 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical specificity in REDOX-responsive materials: the diverse effects of different Reactive Oxygen Species (ROS) on polysulfide nanoparticles†
Damien
Jeanmaire
a,
Jureerat
Laliturai‡
a,
Abdulaziz
Almalik§
a,
Paolo
Carampin
a,
Richard d'Arcy
b,
Enrique
Lallana
b,
Robert
Evans¶
c,
Richard E. P.
Winpenny
d and
Nicola
Tirelli
*bc
aSchool of Pharmacy and Pharmaceutical Sciences, University of Manchester, Oxford Road, Manchester, M13 9PT, UK
bSchool of Medicine, Institute of Inflammation and Repair, University of Manchester, Oxford Road, Manchester, M13 9PT, UK. E-mail: nicola.tirelli@manchester.ac.uk; Fax: +44 (0)161 275 23 96; Tel: +44 (0)161 275 24 80
cSchool of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dSchool of Chemistry, University of Manchester, Brunswick Street, Manchester, M13 9PL, UK
First published on 19th November 2013
Abstract
REDOX responsive (nano)materials typically exhibit chemical changes in response to the presence and concentration of oxidants/reductants. Due to the complexity of biological environments, it is critical to ascertain whether the chemical response may depend on the chemical details of the stimulus, in addition to its REDOX potential, and whether chemically different responses can determine a different overall performance of the material. Here, we have used oxidation-sensitive materials, although these considerations can be extended also to reducible ones. In particular, we have used poly(propylene sulfide) (PPS) nanoparticles coated with a PEGylated emulsifier (Pluronic F127); inter alia, we here present also an improved preparative method. The nanoparticles were exposed to two Reactive Oxygen Species (ROS) typically encountered in inflammatory reactions, hydrogen peroxide (H2O2) and hypochlorite (ClO−); their response was evaluated with a variety of techniques, including diffusion NMR spectroscopy that allowed to separately characterize the chemically different colloidal species produced. The two oxidants triggered a different chemical response: H2O2 converted sulfides to sulfoxides, while ClO− partially oxidized them further to sulfones. The different chemistry correlated to a different material response: H2O2 increased the polarity of the nanoparticles, causing them to swell in water and to release the surface PEGylated emulsifier; the uncoated oxidized particles still exhibited very low toxicity. On the contrary, ClO− rapidly converted the nanoparticles into water-soluble, depolymerized fragments with a significantly higher toxicity. The take-home message is that it is more correct to discuss ‘smart’ materials in terms of an environmentally specific response to (REDOX) stimuli. Far from being a problem, this could open the way to more sophisticated and precisely targeted applications.
Introduction
Variations in REDOX potential can often be associated to specific biological environments and sometimes to pathological conditions,1,2 thus (nano)materials responding to REDOX-active species can be used to perform biological targeting. We are specifically interested in polymeric nanostructures responsive to oxidizing conditions, which are often a signature of inflammatory reactions.3,4 A complete overview of this field is out of the scope of the present paper, but is provided by a number of very recent reviews.5–7 In order to simplify a very complex chemical landscape, in a first approximation we can restrict the field of the biologically occurring, inflammation-related oxidants to only a few of the most common Reactive Oxygen Species (ROS): superoxide anion (O2˙−), hydrogen peroxide (H2O2) and hypochlorite (ClO−).8Our oxidation-sensitive units are hydrophobic organic polysulfides, in particular poly(propylene sulfide) (PPS), which we have previously produced in the form of PEG-containing block-copolymers to yield e.g. micelles9 or vesicles,10 or as PEGylated and cross-linked polymeric nanoparticles.11 Upon exposure to ROS, the sulfide groups can be converted to more polar and thus also more hydrophilic groups (sulfoxides, sulfones). This increase in polarity, leading to water ingress and swelling/solubilization, allows for responsive actions such as the ROS-stimulated release of encapsulated payloads.12,13 However, as recently seen on linear polymers,14 the details of polysulfide oxidation are difficult to explain simply on the basis of a scale of standard reduction potentials. Using water as a final product and a neutral pH, the order of reactivity would be predicted to be O2˙− >H2O2> ClO− (O2˙−–H2O2: 0.94 V;15,16 H2O2 – H2O: 1.32 V,15 1.77 V;17 ClO− – Cl−/H2O 1.08 V (ref. 18)); on the contrary, (a) polysulfides are largely unresponsive to superoxide,19 (b) oxidation leads to sulfur(IV) (sulfoxides) with H2O2,14 while (c) hypochlorite produces both sulfoxide and sulfur(VI) (sulfone)-containing structures which cause the depolymerisation of the polysulfides.14 Therefore factors other than the pure REDOX potential are probably critical in determining the responsiveness of these materials to oxidizing condition, e.g. the solubility of the oxidant in the polysulfide domain.
Here, we have examined the effects of H2O2- and hypochlorite-mediated oxidation on cross-linked polysulfide nanoparticles, a class of particles recently used also for vaccine delivery purposes,20–22 predominantly focusing on the characterization of the structural effects arising from their exposure to the two ROS.
PPS nanoparticles are prepared by combining the emulsion anionic ring-opening polymerization of episulfides23 and a cross-linking based on the reactivity of the PPS terminal thiolate groups.11 For a detailed review of preparative methods for such polysulfide structures, the reader can refer to a specific review,24 while here we rapidly summarize this template preparative method: (a) propylene sulfide (PS) is dispersed in water by the use of a PEGylated emulsifier (generally Pluronic F127) to create droplets sized from tens to hundreds of nm (modulated by the emulsifier-to-monomer ratio); (b) PS polymerization and cross-linking convert the droplets into stable hydrophobic nanoparticles, which are coated by physically but irreversibly entrapped Pluronic (Scheme 1, top left),25 where its poly(propylene glycol) (PPG) middle block remains entangled in the PPS network. The template character of the method allows to precisely control the size of the nanoparticles using different monomer/emulsifier ratios (Scheme 1, top right).
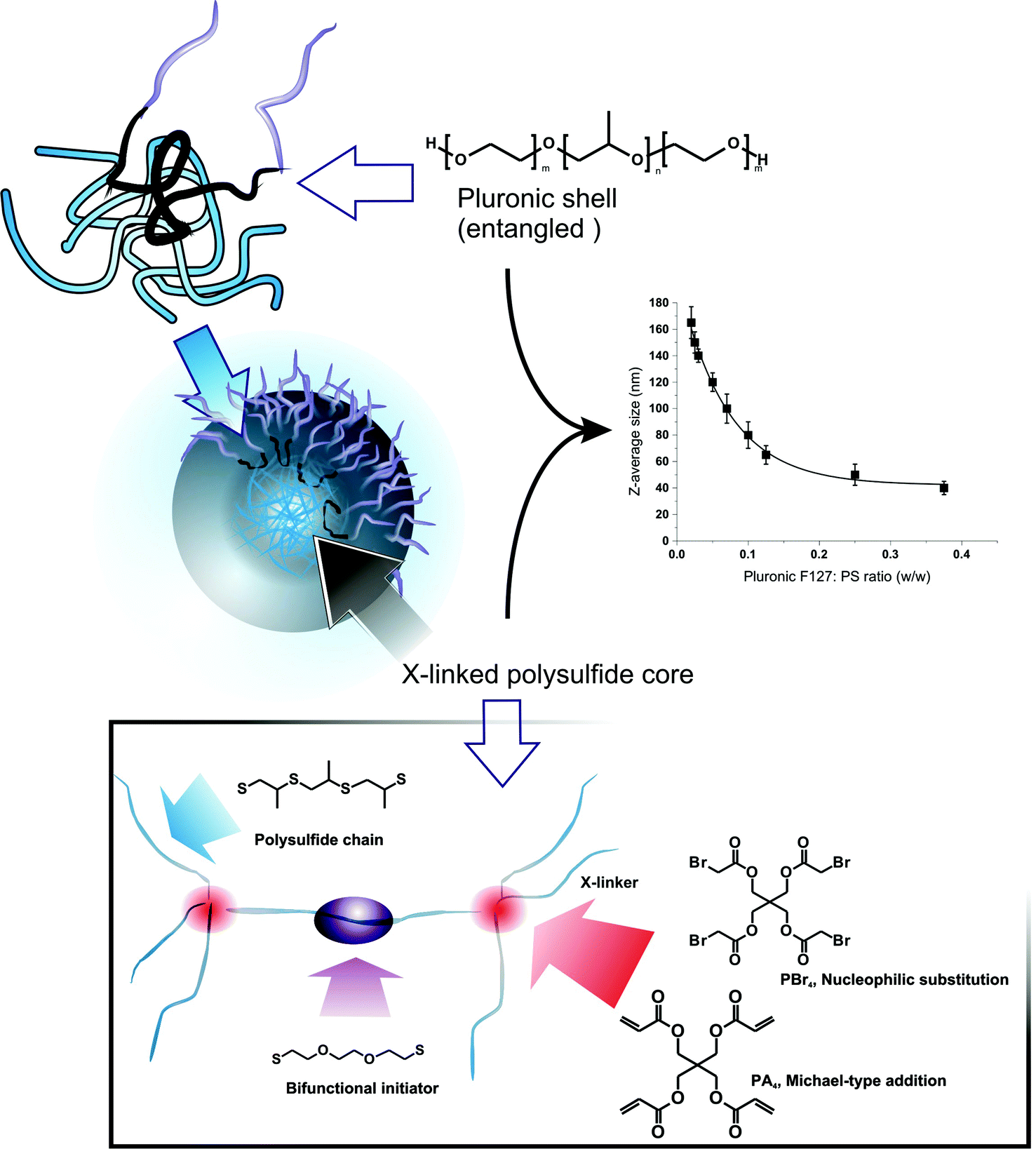 | ||
| Scheme 1 Polysulfide nanoparticles can be prepared via emulsification of PS (Pluronic F127 as PEGylated emulsifier) followed by its polymerization using a bifunctional initiator. The resulting bifunctional PPS chains are then reacted with tetrafunctional cross-linkers: PBr4 and PA4. The hydrophobic PPG block is retained in the network, stabilizing the Pluronic surface layer and providing a permanent PEGylated surface; the dimensions of the nanoparticles are templated on those of the emulsion droplets, which depend on the Pluronic/PS ratio (inset graphic; PPS of overall DP = 50, PA4 as a cross-linker (n = 3)). | ||
In previous studies, we have employed tetrafunctional initiators (in form of thioacetates) to yield tetra-armed star polymers that were eventually cross-linked with difunctional end-cappers, e.g. divinyl sulfone or a diacrylate. Thioacetates allow to minimize the presence of disulfides, which act as chain transfer agents in the episulfide polymerization and compromise the control over molecular weight and end groups.26 This procedure, however, is rather cumbersome, since it requires the synthesis and purification of a protected initiator and also an additional deprotection step prior to polymerization.
