A polymeric chain extension driven by HSCT interaction†
Zhongwei
Ji
,
Jianghua
Liu
,
Guosong
Chen
* and
Ming
Jiang
State Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science, Fudan University, 220 Handan Rd., Shanghai, China. E-mail: guosong@fudan.edu.cn; Tel: +86 21 5566 4275
First published on 24th January 2014
Abstract
In this paper, targeting a high-molecular-weight supramolecular polymer, the chain extension of low-molecular-weight polymers (LMWPs) is achieved via the HSCT (Host-Stabilized Charge Transfer) of CB[8] (cucurbit[8]uril). Here, a combination of ditopic viologen and CB[8] serve as a “supramolecular chain extender” for the first time, to connect the naphthalene (Np)-ended LMWP PDMA or PNIPAm. UV-vis spectra, ITC and NMR prove that the guest Np connected to the LMWP does not affect its complexation with guest viologen and CB[8]. The viscosity measurements clearly demonstrate the formation of a supramolecular polymer, as the viscosity of the LMWP PDMA (DP 52) after complexation exceeds that of PDMA (DP 300). Np-ended LMWP PNIPAm shows a similar chain extension. The resultant PNIPAm supramolecular polymer shows peculiar LCST behavior with a remarkably different variation in concentration from its polymeric precursor.
Introduction
Supramolecular polymers (SPs), as a newly emerging type of polymer architecture, feature non-covalent linkages between monomers instead of the covalent bonds in traditional polymers. In the past decade, the research field of SPs grew dramatically with remarkable achievements.1–11 Compared to traditional polymers, SPs have great advantages in controllable degradability, self-healing and processability because of the dynamic nature of the non-covalent bonds. To date, metal–ligand,12 hydrogen bonding,13,14 π–π15,16 and host–guest interactions17,18 are all involved in constructing SPs. Most SP chains are constructed from small molecules, because of the synthetic accessibility and the significant contrast of the properties caused by polymerization. Various molecules with AB-type or AA/BB-type structure (A and B are complimentary groups which can form non-covalent linkages) have been developed to construct SPs with special designs.19 It has been reported that an appropriate spacer between the reactive groups, together with charge repulsion, can suppress the formation of cyclic species.20However, there is still much room for the development of SPs. The versatility of traditional polymers stems from the tunability of their crystallinity, mechanical strength and thermal stability, which is still unattainable by SPs due to their limited chemical structures. Currently, a practical way to build SPs with more “polymeric” properties has emerged, in which covalent low-molecular-weight polymers (LMWPs), instead of small molecules, are used as building blocks. A few such SPs composed of LMWPs by supramolecular chain extension (i.e. macro-supramolecular polymers, MSPs) have been reported.21–25 For example, a MSP with a multi-block copolymer structure composed of two different kinds of polymeric “monomers” bearing hydrogen bond donors or acceptors was built by Zimmerman et al.26 More recently, a MSP formed by metal–ligand linkage on its main chain, exhibiting phase separation and self-healing properties, was achieved by Rowan et al.27 However, the studies on MSP construction with synthetic polymers as building blocks is still quite limited compared to SPs constructed from small molecules,28–37 and of which, attention is rarely paid to host–guest interactions.23,38,39
Using LMWPs as building blocks of SPs exhibits some advantages over the use of small molecules. It has been shown that the polymeric precursor could facilitate the extension of supramolecular chains by suppressing the possibility of ring closure.26,29 Moreover, SPs constructed from small molecules include a high content of functional groups, resulting in limited solvent solubility.25 This drawback, which may hinder the further application of SPs, is more pronounced in some host–guest systems. For example, SPs resulting from inclusion complexation between CB[8] (cucurbit[8]uril) and two Np (naphthalene) groups could not reach a high concentration in water because of the limited solubility of the Np-containing small molecule and CB[8].40
Among the popular host molecules, CB[n] (n = 5–10), a family of highly symmetrical pumpkin-shaped molecules, has variably-sized cavities accessible by certain guests through hydrophobic and ion–dipole interactions.41 CB[8] is an outstanding member of the family since it can encapsulate two different guest molecules in its cavity at the same time.42–45 In particular, CB[8] can form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ternary complexes with a pair of guests, one electron-deficient and one electron-rich, such as a viologen and 2-naphthol, via a stepwise binding process (Scheme 1a), leading to an enhanced association constant in aqueous buffer. This is named HSCT (Host-Stabilized Charge Transfer). Based on this HSCT, Kim first obtained supramolecular oligomers,46,47 followed by the first SP built from small molecules by Zhang et al.20 and, very recently a crystal structure of CB[8]-mediated SP was obtained by Scherman et al.48 However, all of these remarkable SPs based on HSCT were developed from small molecules, where LMWP was not involved.
