 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of thiol-functionalized amphiphilic polylactide–methacrylic diblock copolymers†
Efrosyni
Themistou‡
*ab,
Giuseppe
Battaglia
bc and
Steven P.
Armes
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, South Yorkshire, S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk; Fax: +44 (0)114 222 9346; Tel: +44 (0)114 222 9342
bDepartment of Biomedical Science, University of Sheffield, Sheffield, South Yorkshire, S10 2TN, UK
cDepartment of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: g.battaglia@ucl.ac.uk
First published on 27th November 2013
Abstract
Biodegradable amphiphilic diblock copolymers based on an aliphatic ester block and various hydrophilic methacrylic monomers were synthesized using a novel hydroxyl-functionalized trithiocarbonate-based chain transfer agent. One protocol involved the one-pot simultaneous ring-opening polymerization (ROP) of the biodegradable monomer (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide, LA) and reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-(dimethylamino)ethyl methacrylate (DMA) or oligo(ethylene glycol) methacrylate (OEGMA) monomer, with 4-dimethylaminopyridine being used as the ROP catalyst and 2,2′-azobis(isobutyronitrile) as the initiator for the RAFT polymerization. Alternatively, a two-step protocol involving the initial polymerization of LA followed by the polymerization of DMA, glycerol monomethacrylate or 2-(methacryloyloxy)ethyl phosphorylcholine using 4,4′-azobis(4-cyanovaleric acid) as a RAFT initiator was also explored. Using a solvent switch processing step, these amphiphilic diblock copolymers self-assemble in dilute aqueous solution. Their self-assembly provides various copolymer morphologies depending on the block compositions, as judged by transmission electron microscopy and dynamic light scattering. Two novel disulfide-functionalized PLA-branched block copolymers were also synthesized using simultaneous ROP of LA and RAFT copolymerization of OEGMA or DMA with a disulfide-based dimethacrylate. The disulfide bonds were reductively cleaved using tributyl phosphine to generate reactive thiol groups. Thiol–ene chemistry was utilized for further derivatization with thiol-based biologically important molecules and heavy metals for tissue engineering or bioimaging applications, respectively.
Introduction
Block copolymers based on the same type of monomer (e.g. either vinyl or cyclic monomers) are traditionally prepared using a single “living” polymerization technique.1 Recently, novel block copolymers with interesting properties have been prepared by combining two or more “living” polymerization chemistries to copolymerize dissimilar monomers.2–33 Sequential polymerizations are most commonly used for such syntheses.2–18 In principle, simultaneous polymerization can also lead to the synthesis of block copolymers. In practice, there are some examples in the literature for which incompatibility problems have been overcome to combine different polymerization techniques for the synthesis of well-defined block copolymers in a single step.19–33The combination of ring-opening polymerization (ROP)34 for the controlled synthesis of biodegradable aliphatic polyesters35,36 and reversible addition-fragmentation chain transfer (RAFT) polymerization,37–40 allows the synthesis of defined block copolymer architectures (for a wide range of vinyl monomers).9,11,15,41–44 This approach bodes well for the synthesis of block copolymers for biomedical applications such as sutures, implants for bone fixation, drug delivery vehicles and tissue engineering scaffolds.45
A typical sequential polymerization strategy for the synthesis of polylactide (PLA)-based block copolymers is either to introduce a RAFT agent after the ROP of lactide (LA)2,10 or to initiate ROP of LA after RAFT polymerization (e.g. from a hydroxyl functionality of a monomer previously polymerized by RAFT).8 An alternative approach is the use of a bifunctional agent, i.e. a RAFT chain transfer agent (CTA) bearing a hydroxyl functional group either in the R- or in the Z-position41 that can initiate both polymerizations. This CTA is either used to mediate the radical process followed by a second step in which the hydroxyl group initiates the ROP of LA,7,13,16 or ROP is performed first, followed by RAFT polymerization.6,12,14,17 Usually, an intermediate purification step is required. Clearly, simplifying the process to just one step where the two polymerizations proceed simultaneously is desirable. Exploring the simultaneous one-pot ROP–RAFT polymerization of LA with a vinyl monomer appears to be promising for the synthesis of new amphiphilic biodegradable PLA-based copolymers. Previous studies have focused on the simultaneous one-pot ROP of other monomers such as ε-caprolactone,21–23,30,33 δ-valerolactone,22,33 trimethylene carbonate,22 β-butyrolactone28 and RAFT polymerization, and also the simultaneous ROP of LA and RAFT polymerization of acrylic monomers.24,29 Simultaneous ROP of LA and RAFT polymerization of methacrylic monomers for the preparation of biodegradable PLA-containing block copolymers using a metal-free approach46 has been previously described, but mainly in the context of the synthesis of hydrophobic diblock copolymers.22 As far as we are aware, such an approach has not been explored for the preparation of amphiphilic PLA-based block copolymers, particularly when the water-soluble block is based on methacrylic repeat units.