Here, we have simplified the nanoparticle preparation, adopting a specular synthetic approach based on a difunctional initiator and tetrafunctional end-cappers. As initiator, we have employed 2,2′-(ethylenedioxy)diethanethiol, which has a low propensity of intramolecular disulfide formation, due both to the low local density of thiol groups (lower than e.g. tetrathiols) and the impossibility to form stable 5- or 6-membered rings, as for 1,3-propanedithiol.23 As tetrafunctional end-cappers, we have evaluated pentaerythritol tetraacrylate (PA4) and tetrabromoacetate (PBr4), which respectively react with PPS terminal thiols through Michael-type addition and nucleophilic substitution (Scheme 1, bottom). We have first applied this procedure to the synthesis of linear polymers (using monofunctional end-cappers), in order to ensure that the polymerization still preserves a controlled character, i.e. the polymers have an acceptably narrow molecular weight distribution. This point is essential to ensure that the final networks have a homogeneous cross-link density (= polymer chains with controlled molecular weight between cross-links).
Experimental section
Materials
All chemicals were used as received from suppliers unless otherwise stated. Propylene sulfide (PS), pentaerythritol tetraacrylate (PA4), Nile Red, triethylamine (TEA), Pluronic F127, sodium hypochlorite (NaOCl) in aqueous solution (available chlorine 10–15%), pentaerythritol, ethyl 2-bromoacetate, 2,2′-(ethylenedioxy)diethanethiol and fungizone were purchased from Sigma-Aldrich (Gillingham, UK). Dichloromethane, acetonitrile and 30% wt. H2O2 in water were purchased from BDH (Leicestershire, UK). Bromoacetyl bromide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were purchased from Fluka (Gillingham, UK). Phosphate buffered saline (PBS) Dulbecco A tablets were purchased from Oxoid (Hampshire, UK). Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum (FBS), L-glutamine 200 mM (×100) and penicillin-streptomycin were purchased from Invitrogen (Paisley, UK). CellTiter 96® AQueous One Solution Cell Proliferation Assay was purchased from Promega (Southampton, UK). All other chemicals were of analytical grade and used without further purification.Physico-chemical characterization
1H NMR spectra were recorded on 1% wt. solutions in deuterated chloroform using a 300 MHz Bruker spectrometer (Bruker UK Limited, UK). FT-IR spectra were recorded in ATR mode (Golden Gate) on a Tensor 27 Bruker spectrometer (Bruker UK Limited, UK) equipped with a 3000 Series TM High Stability Temperature Controller with RS232 Control (Specac, UK). The spectra of nanoparticles upon oxidation with H2O2 or NaOCl were recorded by allowing a drop of the oxidation mixture (typically 20–50 μL) to dry at 50 °C for ca. 3 min on the bottom plate of the Golden Gate ATR accessory. Turbidity measurements were performed by monitoring the optical density of nanoparticle dispersions at 600 nm using a UV/Vis spectrophotometer Perkin Elmer Lambda 25 (Perkin Elmer, UK). Nile Red fluorescence was measured through a Perkin Elmer LS 55 (Perkin Elmer, UK) at a temperature of 25 °C (excitation 540 nm, emission at 620 nm). Gel permeation chromatography (GPC): molecular weight and molecular weight distribution of polymers were determined using a Polymer Laboratories PL-GPC50 integrated GPC (Polymer Laboratories, UK) comprising a PLgel 5 μm Guard and two PolyPore 5 μm columns operating online at 30 °C. THF was used as an eluent at a flow rate of 1.0 mL min−1. A series of near-mono-dispersed linear polystyrene standards (Fluka; Gillingham, UK) was used for calibration with a refractive index detector for the analysis of the polymers. Dynamic light scattering (DLS). Size distributions and scattering intensity of nanoparticles were measured with the help of a Zetasizer Nano ZS Instrument (Model ZEN3500, Malvern Instruments Ltd, UK). All the samples were analyzed at an angle of 173° and a temperature of 25 °C. Pulsed field gradient NMR spectroscopy (PFG NMR, also referred to as diffusion-ordered NMR spectroscopy (DOSY).27 Diffusion NMR data were acquired on a Bruker Avance II 400 MHz spectrometer, without spinning, using a 5 mm insert probe equipped with a gradient coil producing a maximum gradient strength of 1190 G cm−1. All experiments were carried out with active temperature regulation at 25.0 °C. 1H spectra of the samples were acquired with a simple pulse-acquire sequence, recording 32 transients. DOSY data were acquired using a One-Shot28 sequence. Transmission electron microscopy (TEM). Nanoparticle dispersions were pipetted on carbon/formvar coated copper 100 mesh grids and left for 2–3 min; the grids were then washed in distilled water for 3 times, stained with 1% uranyl acetate and air dried. Samples were observed using a Technai 12 electron microscope at 100 kV.Preparative operations
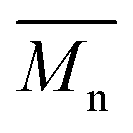 ≈ 12
≈ 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1) was dissolved in 75 mL of degassed MilliQ water (0.2% wt.) in a 250 mL three-neck round bottom flask under argon. The system was continuously stirred at 1000 rpm and purged with argon for 1 h. PS (3.17 mL, 3.0 g, 40.4 mmol) was mixed with an appropriate amount of 2,2′-(ethylenedioxy)diethanethiol (0.246, 0.148, 0.105, 0.082 g, respectively, providing monomer to initiator ratios corresponding to overall DP = 30, 50, 70 and 90, and DP per arm = 15, 25, 35 and 45), the mixture was then added to the Pluronic F127 solution and stirred for another 10 min, followed by the addition of 2.2 equivalents compared to initiator of DBU (0.451, 0.271, 0.194, 0.151 g respectively). The reaction mixture was stirred for 2 h, then the pH was lowered to 9.5 by the addition of acetic acid, and 1.5 equivalents of ethyl 2-bromoacetate (0.676, 0.405, 0.290 0.225 g respectively) were finally added. After 1 minute the pH was brought to 7.4 by adding 5 mL of PBS (50 mM, pH 7.4), in order to minimize the hydrolysis of the end-capper. The mixture was additionally stirred for 1 h before the addition of 20 mL of dichloromethane; the organic phase was decanted and washed with brine three times. The organic layer was dried over Na2SO4 and evaporated under vacuum. The resulting oily material was further washed with methanol (3 × 5 mL), separated by decantation after centrifugation, and dried under high vacuum for 24 h at 40 °C.