:1:1 ternary complexes with a pair of guests, one electron-deficient and one electron-rich, such as a viologen and 2-naphthol, via a stepwise binding process (Scheme 1a), leading to an enhanced association constant in aqueous buffer. This is named HSCT (Host-Stabilized Charge Transfer). Based on this HSCT, Kim first obtained supramolecular oligomers,46,47 followed by the first SP built from small molecules by Zhang et al.20 and, very recently a crystal structure of CB[8]-mediated SP was obtained by Scherman et al.48 However, all of these remarkable SPs based on HSCT were developed from small molecules, where LMWP was not involved.
 | ||
| Scheme 1 Schematic representation of (a) the two-step binding process of cucurbit[8]uril in water. (b) Preparation of MSPs and end-modified LMWPs through the addition of CB[8] and an electron-deficient first guest (viologen dimer and viologen, respectively) to the Np-ended LMWPs. | ||
In this paper, LMWPs are firstly employed to develop MSPs based on HSCT interactions. After the addition of CB[8] and a viologen dimer, LMWPs bearing Np groups at both ends can be extended to MSPs with much larger molecular weight at a relatively high concentration (Scheme 1b). End-modified LMWPs are also prepared, with a monotopic viologen as a control. The newly formed MSPs exhibited aggregation behavior. Interestingly, the MSP originating from PNIPAm shows a remarkably different concentration dependence of the lower critical solution temperature (LCST) from its precursor, which is a pronounced property compared with other SPs made of small molecules.
Results and discussion
To prepare the functional LMWPs, we synthesized a new CTA (chain transfer agent, Scheme S1,† characterization in Fig. S1–S4†) with two Np groups. In the following RAFT (Reversible Addition-Fragmentation Chain Transfer) polymerization, short hydrophilic polymers with two Np ends were obtained. As shown in Scheme 2, the two model LMWPs, Np–PDMA–Np (P1, Scheme S2,†Mn = 5.5 × 103 g mol−1) and Np–PNIPAm–Np (P2, Scheme S3,†Mn = 7.0 × 103 g mol−1) were synthesized with a designed low DP (degree of polymerization) and narrow molecular weight distribution (PDI < 1.2), and characterized by GPC (Gel Permeation Chromatography) with PEG as the standard (Fig. S5 and S6†). The effective incorporation of Np groups at both chain ends of the two homopolymers was assessed by 1H NMR (Fig. S7 and S8†), showing excellent accordance between the DP calculated by 1H NMR (relative integration of polymer backbone protons to that of Np, 52 for P1 and 56 for P2) and the theoretical values. Further structural confirmation was achieved by the aminolysis reaction of P1 or P2 with their chain-cleavage products, characterized by GPC (Fig. S5 and 6†). A small ditopic guest 3 was obtained through the two-step ionization of nitrogen in viologen (Scheme S4,†1H NMR shown in Fig. S9†). For control experiments, the monotopic guest 4 was also synthesized (1H NMR shown in Fig. S10†). It should be noted that the solubility of CB[8] in water is very limited, while P1, P2, 3 and 4 are water-soluble, which can enhance the water solubility of CB[8] after complexation. | ||