Amphiphilic diblock copolymers have attracted the interest of many researchers because they undergo spontaneous self-assembly in aqueous solution. This results in the formation of various morphologies such as spherical micelles, worm-like micelles, vesicles or intermediate structures.47,48 Often the relatively high molecular weight (MW) and/or glass transition temperature can hinder the direct dissolution of amphiphilic diblock copolymers in water, since this makes the formation of micelles or vesicles extremely slow and inefficient.49 These kinetic constrains can be overcome using either thin film rehydration or by prior dissolution using water-miscible co-solvents. This latter approach, commonly known as the ‘solvent switch’ method, is widely used for block copolymer self-assembly.50 Here, the block copolymer is initially dissolved in a water-miscible common organic solvent for both copolymer blocks. Then water is added gradually to the copolymer solution resulting in the formation of various nanostructures after removal of the organic solvent.47 One potential application for amphiphilic copolymers is in drug delivery. Here it is often considered desirable to conjugate peptides, vitamins or sugars so as to confer cell/tissue specificity and targeting.
Amongst the various conjugation approaches, thiol–disulfide chemistry has been widely used for biomedical applications51–54 because of its orthogonality, reversibility and redox activity.55–60 The reduction of a disulfide bond results in the formation of thiol functional groups. The presence of thiol functionalities during a radical-based vinyl polymerization such as RAFT is not desirable since they can act as efficient chain transfer agents.61 We have previously reported post-polymerization formation of thiol-functional copolymers using a disulfide-based dimethacrylate (DSDMA) comonomer.62–66 This disulfide acts as an atom-efficient thiol-protecting group for the synthesis of branched methacrylic copolymers. In relatively dilute solution, DSDMA undergoes predominantly intramolecular cyclization on its statistical copolymerization with a methacrylic monomer.63 Thus cleavage of the lightly branched copolymer results in the formation of near-monodisperse copolymer chains62,64,65 bearing thiol functionality.
The purpose of this work is to investigate the one-pot metal-free ROP–RAFT synthesis of biocompatible linear and branched amphiphilic diblock copolymers based on a biodegradable aliphatic polyester (PLA) and methacrylic monomers. The branched diblock copolymers are prepared using DSDMA as a comonomer for the methacrylic block. Its disulfide bond can be reductively cleaved to produce a thiol-functionalized amphiphilic block copolymer. In principle, such thiols can be useful for conjugation of various unsaturated molecules by thiol–ene chemistry, which has been widely used in polymer science.60,67–69 This approach is especially advantageous for biologically relevant molecules such as oligopeptides since no cytotoxic metal catalysts are required. We also investigate a two-step protocol for preparation of PLA-based amphiphilic block copolymers. This approach can be applied to copolymers that cannot be synthesized via the one-step protocol, such as when the ROP conditions are incompatible with those of RAFT polymerization. For example, hydroxyl-functional monomers can act as ROP initiators, while biomimetic methacrylic monomers such as 2-(methacryloyloxy)-ethyl phosphorylcholine are only soluble in protic solvents, which are unsuitable for ROP. Finally, the self-assembly behavior of selected block copolymers is examined in aqueous solution.