600 g mol−1) was dissolved in 75 mL of degassed MilliQ water (0.2% wt.) in a 250 mL three-neck round bottom flask under argon. The system was continuously stirred at 1000 rpm and purged with argon for 1 h. PS (3.17 mL, 3.0 g, 40.4 mmol) was mixed with an appropriate amount of 2,2′-(ethylenedioxy)diethanethiol (0.246, 0.148, 0.105, 0.082 g, respectively, providing monomer to initiator ratios corresponding to overall DP = 30, 50, 70 and 90, and DP per arm = 15, 25, 35 and 45), the mixture was then added to the Pluronic F127 solution and stirred for another 10 min, followed by the addition of 2.2 equivalents compared to initiator of DBU (0.451, 0.271, 0.194, 0.151 g respectively). The reaction mixture was stirred for 2 h, then the pH was lowered to 9.5 by the addition of acetic acid, and 1.5 equivalents of ethyl 2-bromoacetate (0.676, 0.405, 0.290 0.225 g respectively) were finally added. After 1 minute the pH was brought to 7.4 by adding 5 mL of PBS (50 mM, pH 7.4), in order to minimize the hydrolysis of the end-capper. The mixture was additionally stirred for 1 h before the addition of 20 mL of dichloromethane; the organic phase was decanted and washed with brine three times. The organic layer was dried over Na2SO4 and evaporated under vacuum. The resulting oily material was further washed with methanol (3 × 5 mL), separated by decantation after centrifugation, and dried under high vacuum for 24 h at 40 °C.
1H NMR (CDCl3); δ = 1.26–1.30 (t, 6H, CH3CH2–OOC–), 1.35–1.45 (d, CH3 in PPS chain), 2.55–2.75 (m, PPS chain: 1 diastereotopic CH2), 2.71–2.78 (t, 4H, –SCH2CH2OCH2CH2OCH2CH2S–), 2.85–3.05 (m, PPS chain: CH and 1 diastereotopic CH2), 3.16–3.25 (d, 2H, –S–CH2–COO–: 1 diastereotopic CH2), 3.25–3.37 (d, 2H, –S–CH2–COO–: 1 diastereotopic CH2), 3.61–3.68 (m, 8H, –SCH2CH2–OCH2CH2OCH2CH2S–), 4.15–4.26 ppm (q, 4H, CH3CH2–O(O)C–).
ATR FT-IR (thin film); 2959 (νas CH3), 2917 (νas CH2), 2865 (νs CH3 and νs CH2), 1730 (ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester), 1448 (δs CH2), 1372, 1263, 1221, 1172, 1103 (νas C–O–C), 943, 852 cm−1 (νs C–O–C).
O ester), 1448 (δs CH2), 1372, 1263, 1221, 1172, 1103 (νas C–O–C), 943, 852 cm−1 (νs C–O–C).
Representative 1H NMR and IR spectra are provided in ESI, Fig. 1SI and 2SI.†
:1) and a second one in pure dichloromethane. Yield: 75% wt. (3.39 g, 5.5 mmol; 100% esterification of OH groups by 1H NMR).1H NMR (CDCl3); δ = 3.86 (s, 8H, –O–COCH2–Br), 4.28 ppm (s, 8H, –C–CH2–O–CO–). Pentaerythritol tetraacrylate (PA4). PA4 is commercially available (Sigma Aldrich) and was purified by flash column chromatography on silica gel (230–400 mesh) with dichloromethane/ethyl acetate (10:1) as a mobile phase. Butylated hydroxytoluene (BHT; 3,5-di-tert-butyl-4-hydroxytoluene) was then added (800 ppm) and the resulting waxy solid was stored under Ar at −20 °C. 1H NMR (CDCl3); δ = 4.27 (s, 8H, –C–CH2–O–CO–), 5.86 (dd, 4H, –O–COCHCH2: cis-H), 6.09 (dd, 4H, –O–COCHCH2), 6.39 ppm (dd, 4H, –O–COCHCH2: trans-H).
O), 1246 (νas C–O–C, ester), 1097 (νas C–O–C in Pluronic F-127), 800–600 cm−1 (ν C–S in PPS).
:2500 and 1:25000 Nile Red/thioether molar ratio) and adding it to the reaction mixture. The fluorescence emission of Nile Red in different possible environments was monitored at 620 nm (excitation in the window 540 ± 25 nm, emission in the window 620 ± 40 nm).
(A). DLS and fluorescence analysis. Experiments were carried out with PA4 or PBr4 nanoparticles obtained using a 0.02 Pluronic/PS weight ratio (overall DP = targeted molecular weight between cross-links = 50, corresponding to DP per arm = 25) prepared as described above and with a Z-average size of 160 nm. 1 mL of nanoparticle dispersion in deionized water (concentration ≈ 3.5 mg mL−1 = 0.35% wt.) was added to 10 mL of an aqueous solution of H2O2 or NaOCl (of an adequate concentration) and adjusted to pH = 7.4 by the addition of concentrated NaOH (aq), to obtain a final concentration of ≈ 0.32 mg mL−1 of nanoparticles (= 4.3 mM in thioethers); the reaction was allowed to proceed at 37 °C monitoring the time-dependent changes in turbidity (optical density at a wavelength of 600 nm), particle size (DLS) and Nile Red fluorescence (at 620 nm). Please note that Nile Red's absorption maximum shifts considerably from hydrophobic to polar environments (e.g. from below 500 nm in alkanes to about 530 in alcohols), and the same applies to its emission maximum; therefore, we have chosen spectral parameters (excitation at 540 nm, emission at 620 nm) that provide the highest readings in polar environments with low water content, and negligible fluorescence both in apolar polysulfides and in water-swollen environments.
(B). NMR analysis. Experiments were carried out on PA4 nanoparticles obtained using 0.02 and 0.2 Pluronic/PS weight ratio (overall DP = targeted molecular weight between cross-links = 50, corresponding to DP per arm = 25) prepared as described above and with a Z-average size respectively of 160 and 50 nm. Due to the relatively low sensitivity of the technique when working with cross-linked polymers, the nanoparticle concentrations employed for these experiments were considerably larger than for DLS, turbidity or fluorescence experiments. Diffusion NMR. For diffusion experiments, 200 μL of 10% or 30% wt. H2O2 or 268 mM NaOCl were added to 200 μL of a 20 mg mL−1 nanoparticle dispersion in deionized water to obtain a final concentration of 10 mg mL−1 of nanoparticles (= 134 mM in thioethers). The reaction was allowed to proceed at room temperature for six hours. 100 μL of D2O were then added to the dispersion prior to NMR analysis. For T2 relaxivity experiments, similar conditions were used (200 μL of 10% wt. H2O2 to yield a final 5% wt. H2O2). The experiments were conducted acquiring 256 transients, and using a diffusion delay Δ of 0.01 s and 0.02 s, respectively, and a total diffusion encoding time δ of 0.001 s. 20 gradient levels were acquired, in equal steps of gradient squared from 119 to 890 G cm−1. The maximum gradient was chosen so that ∼75% of the polymer signal (methyl group at δ = 1.47 ppm for PPS, ethylene group at δ = 3.77 ppm and methyl group at δ = 1.19 ppm for Pluronic F127) was attenuated. Due to the broad peaks present in the nanoparticle spectra, neither resolution enhancement nor reference deconvolution was used. T2 spin–spin relaxation measurements. T2 measurements were performed on nanoparticle dispersions in 90% H2O and 10% D2O using a 400 MHz Bruker spectrometer (Bruker UK Limited, UK). The pulse sequence used was the Carr-Purcell-Meiboom-Gill (CPMG) sequence.