| Scheme 2 Chemical structure of the building blocks involved in the construction of MSPs. | ||
With these components in hand, the HSCT interactions at the end of the LMWP chains was first examined by UV-vis spectra. It is known that the CB[8]–Np–viologen ternary complex based on HSCT exhibits a CT absorption band between 400 and 500 nm in UV-vis spectra.43 Indeed, in the current example, as shown in Fig. 1a, the UV-vis spectra of P1 (red line) and 3 (black line) alone had no appreciable absorption bands beyond 400 nm, while their equimolar mixture (P1 + 3, blue line) only showed a slight increase in this area, indicating a very weak CT interaction between P1 and 3. However, after the addition of 2 equiv. of CB[8] to the mixture of P1 + 3 (P1:3:CB[8] = 1:1:2), this CT absorption band was greatly enhanced with the concomitant emergence of another CT band beyond 500 nm (cyan line), indicating the formation of a ternary complex. The complex is denoted as P1+3⊂CB[8], with its stoichiometry of binding determined by a Job’s plot (Fig. 1b). Here, CB[8] was mixed with 3 in a 2:1 molar ratio to form the 3⊂CB[8] complex first, in order to dissolve as much of CB[8] in water as possible. Then 3⊂CB[8] and P1 were mixed together with their total concentration fixed at 0.25 mM in the range of the molar ratio of P1 varying from 0 to 1. The absorption at 495 nm was plotted, showing the maximum absorbance at a molar ratio of 0.5, which indicated that the stoichiometry of binding between P1 and 3⊂CB[8] was 1:1. In a control experiment, 4 was used instead of 3, where 4⊂CB[8] first formed in a 1:1 molar ratio. The result of Job's experiment for 4⊂CB[8] and P1 showed that the maximum absorption appeared when the ratio of P1 reached 0.37, which was close to the calculated 0.33 for the 1:2 stoichiometry of P1 to 4⊂CB[8] (Fig. S11†). The above experimental results show that Np groups connecting to the polymer chain do not affect its complexation ability with viologen in CB[8] even in a quantitative sense.
 | ||
| Fig. 1 (a) UV-vis spectra of P1, 3, P1 + 3 and P1 + 3⊂CB[8] (P1: 0.125 mM, 3: 0.125 mM, CB[8]: 0.25 mM). (b) Job’s plot of P1 and 3⊂CB[8] (total concentration of P1 and 3⊂CB[8] was fixed at 0.25 mM, absorption intensity measured at 495 nm). | ||
The binding behavior forming the ternary complex was further measured by ITC (Isothermal Titration Calorimetry). In the experiments, an aqueous solution of P1 (0.5 mM) was continuously titrated into a solution of 3⊂CB[8] (0.05 mM, calculated as 3). The generated heat fits well to the one-set binding mode (Fig. S12a†) after calculation, showing an experimental n value of 1.0, which is consistent with the result from the Job's plot (Fig. 1b). Meanwhile, the binding constant of P1 and 3⊂CB[8] was calculated as 8.15 × 105 M−1, comparable to the value reported at the small molecular level.49 As a control experiment, the binding of P1 to 4⊂CB[8] was also measured by ITC, which showed an experimental n value of 0.4 (Fig. S12b†), close to the expected n value of 0.5. From the above results, we may expect the supramolecular chain extension of P1 mediated by HSCT interactions.