Experimental section
Materials
2-(Methacryloyloxy)ethyl phosphorylcholine (MPC, >99.9%) was kindly donated by Biocompatibles UK, Ltd. (Farnham, UK). Oligo(ethylene glycol)methacrylate (OEGMA) and glycerol monomethacrylate (GMA, 97%) were kindly donated by Cognis UK Ltd. (Hythe, UK). THF (HPLC grade), DMF (chromatography GPC grade), chloroform (CHCl3, HPLC grade), methanol (CH3OH, HPLC grade), n-hexane (99.8%, Certified ACS), ethyl acetate (HPLC grade) and triethylamine (Et3N, laboratory reagent grade, ≥99%) were purchased from Fisher Scientific (Loughborough, UK). 2-(Dimethylamino)ethyl methacrylate (DMA, 98%), (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide, LA, 98%), 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CADB), 4,4′-azobis(4-cyanovaleric acid) (ACVA), hexane-1,6-diol (99%), dimethylaminopyridine (DMAP), 1,2-dichloroethane (anhydrous, 99.8%), anhydrous ethanol (≥99.5%), tributylphosphine (Bu3P, mixture of isomers 97%), divinyl sulfone (DVS, 97%), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, purum, ≥98.0%), L-glutathione (Glu, reduced form, ≥99%) and indium(III) chloride (InCl3, 99.999% trace metals basis) were purchased from Sigma-Aldrich (UK). N,N′-Dicyclohexylcarbodiimide (DCC, 99%) was purchased from Acros Organics. 2,2′-Azobis(isobutyronitrile) (AIBN) and maleimido-monoamide DOTA (≥94%) were purchased from BDH and Macrocyclics™ (Dallas, TX, USA), respectively.The MPC, OEGMA and GMA monomers were used without further purification. The DMA monomer was passed through a neutral alumina column before use to remove the inhibitor. LA was recrystallized 4–5 times from ethyl acetate prior to use. The solvents THF, DMF, chloroform and methanol were used as the mobile phase in GPC, whereas n-hexane and ethyl acetate were used in column chromatography. Regenerated cellulose dialysis membranes with molecular weight cut-off (MWCO) of 1000 Da were purchased from Spectra/Por. The disulfide-based dimethacrylate (DSDMA) branching monomer was synthesized following a previously reported method.64
Synthesis of hydroxyl-functional RAFT CTA
CADB (3.00 g, 10.74 mmol) and 1,6-hexanediol (6.35 g, 53.70 mmol) were added in a round-bottom flask equipped with a magnetic stir bar. These reagents were dissolved in 75 mL CHCl3 by stirring the contents of the flask for 15 min at 40 °C. The reaction catalysts DCC (2.44 g, 11.81 mmol) and DMAP (131 mg, 1.07 mmol) were dissolved in 15 mL CHCl3 at ambient temperature. The catalyst mixture was subsequently added with a syringe to the round-bottom flask. The reaction mixture was stirred under reflux for 46 h and allowed to cool in the refrigerator overnight. After filtration, CHCl3 was removed under reduced pressure using a rotary evaporator. The resulting oily residue was purified by column chromatography (silica gel/n-hexane–ethyl acetate 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). After removal of the solvents using a rotary evaporator and a vacuum oven, an oily liquid was obtained (1.82 g, 45% yield).
:50). After removal of the solvents using a rotary evaporator and a vacuum oven, an oily liquid was obtained (1.82 g, 45% yield).
1H NMR (CDCl3): δ = 1.32–1.36 (m, 4H), 1.52 (m, 2H), 1.61 (m, 2H), 1.89 (s, 3H), 2.33–2.67 (m, 4H), 3.57 (t, 2H), 4.06 (t, 2H), 7.33–7.87 (m, 5H, aromatic).
13C NMR (CDCl3): δ = 24.2 (CH3), 25.5 (COOCH2CH2CH2), 25.9 (CH2COO), 28.6 (CH2CH2CH2OH), 30.0 (COOCH2CH2), 32.7 (CH2CH2OH), 33.6 (CH2CH2COO), 45.9 (SCCH2), 62.7 (CH2CH2OH), 65.3 (COOCH2), 118.7 (CN), 126.8, 128.7, 133.2, 144.6 (Ph), 171.8 (CO), 222.5 (CS). ESI-MS, m/z (M + H)+ 380.
Synthesis of PLA-based diblock copolymers
The syntheses of other PLA–PDMA and PLA–POEGMA block copolymers were conducted following the same one-step protocol. Representative 1H NMR spectra of diblock copolymers prepared by simultaneous ROP–RAFT processes are given in Fig. 1(c–d).
