Light-harvesting of polymerizable 4-hydroxy-1,3-thiazole monomers by energy transfer toward photoactive Os(II) metal complexes in linear polymers†
Alexander M.
Breul
ab,
Inês
Rabelo de Moraes
c,
Roberto
Menzel
ab,
Michael
Pfeffer
e,
Andreas
Winter
ab,
Martin D.
Hager
ab,
Sven
Rau
e,
Benjamin
Dietzek
bcd,
Rainer
Beckert
*ab and
Ulrich S.
Schubert
*abf
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de; Web: www.schubert-group.com Fax: +49 (0)3641 948202; Tel: +49 (0)3641 948200
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany. E-mail: rainer.beckert@uni-jena.de; Web: www.agbeckert.uni-jena.de Fax: +49 (0)3641 948212; Tel: +49 (0)3641 948230
cLeibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany
dInstitute of Physical Chemistry (IPC) and Abbe Center of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
eInstitute of Inorganic Chemistry I, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany
fDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 14th January 2014
Abstract
A polymer library consisting of a series of three fluorescing monomers (i.e., blue, yellow and red-emitting) based on 4-hydroxy-1,3-thiazole chromophores as well as an Os(II) metal complex containing monomer was synthesized. These materials were characterized using 1H NMR spectroscopy, size-exclusion chromatography (SEC) as well as TGA and DSC measurements. The optical properties were investigated utilizing UV-vis absorption and emission measurements. The tailor-made optical properties of the monomer units facilitate energy transfer from the blue over the yellow and red chromophores to the photoactive Os(II) polypyridyl moiety. For this purpose, the energy transfer was monitored using excitation–emission correlation spectroscopy.
Introduction
Due to its role as one of the primary processes in photosynthesis,1–5 solar cells,6–11 and light-driven catalytic reactions,12–15 light-harvesting emerged into the foreground of scientific research in the last few decades. The efficiency thereof is usually dependent on the structure of the antenna system. In particular, the antenna complex LH2 of the photosynthetic unit in purple bacteria consists of ring-like fashioned cylindrical arranged carotenoids and bacteriochlorophylls.16–19 To understand the function of the antenna system, the energy transfer processes that occurred within pigment rings (LH2 → LH2) between different pigment rings (LH2 → LH1) and the energy trapping by a reaction center (LH1 → RC) had to be investigated.16 For these energy transfer steps, a Förster resonant energy transfer (FRET) mechanism was proven to be active (LH2, LH1 and RC symbolize the different light-harvesting complexes and the reaction center with their adjusted absorption and emission).20 To simulate the embedding of the pigments into proteins, energy transfer studies were carried out on chromophore bearing polymers. Two approaches were investigated: Firstly, the main-chain functionalization,21–23 and secondly, the side-chain functionalization of polymers with different chromophores.24–30 In both cases, the FRET process itself is undirected. To overcome this drawback, the fluorescent dyes were incorporated into block copolymer architectures as side-chains.25 Depending on the polymer architecture, the energy can be transferred from a donor block to an acceptor block along the polymer chain.31–33Substitution of a fluorescent acceptor molecule by a photoactive trap, i.e. metal–organic complexes, enables a directed donation of energy towards a photosensitizer. So far, Fréchet et al. have applied a combination of grafting and copolymerization methods to establish a linear donor–acceptor copolymer containing pendant coumarin and Ru(dmbpy)32+ (dmbpy = 4,4′-dimethyl-2,2′-bipyridine) moieties. The highly efficient FRET process (>95%) between these chromophores enabled an enhanced absorption in the visible region and, consequently, an increased probability of excitation of the Ru(II) complexes compared to the single one.34–36 Moreover, Meyer and coworkers synthesized a p-(chloromethyl)styrene containing polystyrene copolymer as a platform for the grafting of various functional groups by nucleophilic substitution.37 In addition, Happ et al. prepared a statistical PMMA based terpolymer bearing 4-hydroxy-1,3-thiazole derivatives as the chromophores as well as Ru(dmbpy)2(trz-py)2+ (trz-py = 2-(triazol-4-yl)pyridine) complexes as the acceptor.38 In this context, energy transfer rates of over 70% were observed. In this contribution, the latter system is extended. Firstly, the approach of directly polymerizing methacrylate functionalized chromophores was maintained. For this purpose, the reversible addition–fragmentation chain transfer (RAFT) polymerization is applied as a versatile and controlled radical polymerization technique.39 Furthermore, this procedure avoids potential incomplete functionalization of the polymer, which is inevitable in polymer analogous reactions. Secondly, the ligand system was kept, due to its easy availability by the Cu(I)-catalyzed 1,3-cycloaddition/click reaction.40,41 Herein, the Ru(II) metal ion was substituted by an Os(II) metal ion. As a consequence, the spin-forbidden metal-to-ligand charge-transfer (MLCT) absorption band is shifted up to 700 nm due to its spin–orbit coupling by the heavy Os(II) centre.26,42 As a consequence, these complexes are capable of trapping the energy from a wider range of potential donor dyes, even including red emitters. Regarding their straight-forward accessibility, 4-hydroxy-1,3-thiazole-based chromophores provide excellent optical properties, i.e., large Stokes shift (>2000 cm−1) and fluorescence quantum yields up to unity. In addition, thiazole-based compounds can be applied in organic solar cells10,43 and can act as ligands to engineer metallo-supramolecular architectures.11,44,45 Furthermore, a tailor-made absorption and emission behaviour can be obtained with moderate synthetic efforts. Besides the blue29,30 and yellow46,47 emitting monomers of previous contributions, a red emitting one is now provided for the construction of a complete energy transfer cascade from a blue emitter over a yellow and red emitter to a photoactive Os(II) metal complex within a single polymer chain. To follow the sub-steps of this cascade, a polymer library is synthesized that contains all single FRET pairs and a stepwise increasing number of chromophores along the polymer chain as well. To the best of our knowledge, not more than one donor–acceptor FRET pair in a row has been reported so far.