1H NMR was employed to study the molecular state of P1 + 3⊂CB[8]. After the addition of CB[8] to the mixture of P1 and 3 (Fig. 2c), the peaks related to the viologen and Np groups underwent pronounced shifts, indicating that the two groups were captured by CB[8]. The mixture of P1 with 2 equiv. of 4⊂CB[8] was expected to form an end-modified LMWP as a control sample, which is denoted as P1 + 4⊂CB[8]. As shown in Fig. 2, a clear difference in the proton signals was observed in the spectra of P1 + 3⊂CB[8] and P1 + 4⊂CB[8], i.e. the signal in the former was obviously broadened and weakened than that in the latter, indicating the feature of long polymeric chains in the former. A similar phenomenon has been reported for SPs formed from small molecules.20,50
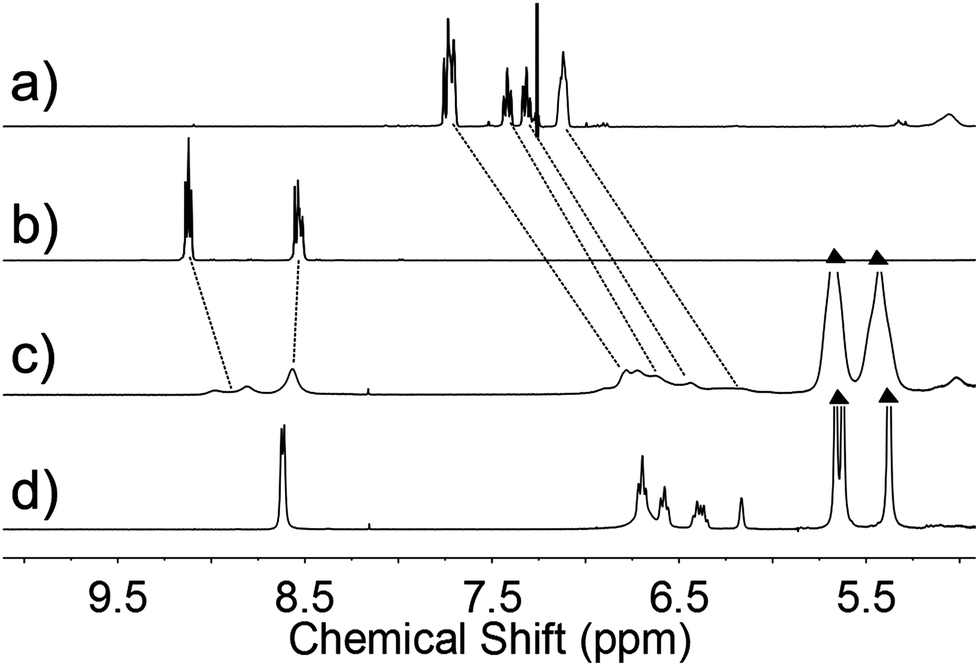 | ||
| Fig. 2 1H NMR spectra of (a) P1 (in CDCl3), (b) 3, (c) P1 + 3⊂CB[8] and (d) P1 + 4⊂CB[8] in D2O (triangles: peaks belonging to CB[8]). Concentrations: P1 (4 mM), 3 (4 mM), CB[8] (8 mM) and 4 (8 mM). | ||
High viscosity is one of the macroscopic characteristics of traditional polymers in solution, thus the formation of MSPs of P1 + 3⊂CB[8] could be directly demonstrated by viscometry. The viscosity of P1 + 3⊂CB[8] was plotted against the concentration of CB[8] (Fig. 3a). When CB[8] was gradually added to the 1:1 mixture of P1 and 3 (4 mM), the relative viscosity of the complex first increased slowly. Then the increase rate became faster after the ratio of CB[8]:P1 exceeded 1. The maximum viscosity appeared at the molar ratio of 2, i.e. equal molar ratios of CB[8], Np and viologen. Thereby, the stoichiometric ratio of the three is a crucial factor leading to long extended chains, which is similar to the feature of condensation polymerization.40 Meanwhile, this process was accompanied by a transmittance decrease (Fig. S13†), suggesting the formation of large particles upon the supramolecular chain extension of LMWPs, which will be discussed in detail later. In Fig. 3b, the relative viscosity of the P1 + 3⊂CB[8] solution was plotted against the concentration of PDMA, with P1 + 4⊂CB[8] as a control. At very low concentrations, the relative viscosity of P1 + 3⊂CB[8] (red line) was comparative to that of P1 + 4⊂CB[8] (black line), indicating that the majority was the unconnected LMWPs in the mixture of P1 + 3⊂CB[8]. When the concentration was increased (>9.4 mg mL−1, calculated as P1), the viscosity increase of the P1 + 3⊂CB[8] solution was much faster than that of P1 + 4⊂CB[8], indicating the formation of extended polymer chains. Furthermore, when 4 was added as a competitive guest to the above MSP solution of P1 + 3⊂CB[8] with a relatively high viscosity, the viscosity drastically decreased, showing the dynamic nature of the P1 + 3⊂CB[8] polymer (Fig. S14†). To semi-quantitatively estimate the Mw and DP of the resultant extended polymeric chain in solution, a traditional