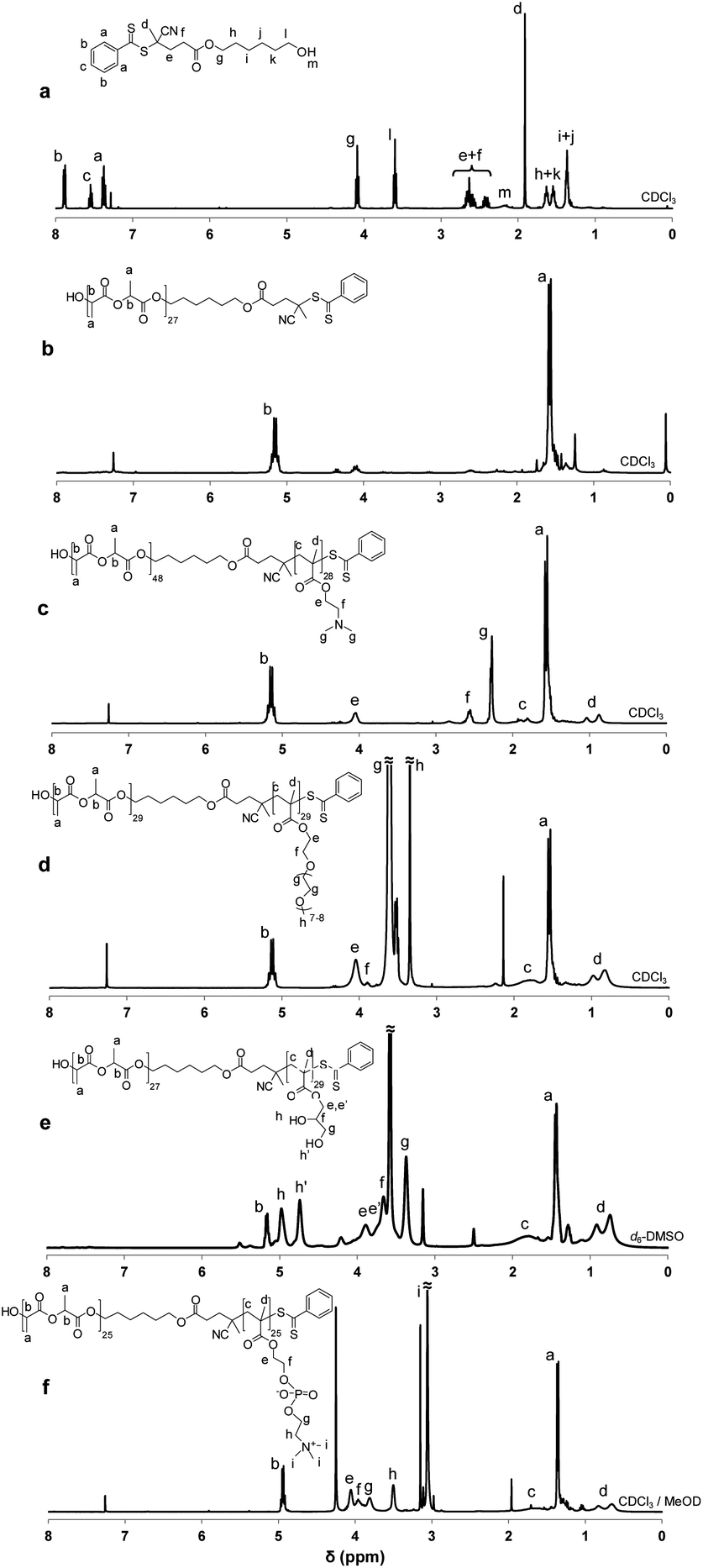 | ||
| Fig. 1 1H NMR spectra of (a) ROP–RAFT dual agent 1, (b) PLA27 macro-CTA, diblock copolymers by simultaneous (c) PLA48–PDMA282 and (d) PLA29–POEGMA292, and by two-step (e) PLA27–PGMA293a and (f) PLA25–PMPC253b ROP–RAFT polymerization. | ||
A similar two-step protocol was followed for the synthesis of a PLA200–PMPC30 block copolymer with a longer hydrophobic PLA block. The same method was applied for the preparation of PLA30–PGMA30 block copolymer and PLA30–PDMA30 block copolymer (for direct comparison with the one-step protocol). 1,2-Dichloroethane was used as a solvent for both polymerization steps. AIBN was the initiator for these two RAFT polymerizations.