Experimental
Materials and instrumentation
N,N-Dimethylacetamide (DMA), dichloromethane, 2,2′-azobis(iso-butyronitrile) (AIBN) and 2-cyano-2-propyl dithiobenzoate (CPDB) were purchased from Sigma-Aldrich. The solvents were of reagent grade, purified using common methods and distilled prior to use. Triethylene glycol monomethylether methacrylate (TEGMA) was passed over a short neutral aluminum oxide column directly before use to remove the stabilizer. AIBN was recrystallized from methanol prior to use. The [Os(dmbpy)2Cl2] precursor complex,48 the HO–C11H22-2-(1H-1,2,3-triazol-4-yl)-pyridine,49 the MB30 and the MY46,47 were synthesized according to the literature. All reactions were performed under a nitrogen atmosphere in glassware equipped with a Teflon™ coated magnetic stirring bar. Preparative size-exclusion chromatography (SEC) was performed on a BioRad S-X1 (size-exclusion limit: 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 with dichloromethane, as an eluent). Size exclusion chromatograms were recorded using a Waters size exclusion chromatogram (SEC) system equipped with a DG-980-50 degasser, a HPLC 1515 pump, a column heater 1500 oven, a photo diode array (PDA) detector 2996, a RI detector 2414, and a Waters pre/Phenomenex Phenogel 103 Å/105 Å column using N,N-dimethylacetamide (DMA) with 0.08 mol% NH4PF6, as the solvent, with a flow rate of 1 mL min−1 at 50 °C. Linear PMMA standards were utilized for the calibration. GC measurements were performed on an Interscience Trace GC with a Trace Column RTX-5 connected to a PAL autosampler. 1H and 13C NMR spectra were recorded using Bruker AC 250 (250 MHz), 300 (300 MHz) and 400 (400 MHz) spectrometers at 298 K, respectively. The chemical shifts are reported in parts per million (ppm, δ scale) relative to signals from the NMR solvents, coupling constants are given in Hz. The melting points were measured with a Galen III apparatus (Boëtius system). Reactions were monitored by TLC on 0.2 mm Merck silica gel plates (60 F254). The mass spectra were measured either with a Finnigan MAT SSQ 710 (EI) or with a MAZ 95 XL (FAB) system. Elemental analyses were carried out using a CHN-932 Automat Leco instrument. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were obtained from an Ultraflex III TOF/TOF mass spectrometer with dithranol or DCTB, as a matrix, in reflector as well as linear mode. The instrument was calibrated prior to each measurement with an external PMMA standard from Polymer Standards Services GmbH (PSS, Mainz, Germany). Electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) measurements were performed in the positive ion mode with a micrOTOF (Bruker Daltonics) mass spectrometer equipped with an automatic syringe pump which was supplied from KD Scientific for sample injection. The standard electrospray ion (ESI) source was used to generate the ions. The concentration of the samples was 10 μg mL−1 and all samples were injected using a constant flow rate (180 μL h−1) of sample solution. ESI solvents used in this study were acetone, dichloromethane, acetonitrile, chloroform or their mixtures. The ESI-Q-TOF-MS instrument was calibrated in the m/z range of 50 to 3000 using an internal calibration standard (Tunemix solution, Agilent). Data were processed via Bruker Data Analysis software version 4.0. HR-MS calculations have been made by using this software. The UV-vis absorption and PL emission spectra were recorded on an Analytik Jena SPECORD 250 and a Jasco FP-6500 spectrometer, respectively, at 298 K. For this purpose, dilute solutions (10−6 M to 10−5 M, 1 cm quartz cuvette) in CH3CN were used. As a reference, a quartz cuvette filled with the pristine solvent was utilized. In the case of the excitation–emission correlation spectra investigation, diluted polymer solutions were prepared in dichloromethane. Quantum yield measurements were performed with a Perkin Elmer Lambda 16 UV-vis spectrometer in perpendicular excitation–emission geometry, while the absorbance in the most red-shifted absorption maximum was <0.05. A detailed description of the respective setup is given in previous contributions.11
000 g mol−1 with dichloromethane, as an eluent). Size exclusion chromatograms were recorded using a Waters size exclusion chromatogram (SEC) system equipped with a DG-980-50 degasser, a HPLC 1515 pump, a column heater 1500 oven, a photo diode array (PDA) detector 2996, a RI detector 2414, and a Waters pre/Phenomenex Phenogel 103 Å/105 Å column using N,N-dimethylacetamide (DMA) with 0.08 mol% NH4PF6, as the solvent, with a flow rate of 1 mL min−1 at 50 °C. Linear PMMA standards were utilized for the calibration. GC measurements were performed on an Interscience Trace GC with a Trace Column RTX-5 connected to a PAL autosampler. 1H and 13C NMR spectra were recorded using Bruker AC 250 (250 MHz), 300 (300 MHz) and 400 (400 MHz) spectrometers at 298 K, respectively. The chemical shifts are reported in parts per million (ppm, δ scale) relative to signals from the NMR solvents, coupling constants are given in Hz. The melting points were measured with a Galen III apparatus (Boëtius system). Reactions were monitored by TLC on 0.2 mm Merck silica gel plates (60 F254). The mass spectra were measured either with a Finnigan MAT SSQ 710 (EI) or with a MAZ 95 XL (FAB) system. Elemental analyses were carried out using a CHN-932 Automat Leco instrument. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were obtained from an Ultraflex III TOF/TOF mass spectrometer with dithranol or DCTB, as a matrix, in reflector as well as linear mode. The instrument was calibrated prior to each measurement with an external PMMA standard from Polymer Standards Services GmbH (PSS, Mainz, Germany). Electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) measurements were performed in the positive ion mode with a micrOTOF (Bruker Daltonics) mass spectrometer equipped with an automatic syringe pump which was supplied from KD Scientific for sample injection. The standard electrospray ion (ESI) source was used to generate the ions. The concentration of the samples was 10 μg mL−1 and all samples were injected using a constant flow rate (180 μL h−1) of sample solution. ESI solvents used in this study were acetone, dichloromethane, acetonitrile, chloroform or their mixtures. The ESI-Q-TOF-MS instrument was calibrated in the m/z range of 50 to 3000 using an internal calibration standard (Tunemix solution, Agilent). Data were processed via Bruker Data Analysis software version 4.0. HR-MS calculations have been made by using this software. The UV-vis absorption and PL emission spectra were recorded on an Analytik Jena SPECORD 250 and a Jasco FP-6500 spectrometer, respectively, at 298 K. For this purpose, dilute solutions (10−6 M to 10−5 M, 1 cm quartz cuvette) in CH3CN were used. As a reference, a quartz cuvette filled with the pristine solvent was utilized. In the case of the excitation–emission correlation spectra investigation, diluted polymer solutions were prepared in dichloromethane. Quantum yield measurements were performed with a Perkin Elmer Lambda 16 UV-vis spectrometer in perpendicular excitation–emission geometry, while the absorbance in the most red-shifted absorption maximum was <0.05. A detailed description of the respective setup is given in previous contributions.11
The acetonitrile used was freshly distilled under argon. The samples for the LED light irradiation experiments were prepared in GC vials (diameter = 45 × 14.75 mm) with a known headspace of 3 mL and a headspace-solution ratio of 3/2. Furthermore the GC vials were charged in the dark and under an argon stream. Hereby 0.6 mL of triethylamine, 0.2 mL of degassed water (containing K2PtCl4) and 1.2 mL of acetonitrile (containing the polymer) were combined and the vials were sealed afterwards. Subsequently, the GC vials were irradiated with blue LED light (470 nm, P = 30 to 40 mW). After irradiation, 200 μL samples were drawn from the headspace and injected immediately into the GC apparatus. The hydrogen evolved was measured by headspace GC on a Bruker Scion GC/MS, with a thermal conductivity detector (column: mol. sieve 5 Å 75 m × 0.53 mm I.D., oven temp. 70 °C, flow rate 22.5 mL min−1, detector temp. 200 °C) with argon as the carrier gas. The GC was calibrated by mixing different volumes of pure hydrogen together with argon into a Schlenk vessel. The obtained signal was plotted against the calibration curve and multiplied accordingly to receive the total produced hydrogen content in the headspace. Irradiation experiments and hydrogen measurements were repeated several times for every reaction time.
Synthesis of the polymers
The required amounts of monomers (for synthetic details, see the ESI†) were transferred into a 5 mL reaction vessel (see Table 1 for exact amounts and reaction conditions). Thereafter, the calculated volumes of stock solutions of 2-cyanopropan-2-yl benzodithioate (CPDB), as a chain-transfer agent, in N,N-dimethylacetamide (DMA) as well as 2,2′-azobis(iso-butyronitrile) (AIBN), as an initiator, in DMA were added. In the case of MOs containing polymers, also a stock solution was prepared to transfer the required amount of polymerizable Os(II) complex into the reaction vessel. The [CPDB] to [AIBN] ratio was always 4:1 and the [overall monomer] to [CPDB] ratio was kept at 120. Subsequently, the reaction mixture was filled-up with DMA (conc. = 1.75 M). Before closing the vial, the reaction solution was purged with a flow of nitrogen for 30 minutes. Subsequently, the reaction was carried out in an oil bath at 70 °C overnight (15 to 17 h, see Table 1). The polymers were purified by preparative SEC (BioRad SX-1 beads and dichloromethane, as an eluent) in order to remove the unreacted monomers. The pure fractions were collected and dried in vacuo.