polymer, PDMA300 (DP 300, PDI 1.21, GPC result in Fig. S15†), was used as a reference. From Fig. 3b, where PDMA300 and P1 + 3⊂CB[8] kept the same concentration of DMA, it was found that the viscosity of PDMA300 was slightly higher than that of P1 + 3⊂CB[8] at low concentrations (<15 mg mL−1), but the latter showed much higher viscosity than PDMA300 at high concentrations (>15 mg mL−1), with the cross point at around 15.5 mg mL−1. Based on this result, the supramolecular chain extension of P1 (DP 52) in the mixture of P1 + 3⊂CB[8] was confirmed, with its viscosity higher than that of the traditional polymer with DP 300. In addition, it is known that the viscosity variation of SPs is generally non-linear with the concentration of its monomers.26 With the current MSPs, the observed viscosity variation of the P1 + 3⊂CB[8] complex with a concentration of 25 mg mL−1 (calculated as DMA) is different from both its covalent counterpart and the control polymer, showing its supramolecular nature. However, at the highest concentration we measured, the viscosity of P1 + 3⊂CB[8] is only slightly higher than that of PDMA300. This is understandable due to the dynamic nature of supramolecularly extended chains, i.e. an equilibrium always exists between the monomer, oligomers and supramolecular long chains.48
 | ||
| Fig. 3 (a) Relative viscosity of the mixture of P1 and 3 (4 mM) with different molar ratios of CB[8] to P1 (20 °C in H2O). (b) Relative viscosity of P1 + 3⊂CB[8], P1 + 4⊂CB[8] and covalent polymer PDMA300vs. the weight concentration of PDMA segments in water at 20 °C. | ||
To examine the structure of the obtained MSP in detail, the solution behavior of the P1 + 3⊂CB[8] ternary complex was measured by DLS. The mixture of P1 and 3 (4 mM each) without CB[8] gave an <Rh> (hydrodynamic radius) value of around 3 nm, that of a common synthetic polymer (Fig. S16†), indicating the absence of interactions between P1 and 3. However, when CB[8] was added, even when the concentration of P1 + 3⊂CB[8] was as low as 0.4 mM, large particles (<Rh> = 60 nm) were found in the system with the co-existence of smaller particles with <Rh> less than 10 nm (Fig. 4a). When the concentration of P1 + 3⊂CB[8] was increased to 1.2 mM, the small peaks disappeared and the large peak remained and grew. This large peak even shifted to a much larger <Rh> value as the P1 + 3⊂CB[8] concentration further increased (Fig. 4b). Meanwhile, data from the turbidity tests supported this DLS result. As the concentration of P1 + 3⊂CB[8] was increased from 2.4 mg mL−1 to 23.6 mg mL−1 (calculated as P1), the transmittance of the solution kept decreasing, indicating the formation of large particles (Fig. 4c). However, the transmittance of the two control samples, i.e. PDMA300 and P1 + 4⊂CB[8], almost remained constant, proving that the formation of large particles was related to the supramolecular chain extension of the LMWPs.