All polymerizations (for both the simultaneous and the two-step protocols) were quenched by allowing the reaction solution to cool to 20 °C. A sample was removed from each reaction solution for 1H NMR and GPC analysis. The final copolymers were purified by dialysis against acetone using membranes with a MWCO of 1000. For PLA–PGMA and PLA–PMPC block copolymers, this was followed by dialysis in methanol. The solvent was evaporated using a rotary evaporator. The copolymers were dried in a vacuum oven for 48 h. The resulting block copolymers were characterized by 1H NMR spectroscopy (Table 1 and Fig. 1) and GPC (Table 1, Fig. 2 and 3).
| Entrya | MRAFT | [LA]0:[MRAFT]0:[1]0 |
Conv.b (%) | DPCalcd | DPNMR | M n (kDa) | M w/Mnd | d (nm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LA | MRAFT | LA | MRAFT | LA | MRAFT | Calcd.c | NMR | GPC | |||||
|
a
Polymerization conditions: for simultaneous ROP–RAFT polymerization (entries 1–7) [1]0:[DMAP]0:[AIBN]0 = 1:4:0.20, 74 °C, 24 h, 55% w/w solids; for two-step ROP–RAFT polymerization (entries 8 and 9) [1]0:[DMAP]0:[AIBN]0 = 1:4:0.20, 74 °C, 24 h, 55% w/w solids; for two-step ROP–RAFT polymerization (entries 10 and 11) [1]0:[DMAP]0:[ACVA]0 = 1:4:0.25, 78 °C, 24 h, 20% w/w solids.
b By 1H NMR analysis (CDCl3 for 1–7 and 8, d6-DMSO for entry 9 and CDCl3/MeOD 1:1 for entries 10 and 11).
c
M
n,Calcd = MWLA × ([LA]0/[1]0) × (conv.LA) + (MW of MRAFT) × ([MRAFT]0/[1]0) × (conv. of MRAFT) + MW1.
d By GPC (eluent THF for entries 1–7 and 8, DMF for entry 9, and CHCl3/CH3OH 3:1 for entries 10 and 11).
e Diameter d is the sphere-equivalent intensity-average diameter measured by DLS in water after a solvent switch protocol.
|
|||||||||||||
| 1 | DMA | 250:30:1 |
81 | 79 | 203 | 24 | 173 | 24 | 33.3 | 29.1 | 12.6 | 1.30 | 214 |
| 2 | DMA | 150:30:1 |
77 | 89 | 116 | 27 | 137 | 27 | 21.2 | 24.4 | 11.1 | 1.30 | 133 |
| 3 | DMA | 60:30:1 |