| Polymer | Molar monomer ratio [TEGMA]:[MB]:[MY]:[MR]:[MOs] |
m TEGMA [mg] | m MB [mg] | m MY [mg] | m MR [mg] | m MOs [mg] | m AIBN [mg] | m CPDB [mg] | V DMA [mL] | time [h] |
|---|---|---|---|---|---|---|---|---|---|---|
| PB | 94/6/—/—/— | 852 | 89.1 | — | — | — | 1.33 | 7.20 | 2.23 | 15 |
| PY | 94/—/6/—/— | 548 | — | 63.8 | — | — | 0.86 | 4.63 | 1.43 | 15 |
| PR | 94/—/—/6/— | 500 | — | — | 67.5 | — | 0.78 | 4.22 | 1.31 | 15 |
| POs | 96/—/—/—/4 | 497 | — | — | — | 110 | 0.76 | 4.11 | 1.27 | 15 |
| PB-Y | 88/6/6/—/— | 409 | 45.7 | 50.8 | — | — | 0.68 | 3.69 | 1.14 | 16 |
| PY-R | 88/—/6/6/— | 400 | — | 49.7 | 57.7 | — | 0.67 | 3.61 | 1.12 | 16 |
| PR-Os | 92/—/—/4/4 | 455 | — | — | 41.9 | 105 | 0.73 | 3.93 | 1.22 | 15.5 |
| PB-Y-R | 82/6/6/6/— | 654 | 78.4 | 87.3 | 101.3 | — | 1.17 | 6.33 | 1.96 | 16 |
| PY-R-Os | 82/—/6/6/6 | 235 | — | 31.4 | 36.4 | 91.3 | 0.42 | 2.28 | 0.71 | 17 |
| PB-Y-R-Os | 84/4/4/4/4 | 435 | 33.9 | 37.8 | 43.8 | 110 | 0.76 | 4.11 | 1.22 | 16.5 |
800 g mol−1, Đ = 1.26. Conv.dye = 87%, conv.TEGMA = 80%. 1H NMR (300 MHz, CD2Cl2): δ (ppm): 8.61 (b, py-H6), 8.13 (b, py-H3), 7.95–7.65 (b, py-H4, Ph-H2,6), 7.54–7.15 (b, py-H5, Ph-H3,4,5), 4.62 (b, OCH2spacer), 4.08 (b, CH2-MA), 3.64 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.38 (b, OCH3TEG), 2.30–1.53 (b, CH2), 1.53–0.32 (b, CH2, CH3). DSC: no Tg. TGA: Td = 211 °C.
400 g mol−1, Đ = 1.29. Conv.dye = 83%, conv.TEGMA = 88%. 1H NMR (250 MHz, CD2Cl2): δ (ppm): 8.59 (b, py-H6), 8.13 (b, py-H3), 7.95–7.65 (b, py-H4, Ph-H2,6), 7.33 (b, Ph-H3,5), 4.62 (b, OCH2spacer), 4.08 (b, CH2-MA), 3.84 (b, OCH3dye), 3.64 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.38 (b, OCH3TEG), 3.15 (b, N-CH3), 2.76–0.32 (b, CH2, CH3). DSC: no Tg. TGA: Td = 231 °C.
700 g mol−1, Đ = 1.26. Conv.dye = 79%, conv.TEGMA = 87%. 1H NMR (250 MHz, CDCl3): δ (ppm): 7.91 (b, CH-(CN)2), 7.77 (b, thiophene-Htrz), 7.61 (b, Ph-H2,6), 7.54 (b, thiophene-HCH), 6.93 (b, Ph-H3,5), 4.56 (b, OCH2spacer), 4.08 (b, CH2-MA), 3.69 (b, OCH2TEG), 3.63 (b, OCH2TEG), 3.56 (b, OCH2TEG), 3.38 (b, OCH3TEG), 3.15 (b, N-CH3) 2.30–1.53 (b, CH2), 1.53–0.32 (b, CH2, CH3). DSC: Tg = 117 °C. TGA: Td = 220 °C.
100 g mol−1, Đ = 1.64. Conv.dye = 78%, conv.TEGMA = 70%. 1H NMR (250 MHz, CD2Cl2): δ (ppm): 8.67 (b, dm-bpy, Ar-H3,3′), 8.22 (b, dm-bpy, Ar-H6,6′), 8.16 (b, trz-py-H), 8.12 (b, trz-py-H), 7.97 (b, trz-py-H), 7.88 (b, trz-H), 7.75 (b, trz-H), 7.49 (b, trz-H), 7.16 (b, dm-bpy, Ar-H5,5′), 7.05 (b, trz-py-H), 4.33 (b, OCH2spacer), 4.12 (b, CH2-MA), 3.84 (b, OCH3dye), 3.64 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.38 (b, OCH3TEG), 2.65 (b, CH3-py), 2.47–0.32 (b, CH2, CH3). DSC: no Tg. TGA: Td = 209 °C.
500 g mol−1, Đ = 1.30. Conv.dyes = 89%, conv.TEGMA = 91%. 1H NMR (300 MHz, CD2Cl2): δ (ppm): 8.58 (b, py-H6), 8.13 (b, py-H3), 7.91–7.55 (b, Ar-H, py-H), 7.45–6.96 (b, Ar-H, py-H), 4.60 (b, OCH2spacer), 4.08 (b, OCH2TEG), 3.64 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.38 (b, OCH3TEG), 3.13 (b, N–CH3), 2.36–0.38 (b, CH2, CH3). DSC: no Tg. TGA: Td = 212 °C.
500 g mol−1, Đ = 1.29. Conv.dyes = 67%, conv.TEGMA = 72%. 1H NMR (250 MHz, CDCl3): δ (ppm): 8.75–7.31 (b, Ar-H, thiophene-H, py-H), 7.02–6.79 (b, Ar-H, py-H), 4.57 (b, OCH2spacer), 4.09 (b, OCH2TEG), 3.86 (b, OCH3thiazole), 3.65 (b, OCH2TEG), 3.56 (b, OCH2TEG), 3.38 (b, OCH3TEG), 3.09 (b, N-CH3), 2.56–0.42 (b, CH2, CH3). DSC: no Tg. TGA: Td = 204 °C.
600 g mol−1, Đ = 1.40. Conv.dyes = 61%, conv.TEGMA = 73%. 1H NMR (300 MHz, CD2Cl2): δ (ppm): 8.7–6.4 (b, Ar-H, trz-H, py-H), 4.60 (b, OCH2spacer), 4.12 (b, OCH2TEG), 3.86 (b, OCH3thiazole), 3.69 (b, OCH2TEG), 3.63 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.37 (b, OCH3TEG), 2.66 (b, CH3-py), 2.14–0.49 (b, CH2, CH3). DSC: no Tg. TGA: Td = 212 °C.
300 g mol−1, Đ = 1.22. Conv.dyes = 77%, conv.TEGMA = 71%. 1H NMR (250 MHz, CDCl3): δ (ppm): 8.54 (b, py-H6), 8.18–6.49 (b, Ar-H, py-H), 4.55 (b, OCH2spacer), 4.09 (b, OCH2TEG), 3.83 (b, OCH3thiazole), 3.64 (b, OCH2TEG), 3.37 (b, OCH3TEG), 3.00 (b, OCH3thiazole), 2.36–0.39 (b, CH2, CH3). DSC: Tg = 117 °C. TGA: Td = 216 °C.
500 g mol−1, Đ = 1.82. Conv.dyes = 58%, conv.TEGMA = 50%. 1H NMR (300 MHz, CD2Cl2): δ (ppm): 8.87–6.38 (b, Ar-H, trz-H, py-H), 4.61 (b, OCH2spacer), 4.35 (b, CH2-MA), 4.11 (b, OCH2TEG), 3.86 (b, OCH3thiazole), 3.69 (b, OCH2TEG), 3.63 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.37 (b, OCH3TEG), 3.03 (b, N-CH3), 2.66 (b, CH3-py), 2.34–0.45 (b, CH2, CH3). DSC: no Tg. TGA: Td = 232 °C.