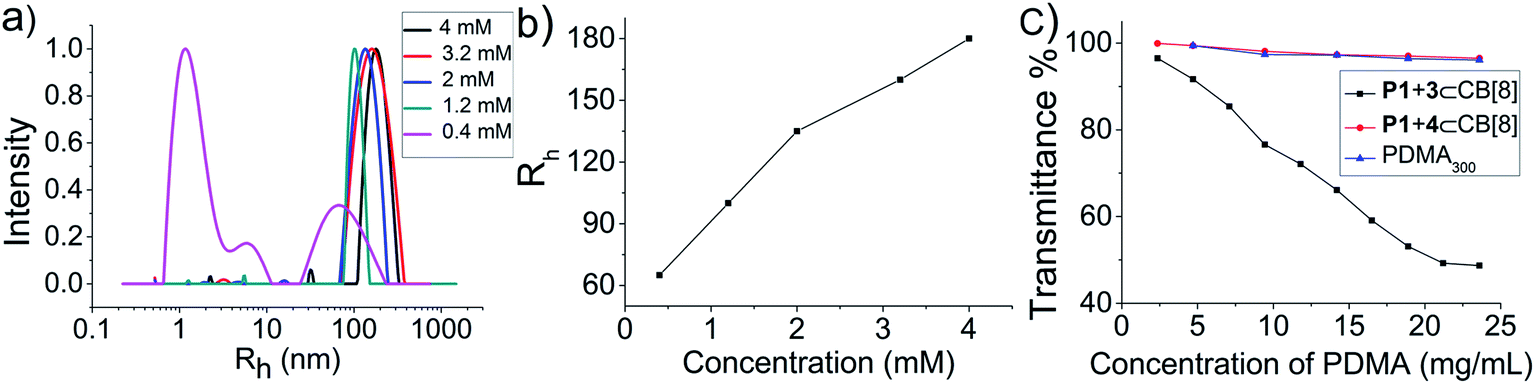 | ||
| Fig. 4 (a) <Rh> distribution of the P1 + 3⊂CB[8] complex at different concentrations. (b) Dependence of <Rh> of the large peak on concentration. (c) Transmittance (data collected at 800 nm) of P1 + 3⊂CB[8], P1 + 4⊂CB[8] and PDMA300 at different concentrations. | ||
Temperature-sensitive supramolecular polymers were constructed via the same strategy. Here, P2, i.e. LMWP PNIPAm as the backbone was employed to form the P2 + 3⊂CB[8] ternary complex. Similarly, an obvious viscosity enhancement of P2 + 3⊂CB[8] vs. concentration was observed (Fig. S17†), which was distinctive to that of the control P2 + 4⊂CB[8] (P2:4:CB[8] = 1:2:2). The relative viscosity of P2 + 3⊂CB[8] even reached as high as 3.0 at a concentration of 4.5 mM (calculated as P2). DLS measurements also supported the formation of aggregates, when concentration of P2 + 3⊂CB[8] reached 2.3 mM (Fig. S18†), which was similar to the case of the P1 + 3⊂CB[8] complex (Fig. 4). The TEM image revealed the particles of P2 + 3⊂CB[8] as random aggregates of MSPs (Fig. S19†). The aggregation could be attributed to the tight association between CB[8] themselves, which was observed previously in crystals and supramolecular gels.20,51 As this aggregation was exposed after the MSPs formed longer polymeric chains, CB[8] units along the long chains should have a higher local concentration and thus promote the aggregation. Furthermore, as the aggregation may cause chain collapse and then result in a viscosity decrease, the real polymerization degree of P1 + 3⊂CB[8] may be much higher than 300 as estimated from Fig. 3.