Results and discussion
Synthesis of model polymers and of nanoparticles
We have first confirmed that the simplified preparative method (dithiol + tetrafunctional end-capper vs. the previously used tetrathioacetate + difunctional end-capper) still offered a sufficient control over molecular weight to allow for homogeneous networks (→ homogeneous diffusion of oxidants): a series of linear polymers was prepared by this emulsion polymerization approach, and GPC, 1H NMR and FT-IR analysis showed a reasonably good agreement between experimental and theoretical degrees of polymerization (Fig. 1 and Table 1). A significant discrepancy between theoretical and spectroscopic values could be seen only at the lowest initiator/monomer ratio (DP = 90), likely due to incomplete polymerization because of the slower polymerization kinetics. The molecular weight dispersity of these model linear polymers is clearly larger than when the disulfide presence is carefully avoided (1.4–1.6 vs. ≈ 1.1 with protected initiators and in situ reducing agents); indeed a high MW shoulder can be easily spotted in the GPC traces (Fig. 1, left): this is typical of polymers with internal disulfides that are a clear visual marker of disulfide-mediated chain transfer activity.26 However, this effect is quantitatively small and we judged the new synthetic method appropriate for the preparation of reasonably controlled cross-linked nanoparticles.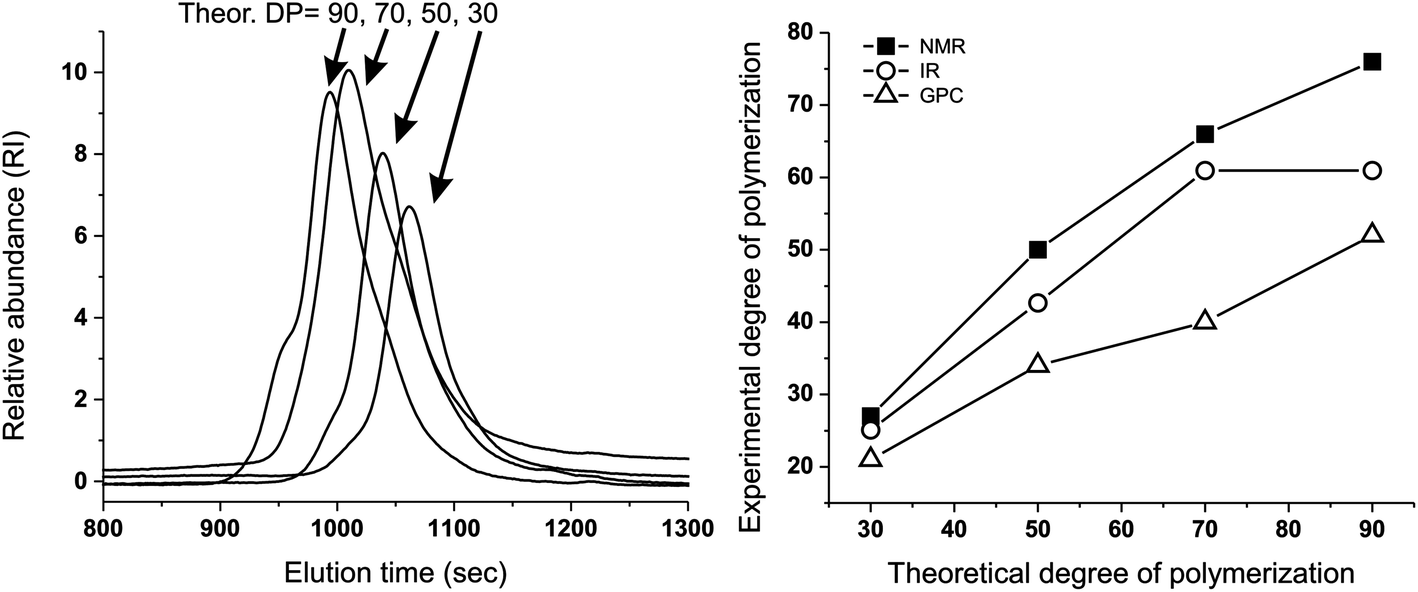 | ||
| Fig. 1 Left: GPC traces showing that the emulsion synthesis of linear PPS from the difunctional initiator 2,2′-(ethylenedioxy)diethanethiol produces polymers (with different DPs) with acceptable molecular weight distributions and relatively small amounts of disulfide impurities: the latter are recognizable because of the presence of a “dimer peak”, i.e. a higher molecular weight shoulder of the GPC trace.26 Right: Experimental vs. theoretical number average DP of linear, difunctional PPS via “unprotected” emulsion polymerization: although numerically different, the results of 1H NMR, FT-IR and GPC clearly showed the proportionality between the theoretical DP (PS/initiator molar ratio in the feed) and that of the actual polymers, which demonstrates the good control of PS polymerization initiated by 2,2′-(ethylenedioxy)diethanethiol, even without the use of thiol protecting groups (e.g. as thioacetate). For typical NMR and FT-IR spectra, see ESI, Fig. 1SI and 2SI,† respectively. Please realize that the discrepancy between GPC and spectroscopic results is probably to ascribe to the use of single detection in the GPC measurements. | ||
| Theoretical overall DP | 1H NMR | GPC | FT-IR | |||
|---|---|---|---|---|---|---|
| Overall DPa | End-cappingb (mol%) |
|
|
Overall DP | Relative DPd | |
|
a Calculated from the ratio between the integrals of protons of the PPS chain (CH3, 1.35–1.45 ppm) and of the initiator (–SCH2CH2–OCH2CH2OCH2CH2S–, 3.61–3.68 ppm). Average values over three different polymerization experiments. For a representative DP, see ESI, Fig. 1SI.
b Calculated from the ratio between the integrals of protons of the PPS chain (CH3, 1.35–1.45 ppm) and of the end-capper (OCH2CH3, δ = 4.15–4.26 ppm). Average values over three different polymerization experiments.
c Average values over three different polymerization experiments.
d Calculated using the integral of the peak at 1130 cm−1 (fingerprint region peak associated to the PPS chain; the area between 1080 and 1200 cm−1 was fitted with four Gaussian peaks to depurate the absorbance readings from the contribution of neighboring peaks) and that of the CO stretching peak at 1735 cm−1 (proportional to the amount of end-capper); these figures are supposedly related to the degree of polymerization assuming a constant end-capping yield. The data were normalized assuming a DP = 27 for PPS with a theoretical DP = 30 (i.e. using the DP calculated through 1H NMR for that sample). FT-IR spectra of the different polymers can be found in ESI, Fig. 2SI. Average values over three different polymerization experiments.
|
||||||
| 30 | 27 ± 1 | 89 | 1900 | 1.39 | 25 ± 1 | 27 ± 2 |
| 50 | 50 ± 1 | 84 | 2900 | 1.48 | 37 ± 3 | 45 ± 3 |
| 70 | 66 ± 1 | 80 | 3300 | 1.61 | 45 ± 2 | 67 ± 6 |
| 90 | 76 ± 4 | 86 | 4200 | 1.51 | 60 ± 3 | 69 ± 6 |
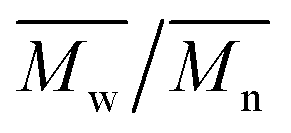
Nanoparticles were therefore prepared by synthesizing a difunctional PPS with overall DP = 50, and using two different tetrafunctional end-cappers, pentaerythritol tetraacrylate (PA4) and pentaerythritol tetrakis(2-bromoacetate) (PBr4) (Scheme 1), in order to investigate possible effects of different cross-linking mechanisms. Often PBr4 yielded slightly smaller nanoparticles but with a larger variability in size than PA4, and among the two we could not detect any statistically relevant difference. Importantly, PPS nanoparticles showed an excellent physical stability, e.g. no agglomeration upon storage (1 mg mL−1 in water; >3 months at room temperature) or freeze-drying (using sucrose as a cryoprotectant; see ESI, Fig. 3SI†).