700 g mol−1, Đ = 1.62. Conv.dyes = 51%, conv.TEGMA = 58%. 1H NMR (250 MHz, CD2Cl2): δ (ppm): 8.08–6.29 (b, Ar-H, trz-H, py-H), 4.62 (b, OCH2spacer), 4.34 (b, CH2-MA), 4.11 (b, CH2-MA), 3.86 (b, OCH3thiazole), 3.69 (b, CH2-MA), 3.63 (b, OCH2TEG), 3.54 (b, OCH2TEG), 3.36 (b, OCH3TEG), 3.02 (b, N-CH3), 2.66 (b, CH3-py), 2.38–0.37 45 (b, CH2, CH3). DSC: no Tg. TGA: Td = 215 °C.
Results and discussion
Synthesis and characterization
A polymer library based on the polymerization of a series of 4-hydroxy-1,3-thiazole-based chromophores as well as one polymerizable photoactive Os(dmbpy)2(trz-py)2+ metal complex (Scheme 1) was generated. To obtain a sufficiently high solubility in a wide range of organic solvents (including water and other hydrophilic organic solvents), triethylene glycol monomethyl ether methacrylate (TEGMA) was selected as a comonomer. For the investigation of the thiazole monomers as an antenna system, three monomers were synthesized that absorb and emit in different regions of visible light. In particular, the blue-emitting monomer (MB) emits in the absorption maximum of the yellow emitting monomer (MY) which again emits in the absorption maximum of the red-emitting monomer (MR). Finally MR emits in the MLCT absorption band of the photoactive monomer MOs (for details, see Fig. 1). As model substances for the characterization of the photophysical properties of each single dye in the polymer matrix, single-functional moiety containing polymers were synthesized: PB, PY, PR and POs. In addition, the interaction of each donor–acceptor pair can be monitored in the terpolymers PB-Y, PY-R, and PR-Os. The extension of the classical two-component donor–acceptor (D–A) system was achieved for the polymers PB-Y-R and PY-R-Os. Therein, the acceptor fluorescence is again quenched by a third chromophore, i.e. the red dye and the metal complex, respectively. Finally, the complete antenna system consisting of the three dyes and the photoactive complex are combined within one polymer: PB-Y-R-Os. For the polymerization, the RAFT technique was applied as a controlled radical polymerization process.39 According to previous publications, all three monomers are stable under the polymerization conditions used.30,38,47 AIBN was applied as a radical initiator, DMA as a solvent and CPDB as a chain transfer agent. As reaction conditions, in particular, the [M]/[CPDB] ratio, the reaction time as well as the temperature, standard procedures known for the polymerization of MMA were applied.50 The content of the single functional moieties was set to 4 to 6% to ensure on the one hand an adequately low content of the dyes along the polymer chain to diminish reabsorbing and self-quenching processes of the chromophores as already described.29 On the other hand, an adequate degree of functionalization of at least one moiety per dye on each single chain is secured.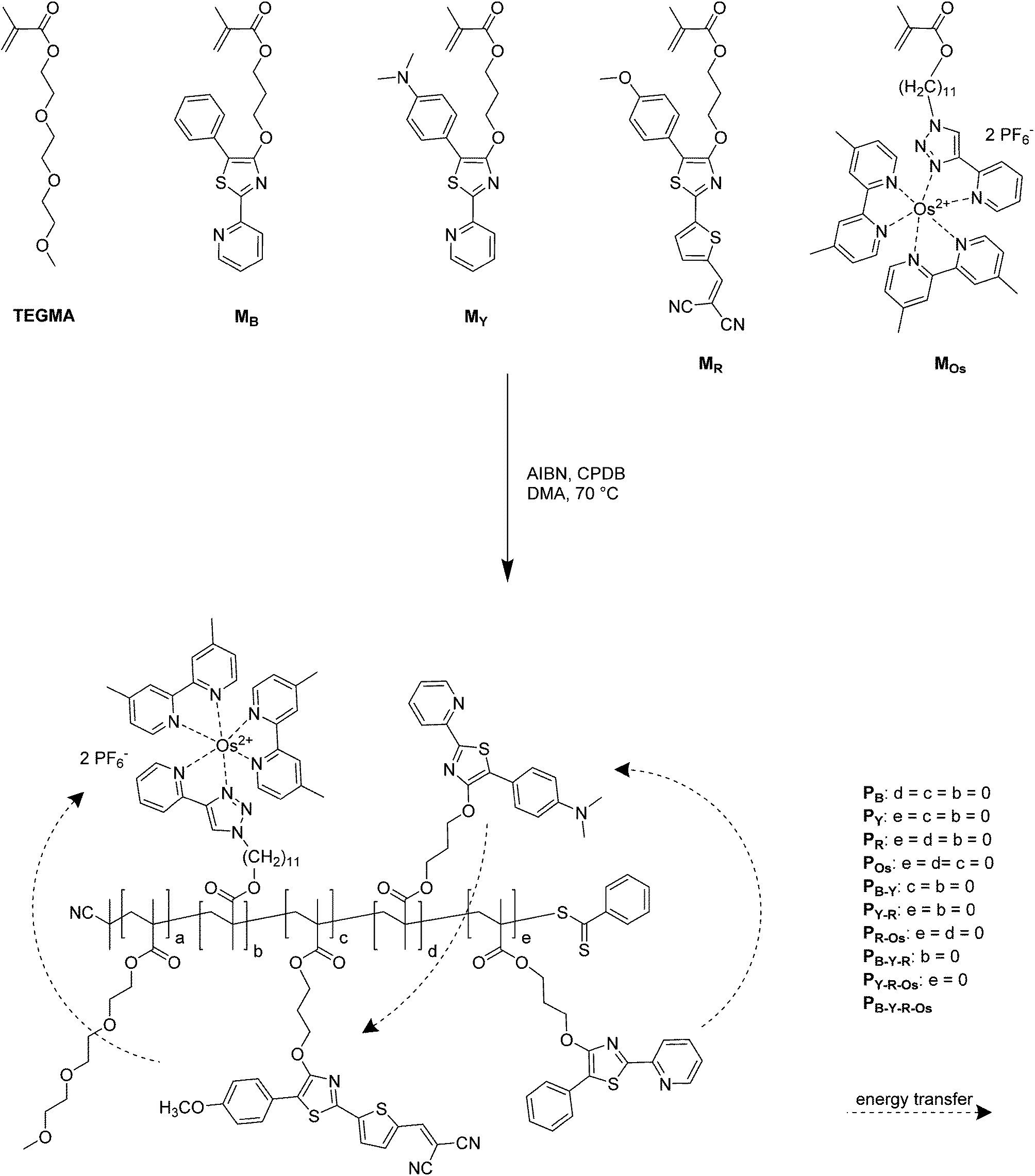 | ||
| Scheme 1 Schematic representation of the polymer syntheses. | ||
 | ||
| Fig. 1 Absorption (solid) and emission spectra (dashed) of the polymerizable thiazole dyes MB, MY, and MR as well as the absorption spectrum of MOs in CH3CN at RT. | ||
Investigations of RAFT polymers showed that the dithiobenzoate end group can potentially interfere with the light-harvesting chromophores attached to the polymer backbone.51,52 According to our previous investigations, the removal of the end group is not required because the optical properties of the thiazole dyes are preserved after RAFT polymerization.29,30 Furthermore, preliminary studies showed the function of diverse chromophores (including thiazoles) for a FRET process with a sufficiently high efficiency in the presence of a RAFT end group attached to the polymer chain.24,38
Statistical incorporation of the thiazole containing monomers is proven by a kinetic study (Fig. 2). For this purpose, samples were taken during the polymerization of PB. In this context, the conversion of the TEGMA was determined by 1H NMR spectroscopy, and the conversion of MB was determined by the integration of the UV-signal of the SEC trace. Using these two independent methods, it can be concluded that both monomers are statistically incorporated: firstly, the Mn plot vs. the conversion of the two monomers is overlapping and reveals a linear increase (Fig. 2a). Secondly, all Đ values are situated below 1.3. Finally, also the ln([M0]/[Mt]) plot over time pictures a linear increasing slope for both monomers. As a consequence, all plots show a first order kinetic for the incorporation of MB. An induction period, which is sometimes observed for RAFT polymerizations, was not detected for the investigated polymerizations (linear regression of ln([M]0/[M]t) = 0.00555 min−1 × t through the origin of the plot).53,54 Due to similar structures of all fluorescent monomers including spatial separation of the chromophore from the methacrylate functionality by a C3-spacer, it is plausible to assume that all monomers undergo statistical incorporation and controlled radical polymerization.