PNIPAm is well-known for its thermo-responsive properties. Thus, MSPs based on PNIPAm seem promising to retain this character. Here, P2 + 4⊂CB[8] was chosen as a control, which had a similar chain composition to P2 + 3⊂CB[8]. Their LCST behavior was measured at different concentrations. At a rather low concentration (0.2 mg mL−1, calculated as P2), P2 + 3⊂CB[8] exhibited a higher LCST temperature (the temperature at 50% transmittance) than that of P2 + 4⊂CB[8] (Fig. 5a). However, when the concentration was increased to 19.8 mg mL−1 (calculated as P2), the LCST temperature of P2 + 3⊂CB[8] was observed to be lower than that of P2 + 4⊂CB[8] (Fig. 5b). Their LCST variation vs. concentration is shown in Fig. 5c, indicating a sharp LCST decrease of P2 + 3⊂CB[8] from 32 °C to 16 °C compared to that of P2 + 4⊂CB[8], with a cross point at around 1.0 mg mL−1. This significant concentration dependence of the LCST is unexpected for covalent PNIPAm, where the molecular weight or polymer concentration did not affect the LCST singnificantly.52 A control experiment showed that the HSCT interaction in P1 + 3⊂CB[8] solution remained at higher temperature, as shown in Fig. S20,† as 90% HSCT absorbance remained when the temperature was increased from 10 °C to 35 °C. In addition, when 3 (2.8 mM, the highest concentration used in the LCST test) was added to P2 solution (2 mg mL−1), the temperature-dependent phase transition process almost remained (Fig. S21†), showing that the positive charge from the viologen guest did not have a significant effect on the phase transition process. Thus our current results indicate very different LCST behavior of the MSPs containing PNIPAm compared to the covalent polymer, which might come from the structural differences between P2 + 3⊂CB[8], P2 + 4⊂CB[8] and covalent PNIPAm. We speculate that the drastic LCST decrease of P2 + 3⊂CB[8] vs. concentration is attributed to the aggregation behavior of CB[8]. As mentioned above, the local concentration of CB[8] increased after the MSPs formed longer polymeric chains, which should facilitate the aggregation of CB[8] together with the PNIPAm. Nevertheless, this is the first LCST behavior observed for supramolecular polymers.
 | ||
| Fig. 5 Temperature dependence of the optical transmittance at 800 nm obtained for ternary complexes P2 + 4⊂CB[8] and P2 + 3⊂CB[8] at various concentrations of P2: (a) 0.2 mg mL−1, (b) 19.8 mg mL−1. (c) Concentration dependence of the LCST of ternary complexes P2 + 4⊂CB[8] and P2 + 3⊂CB[8]. Here LCST is defined as the temperature corresponding to a 50% decrease of the transmittance. | ||
Conclusion
The SP formed in this way from LMWP PDMA (DP 52) shows a higher viscosity than the reference of high MW PDMA (DP 300), which indicates the success of extending polymer chains via such HSCT interactions. Furthermore, when PNIPAm was used as a building block, the resultant SP not only inherited the LCST properties of PNIPAm but also showed an apparent effect on the LCST values. This method is expected to be a general strategy to extend polymer chains.Acknowledgements
The Ministry of Science and Technology of China (no. 2011CB932503), the National Natural Science Foundation of China (no. 91227203 and 51322306) and the Shanghai Rising-Star Program (grant 13QA1400600) are acknowledged for their financial support.Notes and references
- C. Fouquey, J. M. Lehn and A. M. Levelut, Adv. Mater., 1990, 2, 254 CrossRef CAS.
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS PubMed.
- M. Miyauchi and A. Harada, J. Am. Chem. Soc., 2004, 126, 11418 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- F. Wang, C. Han, C. He, Q. ; Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254 CrossRef CAS PubMed.
- G. Gröger, W. Meyer-Zaika, C. Böttcher, F. Gröhn, C. Ruthard and C. Schmuck, J. Am. Chem. Soc., 2011, 133, 8961 CrossRef PubMed.
- L. Zhu, M. Lu, Q. Zhang, D. Qu and H. Tian, Macromolecules, 2011, 44, 4092 CrossRef CAS.
- D. S. Guo and Y. Liu, Chem. Soc. Rev., 2012, 41, 5907 RSC.
- S. L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950 RSC.
- Y. J. Wang and L. M. Tang, Progr. Chem., 2006, 18, 308 CAS.
- F. Wang, S. Y. Dong, B. Zheng and F. H. Huang, Acta Polym. Sin., 2011, 9, 956 CrossRef.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892 CrossRef CAS.
- J. M. Lehn, Polym. Int., 2002, 51, 825 CrossRef CAS.
- M. Fathalla, C. M. Lawrence, N. Zhang, J. L. Sessler and J. Jayawickramarajah, Chem. Soc. Rev., 2009, 38, 1608 RSC.
- Z. Chen, A. Lohr, C. R. Saha-Moller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564 RSC.