80 | 86 | 48 | 26 | 58 | 27 | 11.4 | 13.0 | 7.2 | 1.39 | 240 |
| 4 | DMA | 60:30:1 |
82 | 97 | 49 | 29 | 48 | 28 | 12.0 | 11.7 | 6.0 | 1.34 | 497 |
| 5 | DMA | 45:30:1 |
93 | 97 | 42 | 29 | 41 | 30 | 11.0 | 11.0 | 5.9 | 1.37 | 113 |
| 6 | DMA | 30:30:1 |
89 | 89 | 27 | 27 | 26 | 25 | 8.4 | 8.1 | 5.3 | 1.37 | 685 |
| 7 | OEGMA | 30:30:1 |
96 | 99 | 29 | 30 | 29 | 29 | 18.0 | 17.7 | 19.5 | 1.32 | 550 |
| 8 | DMA | 30:30:1 |
95 | 74 | 29 | 22 | 27 | 21 | 8.0 | 7.6 | 4.1 | 1.41 | 718 |
| 9 | GMA | 30:30:1 |
95 | >99 | 29 | 30 | 27 | 29 | 9.1 | 8.9 | 42.3 | 1.27 | 338 |
| 10 | MPC | 200:30:1 |
92 | 79 | 184 | 24 | 184 | 23 | 34.0 | 33.7 | 70.5 | 1.35 | 1430 |
| 11 | MPC | 30:30:1 |
93 | 85 | 28 | 26 | 25 | 25 | 11.9 | 11.4 | 20.8 | 1.43 | 935 |
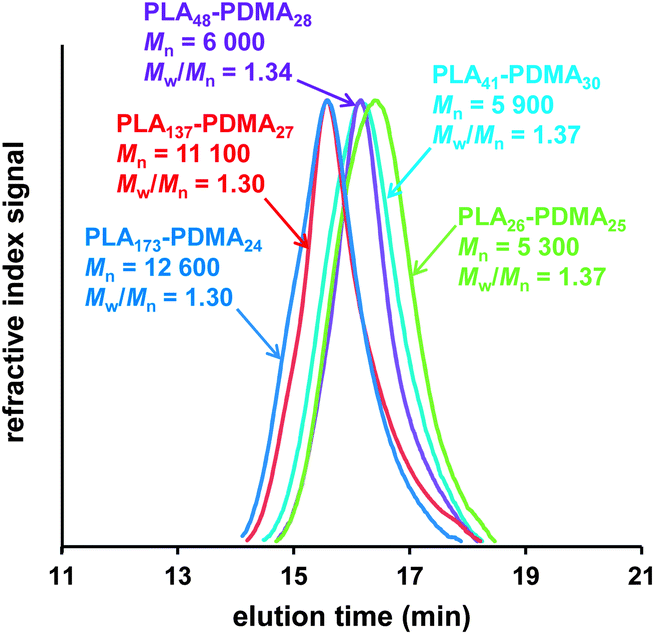 | ||
| Fig. 2 THF GPC curves (vs. poly(methyl methacrylate) standards) obtained for PLAx–PDMAy linear diblock copolymers synthesized via simultaneous ROP–RAFT polymerization in 1,2-dichloroethane at 74 °C, see Table 1 (entries 1, 2, 4, 5, 6). | ||
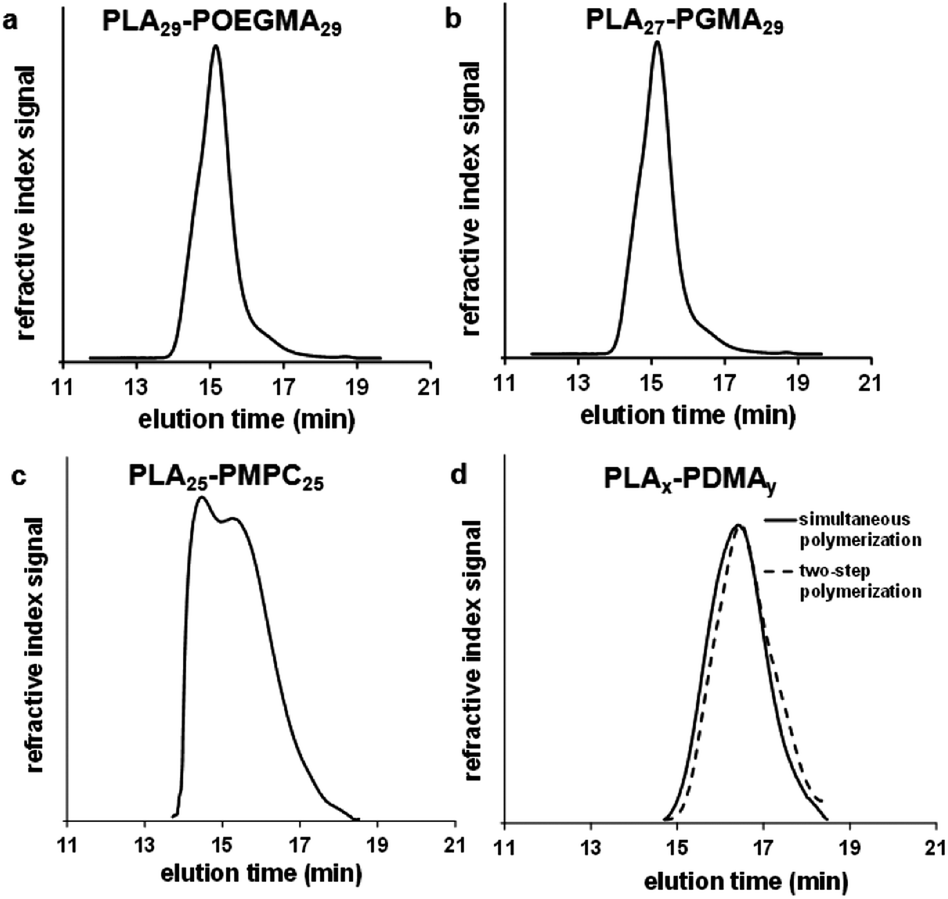 | ||
| Fig. 3 GPC curves (vs. poly(methyl methacrylate) standards) obtained for (a) PLA29–POEGMA29 linear diblock copolymer (THF eluent) synthesized via simultaneous ROP–RAFT polymerization in 1,2-dichloroethane at 74 °C, (b) PLA27–PGMA29 linear diblock copolymer (DMF eluent) synthesized via two-step ROP–RAFT polymerization in 1,2-dichloroethane at 74 °C, (c) PLA25–PMPC25 linear diblock copolymer (CHCl3/CH3OH 3:1 v/v eluent) prepared by two-step ROP–RAFT polymerization (ROP of LA in 1,2-dichloroethane at 74 °C followed by RAFT polymerization of MPC in ethanol at 78 °C) and (d) comparison of PLDMA25 and PLA27–PDMA21 prepared by simultaneous and two-step ROP–RAFT polymerizations, respectively, in 1,2-dichloroethane at 74 °C. | ||
Synthesis of branched PLA-based diblock copolymers
900 g mol−1, Mw = 134000 g mol−1, Mw/Mn = 2.36), see Fig. 4.