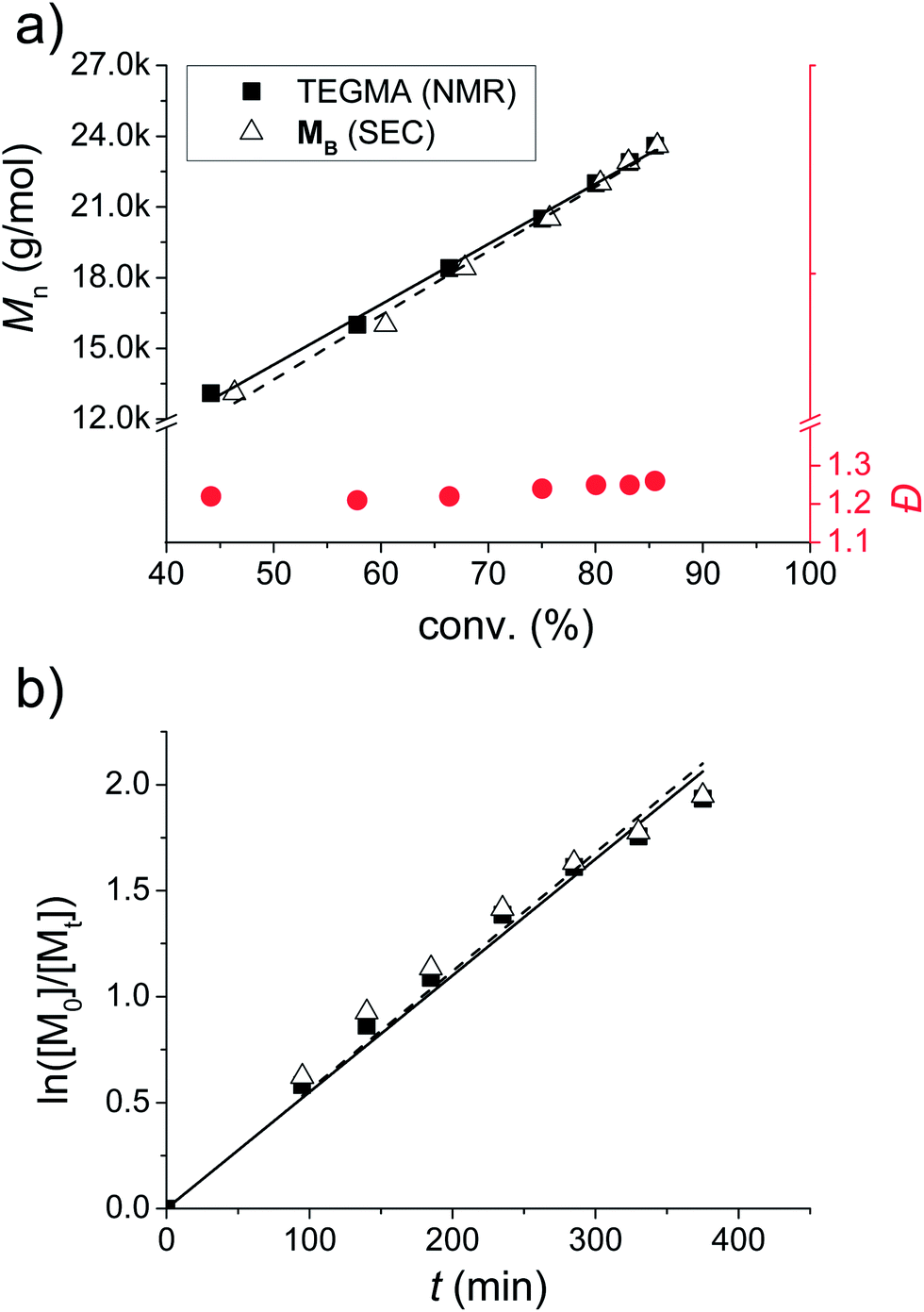 | ||
| Fig. 2 Kinetic study of the polymerization of PB. (a) Plot of the molar mass vs. each monomer conversion as well as the corresponding Đ values. (b) Logarithmic plot vs. time for both monomers. Linear regression for rate constant of the RAFT polymerization: ln([M]0/[M]t) = 0.00555 min−1 × t. | ||
All obtained polymers were characterized by SEC measurements, 1H NMR spectroscopy, TGA and DSC analyses. Fig. 3 depicts the 2D SEC plot of PB-Y-R-Os (for the SEC plots of all other polymers and 1H NMR spectra, see the ESI†). Table 2 lists selected characterization data of the polymers. The 2D SEC-UV-vis plot proves the incorporation of all moieties into the polymer chain, which is expressed by a significantly increased absorption between 350 and 500 nm. The absorption up to 700 nm corresponds to the MLCT of the Os metal complex (for details, see the Optical properties section). The composition of the polymers was estimated by the integration of the characteristic signals from the 1H NMR spectra. In all cases the TEGMA content was calculated from the –OCH2– and –OCH3 groups at 3.54 and 3.36 ppm, respectively. The content of MOs can be calculated from the methyl groups of the pyridine ligand at 2.66 ppm. For MR, the –OCH3 group at 3.86 ppm and for MY, the dimethyl amino functionality at 3.00 ppm was used for the calculation of the corresponding dye content. In contrast MB does not reveal any well-separated significant signals in the 1H NMR spectrum. For this purpose, the overall thiazole-dye content was calculated from the –OCH2– (attached to the chromophore) spacer signal at 4.60 ppm. Taking the values for the other monomers into account, this signal leads to the content of MB after subtraction of the values for MY or MR, respectively. Moreover, the SEC traces of all polymers display a unimodal distribution. However, PY-R-Os revealed a shoulder in the higher molar mass region which can potentially arise from chain-coupling termination reactions.55 The polymers that do not contain any Os(II) metal complexes feature a narrow molar mass distribution with Đ values below 1.3. In the case of the Os(dmbpy)2(trz-py)2+ bearing materials, the Đ value is slightly increased, which might be explained by the sterical demanding side-chain moiety or interactions of the charged complex with the column material. Furthermore, the conversion of TEGMA correlates with the conversion of the dyes which supports the kinetic investigation of a statistical incorporation of the chromophores. The absolute values of the conversion are always situated above 50%. Regarding the applied [M]/[CPDB] ratio of 120, a degree of polymerization of at least 65 to 70 was achieved. The dye contents between 3 and 8% ensure a functionalization approximately of at least three dye units per chain. As a consequence, on average more than a single chromophore is attached to each chain of a polymer. In addition, the theoretical molar mass values, calculated by the conversion of the monomers, also correlate with the measured ones. All polymers are stable up to at least 200 °C proven by the Td values in the range between 204 and 232 °C according to thermogravimetric analysis (TGA). In addition, for polymers PB, PY, POs, PB-Y, PY-R, PR-Os, PY-R-Os and PB-Y-R-Os no Tg values were observed either due to intra- and intermolecular interactions of the dyes and/or the high dye content.27,28 According to previous findings, the incorporation of an Os(tpy)22+ moiety into the polymer can also cause the absence of a glass transition temperature.26
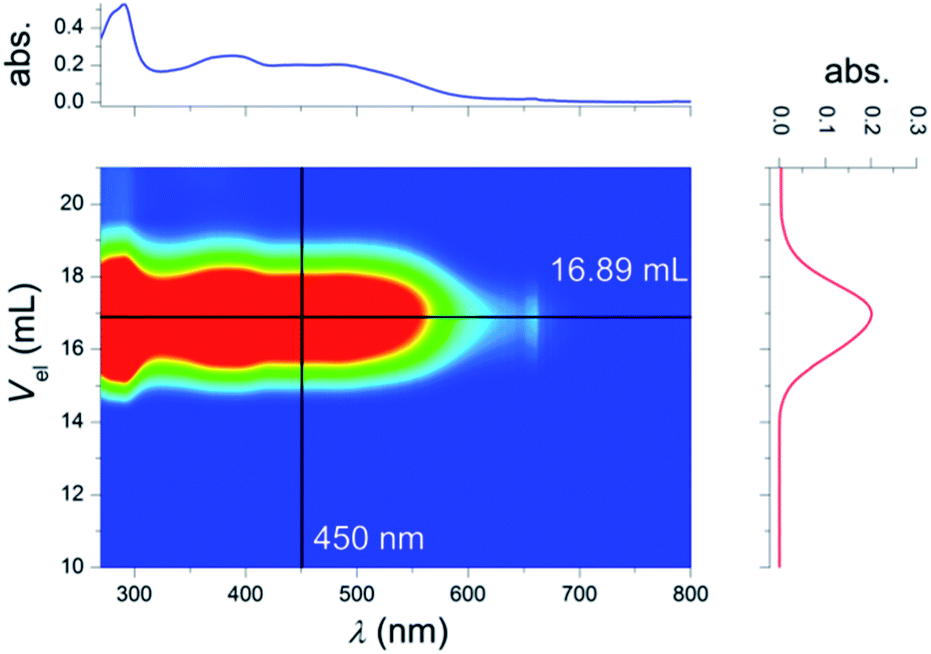 | ||
| Fig. 3 2D-SEC (DMA, NH4PF6, PDA detector) of PB-Y-R-Os. The inserted lines display the corresponding profiles of the absorption spectrum (top) and the SEC trace (right). | ||