Nanoparticle response to hydrogen peroxide
H2O2 caused nanoparticles to swell, independently on the nature of the cross-linker. The turbidity of the nanoparticle suspensions decreased (Fig. 2A) at a rate proportional to the oxidant concentration (Fig. 2B) and their size increased with identical kinetics (Fig. 2B, empty circles). It is important to note that all curves showed the sigmoidal shape typical of auto-accelerating phenomena: the poor solubility of H2O2 in the originally hydrophobic matrix is reflected by a lag phase (a “toe” region), followed by an accelerating phase due to the better solubility of the oxidant in the increasingly polar material.3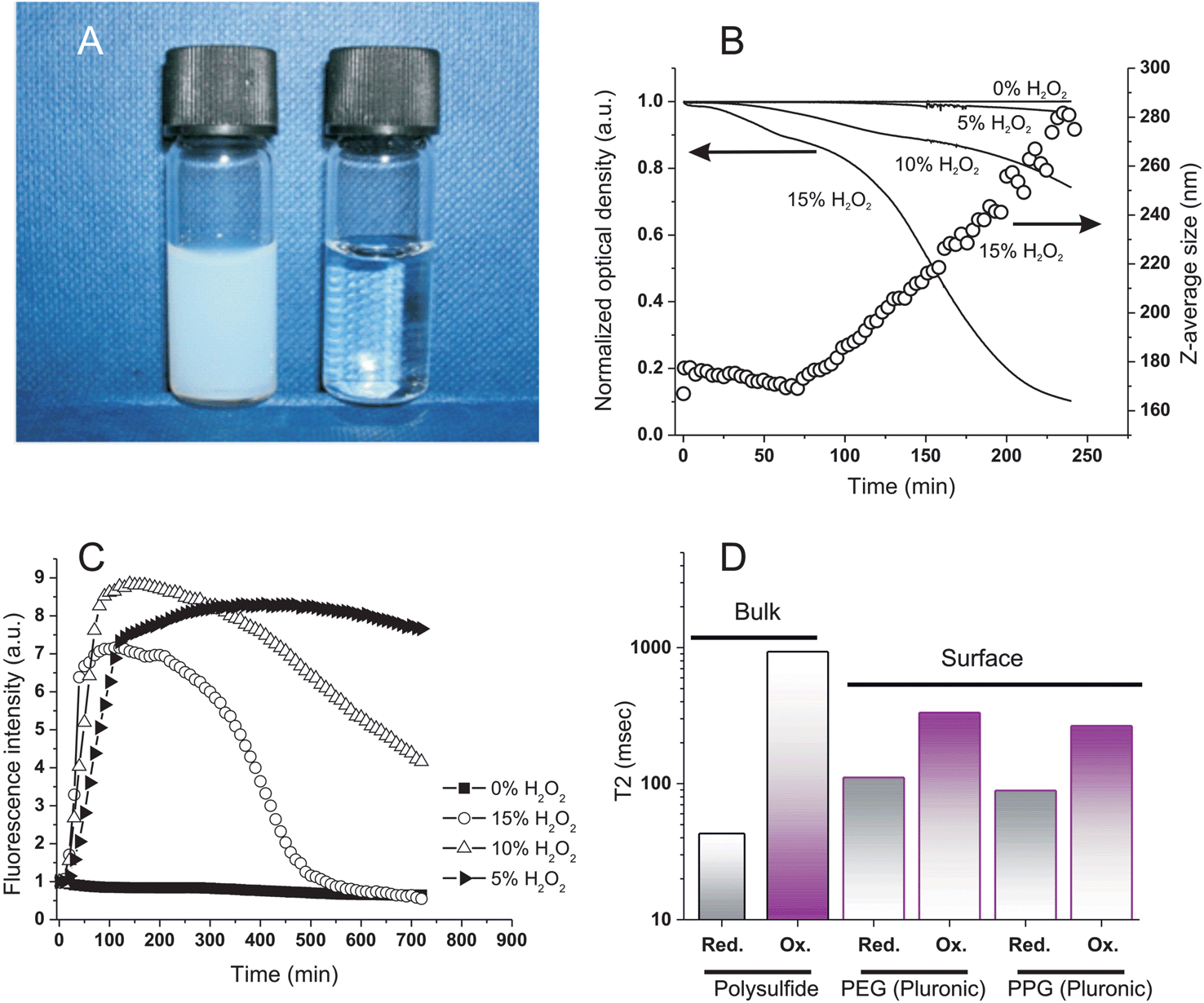 | ||
| Fig. 2 Oxidation of PA4 nanoparticles (Z-average size: 160 nm); 0.32 mg mL−1 in deionized water, 37 °C and pH = 7.4. A: Appearance of the nanoparticle dispersion before and after oxidation with 15% wt. H2O2. B: Optical density at 600 nm (solid lines) and Z-average size (open circles) of nanoparticle dispersion vs. H2O2 concentration and time. The nanoparticle response was marginally affected by the nature of the cross-linker (compare with Fig. 6SIA in ESI;† see also the similarities in Fig. 4SI and 5SI†), although PBr4 particles may undergo a more significant hydrolytical degradation during oxidation. C: Fluorescence emission intensity of Nile Red-loaded nanoparticles (excitation at 540 nm, emission at 620 nm; 1:2500 Nile Red/thioether molar ratio) vs. H2O2 concentration and time. The initial emission intensity is virtually zero due to the choice of the spectral parameters (see text). Similar results were obtained with two largely different Nile Red loadings, excluding therefore any influence of self-quenching effects (see ESI, Fig. 5SI†); PBr4 cross-linked particles appeared to show a marginally quicker first phase, but the effect was not statistically significant (Fig. 5SI†). D: T2 Relaxivity of polysulfide (bulk) and Pluronic (surface) resonances before and after oxidation with 5% wt. H2O2 at room temperature for 3 h; see Table 2SI in ESI† for numerical values. | ||
Infrared spectra showed that sulfoxide (SO) groups were the only major feature introduced during this oxidation, as already shown on soluble polymers14 (stretching vibration peak at 1030 cm−1; see ESI, Fig. 4SI†); they could be detected only after the initial “toe” region (= the time window with relatively little changes) seen in turbidity and Dynamic Light Scattering (DLS) measurements (Fig. 2B).
The polarity of the nanoparticle bulk was monitored via the fluorescence of Nile Red; taking advantage of the solvatochromic shifts of this dye, we have chosen an excitation wavelength (540 nm) that provides negligible emission (620 nm) in a very apolar environment and high fluorescence in moderately polar ones. This allowed to identify a major increase in the nanoparticle polarity (increase in emission) during the “toe” region, before significant swelling (water intake) occurred and caused fluorescence quenching (Fig. 2C); therefore, it would appear some minor oxidative processes to have taken place also in the initial phases of the exposure to H2O2, although not sufficient to produce significant water ingress.
The ingress of water was further confirmed by a dramatic increase of the network molecular mobility after oxidation, as witnessed by a >20-fold increase in the spin–spin relaxation time (T2) of the polysulfide chains (Fig. 2D; see also ESI, Table 1SI†); interestingly, also the Pluronic protons showed a significant (3-fold) increase in T2 values both in the hydrophilic (PEG) and in the hydrophobic (PPG) domains, which would suggest a mobilization also of these water-exposed groups.
To further investigate these transformations we have employed diffusion NMR spectroscopy. This technique has a precision comparable to DLS (Fig. 3), but is chemically selective; for example, it confirms the presence of Pluronic on the surface non-oxidized nanoparticles,25 since the diffusion coefficients associated to both Pluronic and polysulfide resonances are virtually identical, but much different from that of Pluronic in micellar dispersion or in solution.
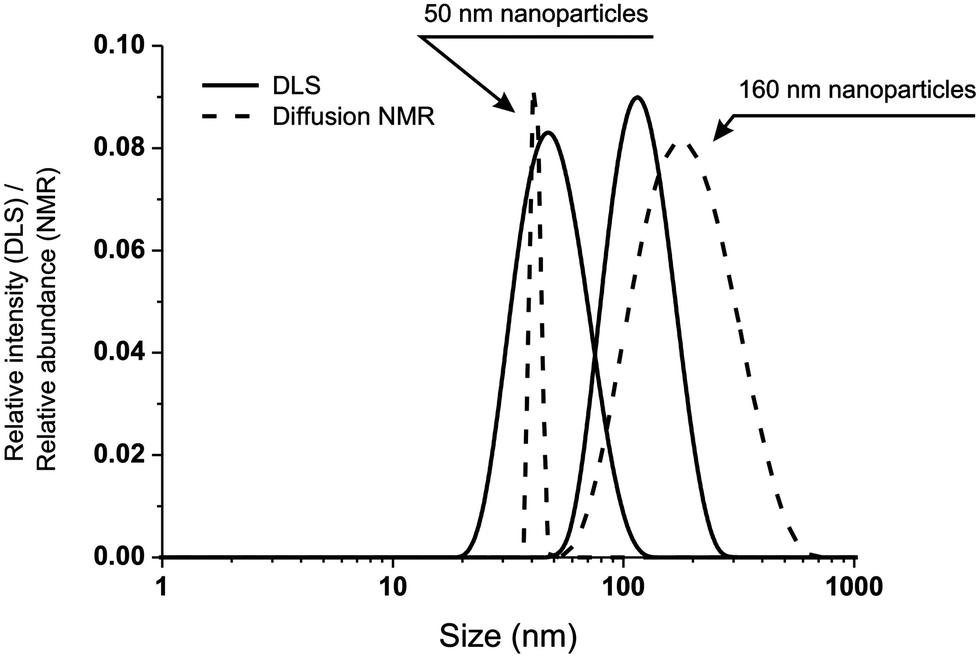 | ||
| Fig. 3 Comparison of size distributions obtained via DLS (solid lines) and diffusion NMR (dashed lines) for two differently sized nanoparticle preparations. Please note that the two techniques provide intensity (DLS) and number (NMR) distributions; considering this, the agreement between the two sets of measurements is remarkable. | ||
The exposure of 50 nm nanoparticles to 5% wt. H2O2 for 6 hours caused roughly 50% oxidation of the sulfide groups (see the broadening of the methyl group resonance highlighted by an arrow in Fig. 4A), which was accompanied by dramatic changes in DOSY spectra (Fig. 4B and C). Before oxidation all resonances presented the same diffusion coefficient, indicating the close association between Pluronic and polysulfides (Fig. 4C); after oxidation Pluronic displayed a much larger diffusion coefficient, which is easy to relate to 10–20 nm micelles (Fig. 4D, purple curve). The increase in mobility associated to the transition from a compressed surface-entrapped state to a micellar one explains why also the hydrophilic PEG blocks showed a decrease in T2 upon oxidation.
