The dried polymer-dye-complex was suspended in MeOH to extract the dye from the polymer scaffold and extensively stirred for 18 h. Subsequently the suspensions were filtered through 0.45 μm Minisart RC 15 syringe filters (Sartorius Stedim Biotech, Germany).Cell preparation for fluorescence microscopy and FACS analysis
For confocal laser scanning microscopy, the epithelial human pancreatic cancer cell line QGP-1, the human lung carcinoma epithelial cell line A549 and the ovarian carcinoma cell line A2780 were routinely propagated as follows: DEMEM medium, with 10% fetal calf serum (FCS), 2% glutamine, and penicillin/streptomycin (all from PAN Biotech) added. Cells were seeded into medium at 1 × 105 cells mL−1, cultured at 37 °C with 5% CO2, and split 1:5 two times a week. For cytochemistry, cells were seeded at 5 × 104 cells mL−1 in a 24-well culture plate on glass coverslips (Sigma), and cultured at 37 °C for 24 h. Thereafter, cells were incubated with normal culture medium or medium containing 10−6 M of dye-labeled test substances for QGP-1 cells and 5.0 × 10−7 M of dye labeled test substances for A2780 and A549 cells at 37 °C for 4 or 24 h. Afterwards, cells were fixed with cold acetone, rinsed and covered with Alexa Fluor 488 Phalloidin (1:300, Molecular Probes) only for QGP-1 cells. 4,6-diamidino-2-phenylindole (DAPI, Abcam) was used for nuclear counterstain. Confocal images were acquired at room temperature with a 63 × 1.4 NA HC PL APO CS2 oil objective suited in a confocal microscope (TCS-SP8, DMI6000 stand; Leica). Images of different groups were acquired with the same laser and detector settings using the Leica LAS AF software. The fluorescence detection was performed sequentially for each channel set with the acousto optical beam splitter between 500 and 530 nm for the Alexa Fluor 488 and between 590 and 630 nm for the ICC dye. Alexa Fluor 488 was excited using the 488 nm Argon laser line, whereas the ICC dye was excited with a 561 nm diode-pumped solid-state laser. DAPI was excited with an UV laser (405 nm) and fluorescence detection was set with the acousto optical beam splitter between and 420 and 470 nm. For FACS analysis, the three different cell lines were routinely propagated as described. The cells were cultured 4 × 104 cells mL−1 cells were cultured in 48-well-plates at 37 °C for 24 h. Thereafter, cells were incubated with normal culture medium or medium containing 10−6 M test substance for 4 or 24 h. Thereafter, cells were washed with PBS, detached with 200 μL per well accutase (PAA), and centrifuged with 350 g, at 4 °C for 5 min. Supernatants were removed and cells were suspended in 50 μL PBS with 0.5% bovine serum albumin (Roth). Cells were immediately analyzed in a FACS Calibur analysis instrument (Becton-Dickinson: FL2 BP 585/42 nm).
Conclusions
Hyperbranched polyglycerol sulfates with hydrophobic cores have been synthesized for the first time. All polymerization reactions were performed in a one-pot batch reaction applying one-step or two-step procedures. The influence of aromatic units as well as the position within the polymer scaffold was determined by comparing block and statistical copolymers. Two block copolymers with bi-aromatic moieties showed transport capacities from <1 wt%. However, the aggregates with stable sizes formed independently without a guest molecule. A statistical distribution obviously provided insufficient amphiphilicity to encapsulate any guest molecules, although stable aggregates did form. The endocytosis of the host–guest complexes and liberation of the free ICC dye indicated that carriers combined with an active sulfate targeting moiety can be developed into potent drug delivery systems for cellular uptake.This study has provided new insight into the prerequisites for potent carriers based on polyglycerol sulfates. For the investigated systems, we can conclude that a core–shell type architecture is much more efficient for guest transport than a statistical distribution of the hydrophobic moieties.
Funding
Financial support from the DFG collaborative SFB 765 research center, the Dahlem Research School, and the HZG portfolio project “Technologie und Medizin” is gratefully acknowledged.Conflict of interest
The authors declare no competing financial interest. All authors have approved the final version of the manuscript.Acknowledgements
We like to thank Cathleen Schlesener and Marlies Graewert from the group of Prof. Dr Schlaad/MPI of Colloids and Interfaces for the GPC measurements, and Dr Pamela Winchester for proof reading the manuscript. mivenion GmbH, namely, Kai Licha and Nicole Wegner, are acknowledged for providing the ICC dye.References
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 CrossRef CAS.
- L. Gros, H. Ringsdorf and H. Schupp, Angew. Chem., Int. Ed., 1981, 20, 305–325 CrossRef.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- M. A. Quadir and R. Haag, J. Controlled Release, 2012, 161, 484–495 CrossRef CAS PubMed.
- A. Sunder, J. Heinemann and H. Frey, Chem.–Eur. J., 2000, 6, 2499–2506 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS PubMed.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- H. Huang, K. L. Wooley and E. E. Remsen, Chem. Commun., 1998, 1415–1416 RSC.
- V. Bütün, A. B. Lowe, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1999, 121, 4288–4289 CrossRef.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- K. R. Gopidas, A. R. Leheny, G. Caminati, N. J. Turro and D. A. Tomalia, J. Am. Chem. Soc., 1991, 113, 7335–7342 CrossRef CAS.
- C. N. Moorefield and G. R. Newkome, C. R. Chim., 2003, 6, 715–724 CrossRef CAS PubMed.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed., 1988, 27, 113–158 CrossRef.