| Polymer | Conv. TEGMAa [%] | Overall dye conv.b [%] | M n,theo [g mol−1] | M n,SEC [g mol−1] | Đ | Dye contente [%] (TEGMA/MB/MY/MR/MOs) | T d [°C] |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy from the reaction mixture. b Determined by integration of the UV-vis trace of the SEC curves from the reaction mixtures. c Calculated from the measured conversion values. d SEC trace: DMA, NH4PF6PMMA standard. e Calculated by integration of the corresponding signals from the 1H NMR spectrum. | |||||||
| PB | 80 | 87 | 23500 |
23800 |
1.26 | 96/4/—/—/— | 211 |
| PY | 88 | 83 | 25800 |
24400 |
1.29 | 96/—/4/—/— | 231 |
| PR | 87 | 79 | 25800 |
25700 |
1.26 | 95/—/—/5/— | 220 |
| POs | 70 | 78 | 23600 |
21100 |
1.64 | 97/—/—/—/3 | 209 |
| PB-Y | 91 | 89 | 27700 |
23500 |
1.30 | 89/6/5/—/— | 212 |
| PY-R | 72 | 67 | 22300 |
19500 |
1.29 | 86/—/6/8/— | 204 |
| PR-Os | 73 | 61 | 25000 |
21600 |
1.40 | 91/—/—/5/4 | 212 |
| PB-Y-R | 71 | 77 | 22800 |
22300 |
1.22 | 85/6/4/5/— | 216 |
| PY-R-Os | 50 | 58 | 19400 |
32500 |
1.83 | 85/—/4/7/4 | 232 |
| PB-Y-R-Os | 58 | 51 | 20700 |
15700 |
1.62 | 85/4/3/4/4 | 215 |
Optical properties
The absorption and emission spectra of the applied monomers are depicted in Fig. 1. The typical π–π* transition of the unsubstituted MB bathochromically shifted by the introduction of a dimethylamino functionality (electron donating) in MY. In the case of MR, also a cyanoacrylic acid group electron accepting moiety was attached to enhance the intramolecular charge transfer (ICT) transition (in this case a methoxy group acts as the electron donor).10 Consequently, the absorption and emission spectra of the different monomers were varied over the entire visible light spectrum. The MLCT absorption is situated in the range of the emission maximum of the red-emitting monomer and, thus, an energy transfer from MB over MY and MR to MOs can be expected.According to the literature, the optical properties of the monomers are preserved after polymerization.24,25,29,30,38,47 To validate this expected behaviour also for the prepared polymers, the absorption spectra of the selected polymers are plotted in Fig. 4. For this purpose, the absorption spectra of the MOs containing polymers POs, PR-Os, PY-R-Os and PB-Y-R-Os were normalized to the MLCT absorption of the Os(dmby)2(trz-py)2+ moiety at 660 nm (Fig. 4e). One can observe that all monomers are incorporated due to the increased absorption of the polymers at the absorption maxima of the corresponding monomers.
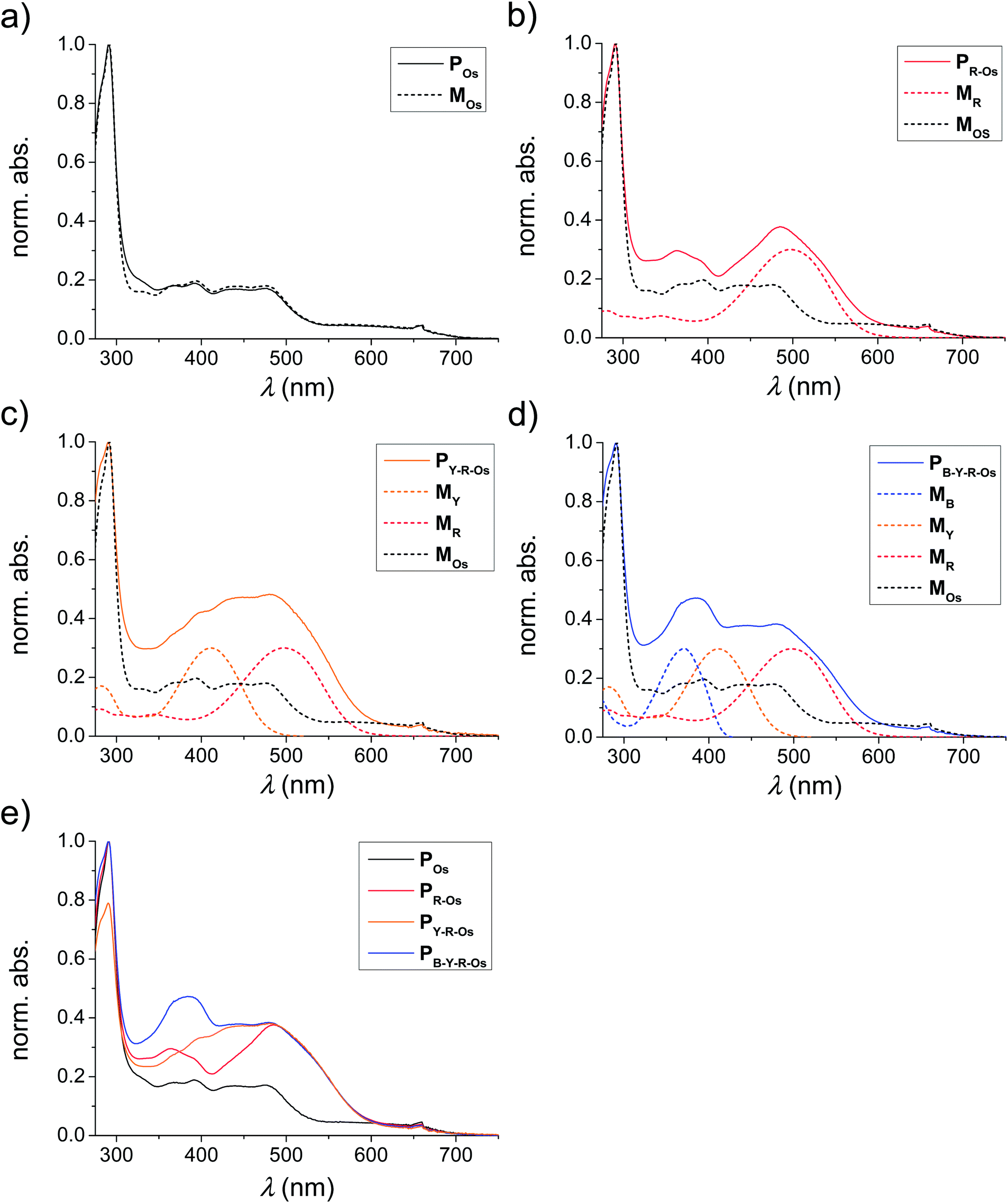 | ||
| Fig. 4 Normalized absorption spectra of the metal complex containing polymers POs, PR-Os, PY-R-Os and PB-Y-R-Os (solid) as well as their corresponding monomers incorporated (dashed). | ||
In recent studies, it was revealed that a mixture of dye-pendant polymers does not undergo any energy transfer processes in dilute solutions.24,27,34,36 As a consequence, the design of a linear polymer chain bearing all chromophores in the side-chain was targeted. To investigate the emission properties of the samples and to test for energy transfer from the antenna dyes to the photoactive Os-complex, the polymers were dissolved in dichloromethane yielding concentrations of Pos = 48.6 mM, PR-Os = 3.9 mM, PY-R-Os = 25.8 mM and PB-Y-R-Os = 10.7 mM, respectively. Emission and excitation spectra of the samples were recorded at room temperature in a 1 cm cuvette. The data were combined to yield two-dimensional excitation–emission correlation plots, which allow for a correlation between the excitation and the emission spectra and enable to deduce energy transfer pathways in the samples in a straightforward manner.