 | ||
| Fig. 4 (A and F) 1H NMR spectra of 160 nm (A) and 50 nm (F) PA4 nanoparticles (10 mg mL−1) recorded before and after 6 h exposure to 15% (A), or 5% wt. H2O2 (F). (B, C and G). DOSY spectra of pristine nanoparticles (C; 160 nm nanoparticles show a similar but more noisy pattern, due to the very low relative intensity of Pluronic resonances -results not shown-) and after oxidation (B and G). Please note the larger scale of diffusion coefficients in (G); the inset allows a better appreciation of the larger species (D < 1 × 10−10 m2 s−1). (D and H). Size distributions obtained by applying a CONTIN algorithm to DOSY data. For the pristine nanoparticles (black lines), the size distributions obtained using the polysulfide and the Pluronic signals overlap (only Pluronic signal showed). (E): Z-average size (DLS) of 160 nm and 50 nm nanoparticles (10 mg mL−1) during their oxidation with different H2O2 concentrations as a function of time. Please note that the oxidation of 160 nm nanoparticles with 5% wt. H2O2 proceeds very slowly in this time window (see Fig. 1B), while that of 50 nm nanoparticles with 15% wt. H2O2 is too rapid to be easily monitored. | ||
Further, the Pluronic loss from the nanoparticle surface is likely to cause aggregation in the yet not completely oxidized nanoparticles, which explains the presence of objects with a size too large (up to 1 μm) to be due only to swelling (Fig. 4D, red curve).
We have also analyzed the diffusion behavior of nanoparticles with different size, because their dimensions may influence the rapidity of the oxidation response: indeed, 50 nm nanoparticles treated with a lower amount of H2O2 increased in size (swelled) more rapidly than 160 nm nanoparticles with a larger excess of the oxidant (5% wt. H2O2, ≈ 10-fold stoichiometric excess vs. thioethers as opposed to 15% wt. H2O2, ≈ 30-fold stoichiometric excess, Fig. 4E). The accelerated response of the smaller colloids is likely due to a kinetic effect: their higher surface/volume ratio would allow a more rapid equilibration of the H2O2 concentration in the nanoparticle bulk. Despite this kinetic difference, larger and smaller nanoparticles behaved similarly from a structural point of view: for example, DOSY spectra (Fig. 4B and G) and size distributions (Fig. 4D and H) showed that also in this case the oxidation implied the loss of the Pluronic coating (always present in micellar form after oxidation) and that the nanoparticles increased in size. However it is noteworthy that the 160 nm nanoparticles produced also some high diffusion coefficient (ca. 2–20 nm sized) polysulf(ox)ide-containing compounds (<10% in mol of the dispersed material); due to the high degree of oxidation, they are probably produced by the hydrolysis of the cross-liker's ester groups, which are no longer protected by a hydrophobic environment.
Nanoparticle response to hypochlorite
Hypochlorite almost instantaneously decreased the turbidity of nanoparticle dispersions, and it is noticeable that this reached a minimum at the stoichiometric equivalence between sulfides and hypochlorite (Fig. 5A and B): at a 4.3 mM thioether concentration, the opacity was rapidly reduced to less than a half at 2.5 mM ClO− and down to zero at 10 mM ClO−. DLS analysis suggested this effect to be due to a reduction in particle number, since the scattering intensity decreased but the particle size remained substantially unaltered (Fig. 5A and B, empty circles); the reduction in particle number without significant changes in dimension was also confirmed by TEM (Fig. 5C). Also the ClO−-mediated oxidation may proceed through an autocatalytic mechanism, although much more rapid than for H2O2: this would explain the persistence of apparently pristine nanoparticles (= unaltered size and negligible Nile Red fluorescence) at intermediate oxidant concentrations. Diffusion NMR experiments supported this point, showing that at a 1:1 sulfide/hypochlorite molar ratio, only micellar Pluronic and oxidized low molecular weight compounds could be detected, without any larger colloidal object being present (Fig. 6). The nanoparticle solubilization is to ascribe to polymer fragmentation occurring at sulfone sites and producing fragments containing carbonyl, hydroxyl and sulfone groups (→ band at 1350 cm−1, see ESI, Fig. 6SIB†), as previously shown on linear polysulfides.14 Nile Red fluorescence at 620 nm remained very low at any time point for any ClO− concentration (data not shown), suggesting that the fluorophore did not experience environments with intermediate polarity (as opposed to with H2O2) for significant periods of time during the oxidation process.
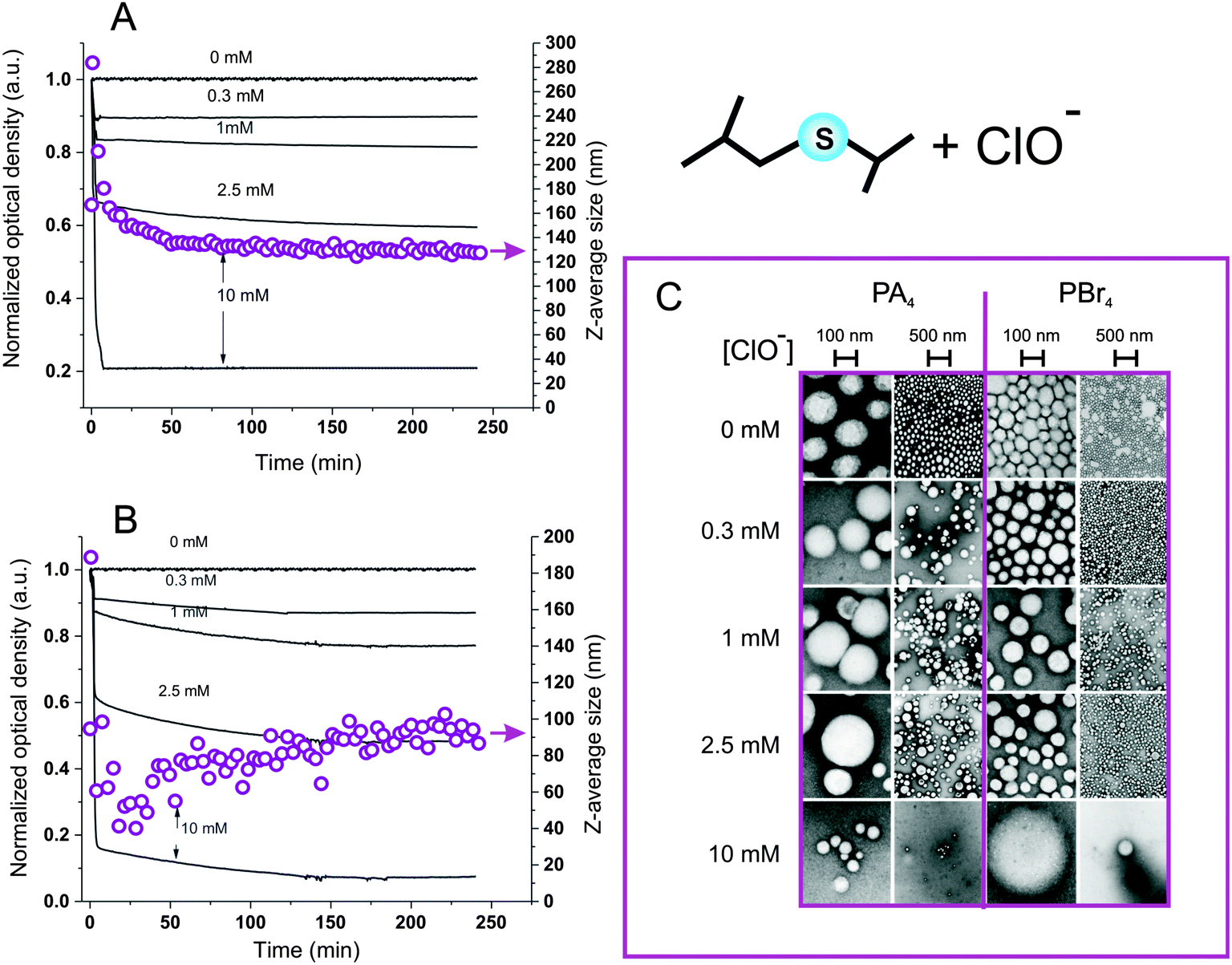 | ||
| Fig. 5 A) Optical density at 600 nm (solid lines) and Z-average size (open circles) of 0.32 mg mL−1 PA4-based nanoparticle dispersions vs. tune for different ClO− concentrations; please note a sudden and transient increase in the nanoparticle size at initial reaction times (<3 min), which is possibly related to the initial swelling of degrading particles. (B) As in graph A, but using PBr4-based nanoparticles. (C): TEM images of PA4 and PBr4 nanoparticles oxidized with different concentrations of NaOCl for 240 min (at 37 °C and pH = 7.4) and then purified via dialysis. Although in some cases a small increase in nanoparticle dimensions could be noticed, the most striking result of this analysis is the dramatic reduction in the number of nanoparticles when approaching the stoichiometric thioether/ClO− equivalence. | ||