The branched PLA30–P(DMA30-stat-DSDMA1) block copolymer was synthesized using a similar protocol. This resulted in the formation of a gel.
Disulfide cleavage of branched PLA30–P(OEGMA30-stat-DSDMA1) diblock copolymer, functionalization of the resulting PLA30–P(OEGMA30-stat-TEMA2) block copolymer with divinyl sulfone and its subsequent conjugation with L-glutathione
500 mg (275 mg solids content, 30.6 μmol disulfide bonds) of the branched PLA30–P(OEGMA30-stat-DSDMA1) polymer were added in a 50 mL round-bottom flask. Subsequently, 20 mL DMF (1.4% w/v) was added to the flask to dissolve the reagents. The flask was equipped with a magnetic stir bar and sealed with a rubber septum. The mixture was purged with nitrogen gas for 20 min and placed in an oil bath at 30 °C. A solution of Bu3P (91.9 μmol, 18.6 mg, 3.0 eq. relative to disulfide bonds) and Et3N (64.3 μmol, 6.5 mg, 2.1 eq. relative to disulfide bonds) in 5 mL DMF was added under a nitrogen atmosphere via syringe to the round-bottom flask. This resulted in disulfide cleavage, allowing the conversion of 1 eq. of DSDMA to 2 eq. of TEMA. The reaction was left in an oil bath for 2 h at 30 °C to afford a thiol-functionalized linear PLA30–P(OEGMA30-stat-TEMA2) block copolymer solution. A sample was extracted from the reaction flask for DMF GPC analysis (Mn = 38800 g mol−1, Mw = 53100 g mol−1, Mw/Mn = 1.37). A degassed solution of divinyl sulfone (0.92 mmol, 108.6 mg, 15 eq. relative to the thiol –SH group) in 5 mL DMF was added to the block copolymer. The resulting mixture was stirred at 30 °C for 15 h to convert the TEMA monomer units into VSTEMA monomer units. The final solution was dialyzed (MWCO 1000 Da) against acetone (5 times/3 days). The solvent was removed by rotary evaporator, followed by vacuum oven drying. The dried PLA30–P(OEGMA30-stat-VSTEMA2) was characterized by DMF GPC (Mn = 37800 g mol−1, Mw = 52800 g mol−1, Mw/Mn = 1.40) and 1H NMR (Fig. 4).