- G. R. Newkome, C. N. Moorefield, G. R. Baker, M. J. Saunders and S. H. Grossman, Angew. Chem., Int. Ed., 1991, 30, 1178–1180 CrossRef.
- C. J. Hawker, K. L. Wooley and J. M. J. Frechet, J. Chem. Soc., Perkin Trans. 1, 1993, 1287–1297 RSC.
- A. Sunder, R. Mülhaupt, R. Haag and H. Frey, Adv. Mater., 2000, 12, 235–239 CrossRef CAS.
- H. Frey and R. Haag, Rev. Mol. Biotechnol., 2002, 90, 257–267 CrossRef CAS.
- D. Gröger, F. Paulus, K. Licha, P. Welker, M. Weinhart, C. Holzhausen, L. Mundhenk, A. D. Gruber, U. Abram and R. Haag, Bioconjugate Chem., 2013, 24, 1507–1514 CrossRef PubMed.
- R. K. Kainthan, S. R. Hester, E. Levin, D. V. Devine and D. E. Brooks, Biomaterials, 2007, 28, 4581 CrossRef CAS PubMed.
- R. K. Kainthan and D. E. Brooks, Biomaterials, 2007, 28, 4779–4787 CrossRef CAS PubMed.
- M. Imran ul-haq, R. A. Shenoi, D. E. Brooks and J. N. Kizhakkedathu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2614–2621 CrossRef.
- R. Haag, J.-F. Stumbé, A. Sunder, H. Frey and A. Hebel, Macromolecules, 2000, 33, 8158–8166 CrossRef CAS.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2009, 43, 129–141 CrossRef PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- M. Weinhart, D. Gröger, S. Enders, J. Dernedde and R. Haag, Biomacromolecules, 2011, 12, 2502–2511 CrossRef CAS PubMed.
- M. Weinhart, D. Gröger, S. Enders, S. B. Riese, J. Dernedde, R. K. Kainthan, D. E. Brooks and R. Haag, Macromol. Biosci., 2011, 11, 1088–1098 CrossRef CAS PubMed.
- H. Türk, A. Shukla, P. C. Alves Rodrigues, H. Rehage and R. Haag, Chem.–Eur. J., 2007, 13, 4187–4196 CrossRef PubMed.
- I. N. Kurniasih, H. Liang, J. P. Rabe and R. Haag, Macromol. Rapid Commun., 2010, 31, 1516–1520 CrossRef CAS PubMed.
- I. N. Kurniasih, H. Liang, V. D. Moschwitzer, M. A. Quadir, M. Radowski, J. P. Rabe and R. Haag, New J. Chem., 2012, 36, 371–379 RSC.
- F. Paulus, M. E. R. Weiss, D. Steinhilber, A. N. Nikitin, C. Schütte and R. Haag, Macromolecules, 2013, 46, 8458–8466 CrossRef CAS.
- E. Gilbert and B. Veldhuis, J. Am. Oil Chem. Soc., 1960, 37, 298–300 CrossRef CAS.
- R. A. Al-Horani and U. R. Desai, Tetrahedron, 2010, 66, 2907–2918 CrossRef CAS PubMed.
- E. Fleige, R. Tyagi and R. Haag, in Nanocarriers, 2013, vol. 1, pp. 1–9 Search PubMed.
- A. L. Sisson, D. Steinhilber, T. Rossow, P. Welker, K. Licha and R. Haag, Angew. Chem., Int. Ed., 2009, 48, 7540–7545 CrossRef CAS PubMed.
- S. Reichert, P. Welker, M. Calderón, J. Khandare, D. Mangoldt, K. Licha, R. K. Kainthan, D. E. Brooks and R. Haag, Small, 2011, 7, 820–829 CrossRef CAS PubMed.
- K. Licha, P. Welker, M. Weinhart, N. Wegner, S. Kern, S. Reichert, I. Gemeinhardt, C. Weissbach, B. Ebert, R. Haag and M. Schirner, Bioconjugate Chem., 2011, 22, 2453–2460 CrossRef CAS PubMed.
- S. Biffi, S. Dal Monego, C. Dullin, C. Garrovo, B. Bosnjak, K. Licha, P. Welker, M. M. Epstein and F. Alves, PLoS One, 2013, 8, 1–9 Search PubMed.
- P. J. Sims, A. S. Waggoner, C. H. Wang and J. F. Hoffman, Biochemistry, 1974, 13, 3315–3330 CrossRef CAS.
- E. M. Pearce, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1797 CrossRef.
- D. Steinhilber, F. Paulus, A. T. Zill, S. C. Zimmerman and R. Haag, MRS Proceedings, 2012, 1403, mrsf11-1403-v09-01 DOI:10.1557/opl.2012.419.
- F. Paulus, R. Schulze, D. Steinhilber, M. Zieringer, I. Steinke, P. Welker, K. Licha, S. Wedepohl, J. Dernedde and R. Haag, Macromol. Biosci., 2014, 14, 643–654 CrossRef CAS PubMed.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS.
- H. Iguchi, I. Hayashi and A. Kono, Cancer Res., 1990, 50, 3691–3693 CAS.
- D. S. Bose, A. V. N. Reddy and S. W. Chavhan, Synthesis, 2005, 2345–2348 CrossRef CAS PubMed.
- E. Fullam, A. Abuhammad, D. L. Wilson, M. C. Anderton, S. G. Davies, A. J. Russell and E. Sim, Bioorg. Med. Chem. Lett., 2011, 21, 1185–1190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of unsulfated and sulfated copolymers. See DOI: 10.1039/c4py00430b |
| This journal is © The Royal Society of Chemistry 2014 |