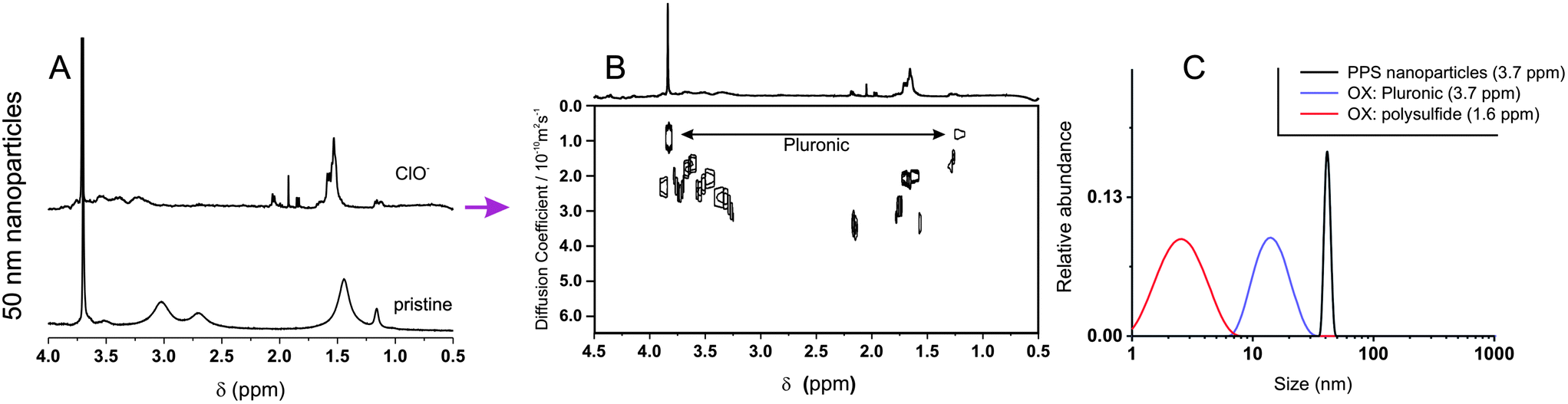 | ||
| Fig. 6 (A) 1H NMR spectra of 50 nm PA4 nanoparticles (10 mg mL−1) recorded before and after exposure to 135 mM NaOCl (stoichiometric equivalence to sulfides). (B). DOSY spectrum of the oxidized nanoparticles. (C). Size distributions obtained by applying a CONTIN algorithm to DOSY data. | ||
It is therefore apparent that the ROS-dependent chemical response was mirrored by a ROS-dependent evolution of the material morphology, as graphically summarized in Scheme 2.
 | ||
| Scheme 2 Summary of the main effects of hydrogen peroxide and hypochlorite on polysulfide nanoparticles. | ||
It is noticeable that, despite the known sensitivity of polyether chains to oxidizing conditions,29 both hydrogen peroxide and hypochlorite did not appear to dramatically affect Pluronic behavior: (1) if oxidized groups (e.g. carbonyls) were introduced in large amounts in Pluronic chains without cleaving them, their resonances should be recorded in correspondence of the Pluronic micelles' diffusion coefficient; however, the only intense signals that we could recognize in NMR are those of ethylene glycol units and propylene glycol methyl groups (Fig. 4B and G, 6B). (2) If Pluronic chains were significantly cleaved, this would have predominantly occurred in PEG blocks, leading to a lower hydrophilic/hydrophobic balance and to the formation of larger aggregates associated by the Pluronic resonances; however, the only diffusion coefficient associated to PEG resonances was indeed that of the usual Pluronic micelles, both for oxidation with hydrogen peroxide and hypochlorite. This does not imply the complete absence of Pluronic oxidation, but indicates a protective effect of polysulfides.
Biological effects of the differential oxidation response
Due to the morphologically and chemically different nature of the H2O2 and ClO− oxidation products, it is reasonable to assume them to have different effects on a cellular environment. The cytotoxicity of nanoparticles was assessed using two common cell lines: L929 mouse fibroblasts and J774.2 mouse macrophages (Fig. 7A); the latter phagocytic cells are typically characterized by a higher sensitivity to toxic stimuli and were used as a worst case scenario. PPS nanoparticles had already shown a benign character in vivo,11 therefore the absence of significant effects on the viability of both cell lines also at concentrations as high as 5 mg mL−1 is not surprising. More importantly, the H2O2-oxidized nanoparticles showed an remarkably benign behavior, which confirms previous data of low cytotoxicity of the oxidized linear PPS (IC50 > 30–40 mg mL−1 on L929 mouse fibroblasts14).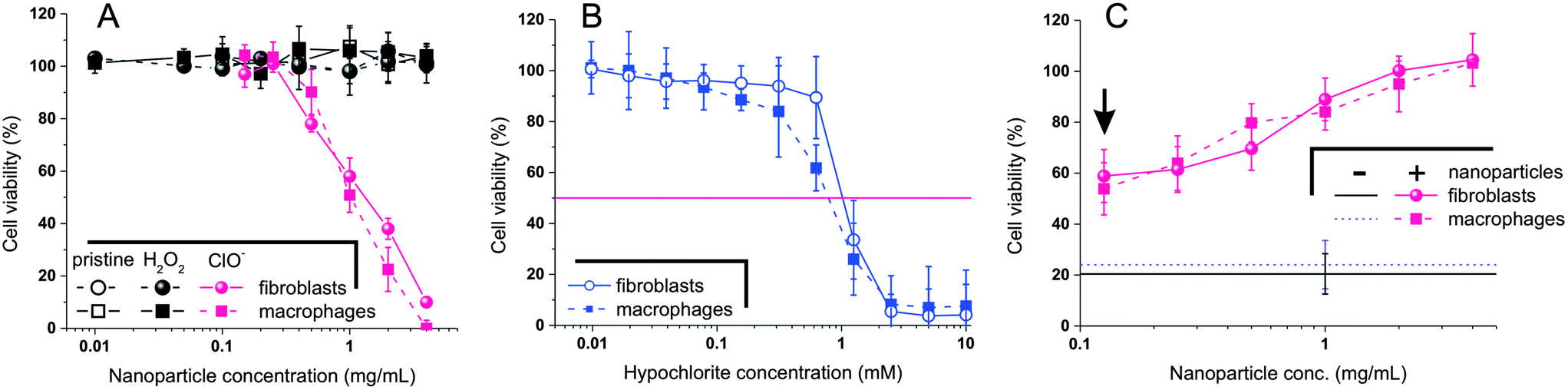 | ||
| Fig. 7 (A) Viability of L929 fibroblasts and J774.2 macrophages (mitochondrial activity measured via MTS and normalized against the protein content measured via BCA) upon incubation with nanoparticles before and after oxidation with H2O2 and ClO− (15% H2O2, 10 mM ClO−; 48 h; 37 °C; pH = 7.4). Please realize that the nanoparticles were purified via dialysis and very low MW compounds (<1 kDa) were eliminated. (B) Viability of the two cell lines upon a 48 hours incubation with different concentrations of hypochlorite. (C) Viability of the two cell lines upon a 48 hours incubation with 1.25 mM hypochlorite and different concentrations of nanoparticles. The arrow indicated the results for the nanoparticle concentration with a stoichiometric equivalence between hypochlorite and sulfide groups. The two horizontal lines indicate the viability of the two cell lines in the absence of nanoparticles. | ||
The products of ClO−-mediated oxidation provided a pretty different picture. They considerably reduced the viability of both cell lines already at a concentration of 0.3 mg mL−1, with IC50s located around 1 mg mL−1. Although the sulfone-containing fragments were considerably more toxic from their precursors, these IC50 values are similar to those of several polymers generally considered ‘biocompatible’ such as chitosan, and indicate also a lower toxicity than hypochlorite: the IC50 of NaOCl in L929 mouse fibroblasts (24 h exposure) is reported to be lower than 0.05 mg mL−1 = 0.7 mM.30 The polysulfide nanoparticles may therefore provide a remarkable detoxification effect; indeed, following a analogous concept, similar nanoparticles have been used to improve the stability of enzymes against hypochlorite.31
Using both cell lines we have confirmed the hypochlorite IC50 to be in the range of 0.7–1 mM for fibroblasts (Fig. 7B). Exposing then the cells to hypochlorite at a concentration > IC50 (1.25 mM, with a residual cell viability of ≈ 20%), we have recorded a 3-fold improvement of viability in the presence of nanoparticles at the concentration of the hypochlorite:sulfide stoichiometric equivalence (arrow in Fig. 7C).