Fig. 5 displays the excitation–emission correlation spectra of the polymers: (a) POs, (b) PR-Os, (c) PY-R-Os, and (d) PB-Y-R-Os. The contour plots are generated by normalization of the spectra to the maximum. Subsequently, ten equally spaced contours are plotted.
 | ||
| Fig. 5 Excitation–emission correlation spectra of the polymers: (a) POs, (b) PR-Os, (c) PY-R-Os, and (d) PB-Y-R-Os. The grid lines are inserted for easier readability: the vertical lines correspond to the absorption maxima of the corresponding monomers, the horizontal lines correspond to the emission maxima of the corresponding dye monomers. | ||
The polymer containing only the Os-complex reveals the characteristic emission of the complex at around 700 nm irrespective of the excitation wavelength. In particular, the elongated emission ridge in the emission range between 675 and 800 nm (1) upon excitation at 320 nm turns out to be characteristic for emission of the complex. This feature remains visible in all polymers though to a much reduced extent, indicating Os-complex emission in all of the polymers. Upon inclusion of the organic dyes into the polymer structure, the excitation–emission correlation spectra become much more complex indicating contributions to both the excitation and the emission of the polymers from the organic dyes. Particularly, PR-Os, PY-R-Os and PB-Y-R-Os display emission at 620 nm, which stems from the presence of the red dye (MR). The emission is both visible upon direct excitation of the dye at around 515 nm and upon excitation of the yellow and blue dyes in PY-R-Os and PB-Y-R-Os. The energy transfer from the latter dyes to the red dye can be seen by the ridge forming at an emission at 650 nm upon excitation in the range between 275 and 450 nm (2). Furthermore, indications of energy transfer from the organic antenna dyes to the Os-complex can be found as the red-wing of the emission spectrum of the polymers PR-Os, PY-R-Os and PB-Y-R-Os upon excitation between 400 and 550 nm extents up to about 700 nm (3). Additional indications for energy transfer between the different organic dyes in the polymers PY-R-Os and PB-Y-R-Os can be found when considering the complex spectral shape in the excitation (emission) region 320 to 500 (400 to 575) nm (4). Here, the spectral characteristics change upon inclusion of the blue dye, i.e. going from PY-R-Os and PB-Y-R-Os, which indicates different (additional) interactions in the polymer chains. Moreover, all plots display a diagonal line of punctiform maxima (5) which stem from scattered excitation light.
For the investigation of a side-chain pendant antenna system connected to a potential reaction center, POs was tested for its photosensitizing properties in photochemical hydrogen production (see ESI†).14,56 In this context, the Os(II) model system proved to be a functioning polymer-based photosensitizer and electron relay for light-driven hydrogen evolution induced by LED light on colloidal platinum.
Conclusion
A series of three fluorescent monomers (i.e., blue, yellow and red fluorescing) bearing an alkylated 4-hydroxy-1,3-thiazole chromophore were utilized for the creation of a polymer library using the RAFT polymerization procedure. The optical properties of these monomers were altered by the variation of the push–pull system attached to the thiazole heterocycle. In this context, a spectral overlap between the emission and absorption behaviour of the different chromophores was guaranteed. In addition, a – with respect to hydrogen production – photochemically active Os(dmbpy)2(trz-py)2+ bearing monomer was prepared for copolymerization with these dyes. The MLCT absorption of this metal complex containing monomer also features an overlap between the emissions of the longest wavelength emitting thiazole (i.e., the red fluorescing monomer). Consequently, an energy transfer from the blue over the yellow and red emitting moieties to the metal complex should be possible. To follow the sub-steps of this antenna cascade, the number of functional pendants along the polymer chain was increased stepwise from one to four. The energy transfer within the polymers was monitored by excitation–emission spectroscopy.Future investigations will focus on block copolymer architectures for the antenna system that can potentially enhance the efficiency of the energy transfer between the dyes. Moreover, the design of block copolymers might enable a micelle formation in solution by the choice of backbone materials with different polarities, e.g., styrene and PEGMA. Therefore, a more precisely directed energy transfer might be achieved.
Acknowledgements
The authors thank the Thuringian Ministry for Education, Science and Culture [grant #B514-09049, Photonische Mizellen (PhotoMIC)] for financial support. B.D. acknowledges financial support by the Fonds der Chemischen Industrie, M.P. and S.R. acknowledge the German research Association (DFG, GRK 1626). Additionally, the Dutch Polymer Institute (technology area HTE) is acknowledged for financial support.Notes and references
- Z. F. Liu, H. C. Yan, K. B. Wang, T. Y. Kuang, J. P. Zhang, L. L. Gui, X. M. An and W. R. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- E. Collini, C. Y. Wong, K. E. Wilk, P. M. G. Curmi, P. Brumer and G. D. Scholes, Nature, 2010, 463, 644–647 CrossRef CAS PubMed.
- X. P. Li, O. Bjorkman, C. Shih, A. R. Grossman, M. Rosenquist, S. Jansson and K. K. Niyogi, Nature, 2000, 403, 391–395 CrossRef CAS PubMed.
- V. S. Y. Lin, S. G. Dimagno and M. J. Therien, Science, 1994, 264, 1105–1111 CAS.
- R. Takahashi and Y. Kobuke, J. Am. Chem. Soc., 2003, 125, 2372–2373 CrossRef CAS PubMed.
- C. L. Ho and W. Y. Wong, Coord. Chem. Rev., 2013, 257, 1614–1649 CrossRef CAS PubMed.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. C. Du, R. Katoh, A. Furube and D. L. Officer, J. Am. Chem. Soc., 2009, 131, 15621–15623 CrossRef CAS PubMed.