- M. Hasegawa and M. Iyoda, Chem. Soc. Rev., 2010, 39, 2420 RSC.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6576 CrossRef CAS PubMed.
- W. H. Binder, L. Petraru, T. Roth, P. W. Groh, V. Pálfi, S. Keki and B. Ivan, Adv. Funct. Mater., 2007, 17, 1317 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874 CrossRef CAS.
- Z. Ge, J. Hu, F. Huang and S. Liu, Angew. Chem., Int. Ed., 2009, 48, 1798 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS.
- H. Hofmeier, R. Hoogenboom, M. E. L. Wouters and U. S. Schubert, J. Am. Chem. Soc., 2005, 127, 2913 CrossRef CAS PubMed.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS PubMed.
- S. Abed, S. Boileau and L. Bouteiller, Macromolecules, 2000, 33, 8479 CrossRef CAS.
- G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2004, 127, 810 CrossRef PubMed.
- C. P. Lillya, R. J. Baker, S. Hutte, H. H. Winter, Y. G. Lin, J. Shi, L. C. Dickinson and J. C. W. Chien, Macromolecules, 1992, 25, 2076 CrossRef CAS.
- S. Zou, H. Schönherr and G. J. Vancso, Angew. Chem., Int. Ed., 2005, 44, 956 CrossRef CAS PubMed.
- D. Knapton, S. J. Rowan and C. Weder, Macromolecules, 2005, 39, 651 CrossRef.
- J. R. Kumpfer, J. Jin and S. J. Rowan, J. Mater. Chem., 2010, 20, 145 RSC.
- J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed., 2004, 116, 4560 CrossRef.
- S. Schmatloch, A. M. J. van den Berg, A. S. Alexeev, H. Hofmeier and U. S. Schubert, Macromolecules, 2003, 36, 9943 CrossRef CAS.
- S. K. Yang, A. V. Ambade and M. Weck, Chem. – Eur. J., 2009, 15, 6605 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051 CrossRef CAS PubMed.
- Z. Ge, H. Liu, Y. Zhang and S. Liu, Macromol. Rapid Commun., 2011, 32, 68 CrossRef CAS PubMed.
- Y. Hasegawa, M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2005, 38, 3724 CrossRef CAS.
- Y. Liu, R. Fang, X. Tan, Z. Wang and X. Zhang, Chem. – Eur. J., 2012, 18, 15650 CrossRef CAS PubMed.
- Y. Xu, M. Guo, X. Li, A. Malkovskiy, C. Wesdemiotis and Y. Pang, Chem. Commun., 2011, 47, 8883 RSC.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, J. Am. Chem. Soc., 2009, 131, 2408 CrossRef CAS PubMed.
- Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305 RSC.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251 CrossRef CAS PubMed.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690 CrossRef CAS PubMed.
- K. Kim, D. Kim, J. W. Lee, Y. H. Ko and K. Kim, Chem. Commun., 2004, 848 RSC.
- Y. H. Ko, K. Kim, J. K. Kang, H. Chun, J. W. Lee, S. Sakamoto, K. Yamaguchi, J. C. Fettinger and K. Kim, J. Am. Chem. Soc., 2004, 126, 1932 CrossRef CAS PubMed.
- J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760 CrossRef CAS PubMed.
- U. Rauwald, F. Biedermann, S. P. Deroo, C. V. Robinson and O. A. Scherman, J. Phys. Chem. B, 2010, 114, 8606 CrossRef CAS PubMed.
- Q. Yan, A. Feng, H. Zhang, Y. Yin and J. Yuan, Polym. Chem., 2013, 4, 1216 RSC.
- I. Hwang, W. S. Jeon, H. J. Kim, D. Kim, H. Kim, N. Selvapalam, N. Fujita, S. Shinkai and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 210 CrossRef CAS PubMed.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details, including the preparation and characterization of CTA, P1, P2, 3 and 4. See DOI: 10.1039/c3py01763j |
| This journal is © The Royal Society of Chemistry 2014 |