Keeping fixed the concentration of hypochlorite, and considering the apparently stoichiometric character of its oxidation, the same amount of oxidized fragments should be produced irrespectively of the overall concentration of nanoparticles, which would therefore be expected not to influence the cell viability. On the contrary, the viability increased with increasing nanoparticle concentration, above all above 1 mg mL−1, which indicates the presence of further protecting effects of the polysulfides.
Conclusions
Polysulfide nanoparticles can be produced with a controlled size (through the Pluronic/PS ratio) and a homogeneous cross-linking density (through the control over PPS molecular weight); we have showed that they feature different oxidation-responsive mechanisms in response to the presence of different ROS, and that this point does not depend on size and nature of the cross-linker. In particular, the exposure to H2O2 induces swelling and loss of PEGylated surface layer without however significantly affecting the nanoparticle cytotoxicity; on the contrary, ClO− causes instantaneous solubilization and production of medium-toxicity fragments, in addition to the concomitant release of Pluronic (as summarized in Scheme 2).This differential response can open the way to ROS-specific therapeutic actions e.g. release of encapsulated drugs. The remarkable sensitivity to ClO− could make them particularly attractive to perform rapid and efficient actions in response to pathological neutrophil respiratory bursts,32 or to other pathologies based on myeloperoxidase over-activation.33 On the other hand, their slower response to H2O2 and lower toxicity after oxidation appear more suited for sustained release actions.
It is noteworthy that similar general considerations could be applied to materials sensitive to reducing agents; for example, disulfide-containing materials are notoriously responsive to the presence of thiols,34,35 but chemical details unrelated to the REDOX potential may significantly affect the material response, e.g. the tendency of the thiol to produce homo- or heterodisulfides or the kinetics of the thiol-disulfide exchange process.
Acknowledgements
Dr Jureerat Laliturai gratefully acknowledges a studentship from the Government Pharmaceutical Organization of Thailand. Mr Damien Jeanmaire and Mr Richard d'Arcy are indebted to EPSRC for their studentships in the NoWNano Doctoral Training Centre. Financial support from EPSRC (grant no. EP/C543564/1 and Advanced Research Fellowship for Nicola Tirelli) for the initial part of this work and from the Knowledge Centre of Materials Chemistry (University of Manchester) is also gratefully acknowledged.References
- V. Adler, Z. M. Yin, K. D. Tew and Z. Ronai, Oncogene, 1999, 18, 6104–6111 CrossRef CAS PubMed
.
- I. Rahman and W. MacNee, Eur. Respir. J., 2000, 16, 534–554 CrossRef CAS
- V. V. Khutoryanskiy and N. Tirelli, Pure Appl. Chem., 2008, 80, 1703–1718 CrossRef CAS
- C. D. Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef PubMed
- E. Lallana and N. Tirelli, Macromol. Chem. Phys., 2013, 214, 143–158 CrossRef CAS
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2013 10.1039/c1033py01192e
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H. J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS
- B. M. Babior, Am. J. Med., 2000, 109, 33–44 CrossRef CAS
- P. Hu and N. Tirelli, React. Funct. Polym., 2011, 71, 303–314 CrossRef CAS PubMed
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed
- A. Rehor, H. Schmoekel, N. Tirelli and J. A. Hubbell, Biomaterials, 2008, 29, 1958–1966 CrossRef CAS PubMed
- B. L. Allen, J. D. Johnson and J. P. Walker, ACS Nano, 2011, 5, 5263–5272 CrossRef CAS PubMed
- A. Rehor, N. E. Botterhuis, J. A. Hubbell, N. Sommerdijk and N. Tirelli, J. Mater. Chem., 2005, 15, 4006–4009 RSC
- P. Carampin, E. Lallana, J. Laliturai, S. C. Carroccio, C. Puglisi and N. Tirelli, Macromol. Chem. Phys., 2012, 213, 2052–2061 CrossRef CAS
- W. H. Koppenol and J. Butler, Adv. Free Radical Biol. Med., 1985, 1, 91–131 CrossRef CAS
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS
- W. H. Koppenol, FEBS Lett., 1994, 347, 5–8 CrossRef CAS
- P. Hu and N. Tirelli, Bioconjugate Chem., 2012, 23, 438–449 CrossRef CAS PubMed
- S. Hirosue, I. C. Kourtis, A. J. van der Vlies, J. A. Hubbell and M. A. Swartz, Vaccine, 2010, 28, 7897–7906 CrossRef CAS PubMed
- A. Stano, A. J. van der Vlies, M. M. Martino, M. A. Swartz, J. A. Hubbell and E. Simeoni, Vaccine, 2011, 29, 804–812 CrossRef CAS PubMed
- S. N. Thomas, A. J. van der Vlies, C. P. O'Neil, S. T. Reddy, S. S. Yu, T. D. Giorgio, M. A. Swartz and J. A. Hubbell, Biomaterials, 2011, 32, 2194–2203 CrossRef CAS PubMed
- A. Rehor, N. Tirelli and J. A. Hubbell, Macromolecules, 2002, 35, 8688–8693 CrossRef CAS
- C. D. Vo, G. Kilcher and N. Tirelli, Macromol. Rapid Commun., 2009, 30, 299–315 CrossRef CAS PubMed
- M. Valentini, A. Vaccaro, A. Rehor, A. Napoli, J. A. Hubbell and N. Tirelli, J. Am. Chem. Soc., 2004, 126, 2142–2147 CrossRef CAS PubMed
- G. Kilcher, L. Wang and N. Tirelli, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2233–2249 CrossRef CAS
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS
- M. D. Pelta, G. A. Morris, M. J. Stchedroff and S. J. Hammond, Magn. Reson. Chem., 2002, 40, S147–S152 CrossRef CAS
- M. Lange, S. Braune, K. Luetzow, K. Richau, N. Scharnagl, M. Weinhart, A. T. Neffe, F. Jung, R. Haag and A. Lendlein, Macromol. Rapid Commun., 2012, 33, 1487–1492 CrossRef CAS PubMed
- G. Simbula, C. Dettori, T. Camboni and E. Cotti, J. Endod., 2010, 36, 1516–1520 CrossRef PubMed
- B. L. Allen, J. D. Johnson and J. P. Walker, Nanotechnology, 2012, 23, 294009 CrossRef PubMed
- M. Heinzelmann, M. A. Mercer-Jones and J. C. Passmore, Am. J. Kidney Dis., 1999, 34, 384–399 CrossRef CAS
- B. S. van der Veen, M. P. J. de Winther and P. Heeringa, Antioxid. Redox Signalling, 2009, 11, 2899–2937 CrossRef CAS PubMed
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py01475d |
| ‡ Current address: The Government Pharmaceutical Organization, 75/1 Rama 6 Road, Ratchathewi, Bangkok 10400, Thailand. |
| § Current address: King Abdulaziz City for Science and Technology Kingdom of Saudi Arabia, P.O Box 6086, Riyadh 11442. |
| ¶ Current address: Chemical Engineering and Applied Chemistry, Aston University, Aston Triangle, Birmingham, B4 7 ET. |
| This journal is © The Royal Society of Chemistry 2014 |