- C. Y. Chen, S. J. Wu, C. G. Wu, J. G. Chen and K. C. Ho, Angew. Chem., Int. Ed., 2006, 45, 5822–5825 CrossRef CAS PubMed.
- Z. S. Wang, Y. Cui, K. Hara, Y. Dan-Oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- R. Menzel, D. Ogermann, S. Kupfer, D. Weiss, H. Goerls, K. Kleinermanns, L. Gonzalez and R. Beckert, Dyes Pigm., 2012, 94, 512–524 CrossRef CAS PubMed.
- J. Schaefer, R. Menzel, D. Weiss, B. Dietzek, R. Beckert and J. Popp, J. Lumin., 2011, 131, 1149–1153 CrossRef CAS PubMed.
- S. Rau, D. Walther and J. G. Vos, Dalton Trans., 2007, 915–919 RSC.
- M. Graetzel, Energy resources through photochemistry and catalysis, Elsevier, Munich, Germany, 1983 Search PubMed.
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. Gonzalez, M. Schmitt, S. Rau and J. Popp, Angew. Chem., Int. Ed., 2010, 49, 3981–3984 CrossRef CAS PubMed.
- K. Okeyoshi and R. Yoshida, Soft Matter, 2009, 5, 4118–4123 RSC.
- V. Sundstroem, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- A. Schubert, A. Stenstam, W. J. D. Beenken, J. L. Herek, R. Cogdell, T. Pullerits and V. Sundstroem, Biophys. J., 2004, 86, 2363–2373 CrossRef CAS.
- T. Foerster, Ann. Phys., 1948, 2, 55–75 CrossRef.
- I. G. Scheblykin, A. Yartsev, T. Pullerits, V. Gulbinas and V. Sundstrom, J. Phys. Chem. B, 2007, 111, 6303–6321 CrossRef CAS PubMed.
- K. G. Jespersen, W. J. D. Beenken, Y. Zaushitsyn, A. Yartsev, M. Andersson, T. Pullerits and V. Sundstrom, J. Chem. Phys., 2004, 121, 12613–12617 CrossRef CAS PubMed.
- M. M. L. Grage, Y. Zaushitsyn, A. Yartsev, M. Chachisvilis, V. Sundstrom and T. Pullerits, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205207 CrossRef.
- J. Schaefer, A. Breul, M. D. Hager, U. S. Schubert and B. Dietzek, ChemPhysChem, 2013, 14, 170–178 CrossRef CAS PubMed.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- A. M. Breul, J. Schaefer, E. Altuntas, M. D. Hager, A. Winter, B. Dietzek, J. Popp and U. S. Schubert, J. Inorg. Organomet. Polym. Mater., 2013, 23, 74–80 CrossRef CAS.
- A. M. Breul, J. Schaefer, G. M. Pavlov, A. Teichler, S. Hoeppener, C. Weber, J. Nowotny, L. Blankenburg, J. Popp, M. D. Hager, B. Dietzek and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3192–3205 CrossRef CAS.
- A. M. Breul, J. Schaefer, C. Friebe, F. Schluetter, R. M. Paulus, G. Festag, M. D. Hager, A. Winter, B. Dietzek, J. Popp and U. S. Schubert, Macromol. Chem. Phys., 2012, 213, 808–819 CrossRef CAS.
- A. M. Breul, C. Pietsch, R. Menzel, J. Schaefer, A. Teichler, M. D. Hager, J. Popp, B. Dietzek, R. Beckert and U. S. Schubert, Eur. Polym. J., 2012, 48, 1339–1347 CrossRef CAS PubMed.
- R. Menzel, A. Breul, C. Pietsch, J. Schaefer, C. Friebe, E. Taeuscher, D. Weiss, B. Dietzek, J. Popp, R. Beckert and U. S. Schubert, Macromol. Chem. Phys., 2011, 212, 840–848 CrossRef CAS.
- C. H. Li, Y. X. Zhang, J. M. Hu, J. J. Cheng and S. Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS PubMed.
- S. Y. Liu, J. Yin, C. H. Li and D. Wang, J. Phys. Chem. B, 2010, 114, 12213–12220 CrossRef PubMed.
- S. Y. Liu and J. M. Hu, Macromolecules, 2010, 43, 8315–8330 CrossRef.
- X. Schultze, J. Serin, A. Adronov and J. M. J. Frechet, Chem. Commun., 2001, 1160–1161 RSC.
- J. M. Serin, D. W. Brousmiche and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124, 11848–11849 CrossRef CAS PubMed.
- J. Serin, X. Schultze, A. Adronov and J. M. J. Frechet, Macromolecules, 2002, 35, 5396–5404 CrossRef CAS.
- M. H. V. Huynh, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2005, 249, 457–483 CrossRef CAS PubMed.
- B. Happ, J. Schaefer, R. Menzel, M. D. Hager, A. Winter, J. Popp, R. Beckert, B. Dietzek and U. S. Schubert, Macromolecules, 2011, 44, 6277–6287 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed.
- W. Y. Wong, X. Z. Wang, Z. He, K. K. Chan, A. B. Djurisic, K. Y. Cheung, C. T. Yip, A. M. C. Ng, Y. Y. Xi, C. S. K. Mak and W. K. Chan, J. Am. Chem. Soc., 2007, 129, 14372–14380 CrossRef CAS PubMed.
- R. Menzel, E. Taeuscher, D. Weiss, R. Beckert and H. Goerls, Z. Anorg. Allg. Chem., 2010, 636, 1380–1385 CrossRef CAS.
- W. Y. Wong, K. H. Choi, G. L. Lu and Z. Y. Lin, Organometallics, 2002, 21, 4475–4481 CrossRef CAS.
- A. Vollrath, D. Pretzel, C. Pietsch, I. Perevyazko, S. Schubert, G. M. Pavlov and U. S. Schubert, Macromol. Rapid Commun., 2013, 33, 1791–1797 CrossRef PubMed.
- A. Vollrath, D. Pretzel, C. Pietsch, I. Perevyazko, R. Menzel, S. Schubert, G. M. Pavlov, D. Weiss, R. Beckert and U. S. Schubert, Macromol. Rapid Commun., 2013, 34, 280 CrossRef CAS.
- J. W. Gallaway and S. A. C. Barton, J. Am. Chem. Soc., 2008, 130, 8527–8536 CrossRef CAS PubMed.
- B. Happ, C. Friebe, A. Winter, M. D. Hager, R. Hoogenboom and U. S. Schubert, Chem. – Asian J., 2009, 4, 154–163 CrossRef CAS PubMed.
- M. W. M. Fijten, R. M. Paulus and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3831–3839 CrossRef CAS.
- J. P. S. Farinha, P. Relogio, M. T. Charreyre, T. J. V. Prazeres and J. M. G. Martinho, Macromolecules, 2007, 40, 4680–4690 CrossRef CAS.
- A. D. Kitchin, S. Velate, M. Chen, K. P. Ghiggino, T. A. Smith and R. P. Steer, Photochem. Photobiol. Sci., 2007, 6, 853–856 CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- C. Weber, M. Wagner, D. Baykal, S. Hoeppener, R. M. Paulus, G. Festag, E. Altuntas, F. H. Schacher and U. S. Schubert, Macromolecules, 2013, 5107–5116 CrossRef CAS.
- Y. Kwak, A. Goto and T. Fukuda, Macromolecules, 2004, 37, 1219–1225 CrossRef CAS.
- S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundstroem, M. Karnahl, M. Schwalbe, B. Schaefer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15, 7678–7688 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis (including relevant spectra) of the monomers PR and POs; 1H NMR spectra and 2D SEC plots for all polymers; hydrogen production test of POs. See DOI: 10.1039/c3py00915g |
| This journal is © The Royal Society of Chemistry 2014 |