202 mg of the above dried vinyl sulfone-functionalized block copolymer (218.4 mmol vinyl sulfone, assuming 2 vinyl sulfones per polymer chain) were placed in a 10 mL glass vial equipped with a magnetic stir bar and sealed with a rubber septum. A mixture of 7.0 mg Glu (229.3 mmol Glu or free thiol group, 1.05 eq. relative to vinyl sulfone), 0.6 mg TCEP (0.1 eq. relative to vinyl sulfone) and 4 mL water was purged with nitrogen gas for 20 min. This solution was then added to the vial containing the copolymer. The resulting mixture was purged with nitrogen for another 10 min. The reaction was allowed to proceed a 20 °C with stirring for 4 h. The final copolymer solution was dialyzed (MWCO 1000 Da) against water (6 times/1 day). The purified PLA30–P(OEGMA30-stat-GluVSTEMA2) was freeze-dried. The dried copolymer was dissolved in the appropriate solvent and characterized by DMF GPC (Mn = 23600 g mol−1, Mw = 34100 g mol−1, Mw/Mn = 1.45) and 1H NMR (Fig. 4 in CDCl3 and Fig. S2† in D2O).
Disulfide cleavage of the branched PLA30–P(DMA30-stat-DSDMA1) diblock copolymer, functionalization of the resulting PLA30–P(DMA30-stat-TEMA2) block copolymer with maleimido-monoamide-DOTA and subsequent In conjugation
182 mg (100 mg solids content, 10.3 μmol disulfide bonds) of the PLA30–P(DMA30-stat-DSDMA1) gel was placed in a 14 mL glass vial containing 5 mL DMF (2.0% w/v). The flask was equipped with a magnetic stir bar and sealed with a rubber septum. The mixture was placed in an oil bath at 30 °C after purging with nitrogen for 20 min. A solution of Bu3P (30.9 μmol, 6.3 mg, 3.0 eq. relative to disulfide bonds) and Et3N (21.6 μmol, 2.2 mg, 2.1 eq. relative to disulfide bonds) in 1 mL DMF was prepared and added under nitrogen atmosphere to the glass vial using a syringe. The reaction mixture was left at 30 °C for 2 h, resulting in cleavage of the disulfide bonds. With this reaction the gel network was converted to a thiol-functionalized linear PLA30–P(DMA30-stat-TEMA2) block copolymer solution. Maleimido-monoamide-DOTA was reacted with the TEMA residues to produce DOTA-functionalized TEMA (DOTATEMA) residues. Specifically, a degassed solution of maleimido-monoamide-DOTA (21.6 μmol, 17.0 mg, 2.1 eq. relative to disulfide bonds) was added. The reaction was allowed to proceed at 30 °C overnight. Then InCl3 (4.6 mg, 20.6 μmol, 2.0 eq. relative to disulfide bonds) were added and the mixture was stirred at 90 °C for 1 h. The solution was dialyzed against acetone (MWCO 1000 Da/5 times/3 days) and the solvent was removed using rotary evaporator. The resulting In-conjugated PLA30–P(DMA30-stat-InDOTATEMA2) block copolymer was dried in a vacuum oven overnight and characterized by ICP-AES (1.16 In atoms per copolymer chain).Self-assembly of amphiphilic block copolymers in dilute aqueous solution
Self-assembly of the block copolymers was carried out using the solvent switch method. Briefly, PLA–PDMA and PLA–POEGMA block copolymers were dissolved in acetone to give 50 mg per mL of copolymer solutions. 4.8 mL deionized water was added drop by drop in 200 μL of the above solutions under stirring giving copolymer solutions with a final concentration of 2.5 mg mL−1. Similarly, an acetone–methanol mixture (3:1 v/v) was used to solubilize the PLA–PGMA block copolymer. These solutions were stirred vigorously at room temperature for 5 h, allowing acetone evaporation. For more efficient solvent evaporation the solution was left on a rotary evaporator for 1 min. Following a similar method, the PLAx–PMPCy block copolymers were dissolved in a 3:1 v/v chloroform–methanol mixture. This was followed by addition of water and stirring for 2 h to allow solvent evaporation. Dialysis was performed against deionized water for 4 h (5 times) to remove trace organic solvents. Finally, all copolymer solutions were stored at 4 °C overnight to avoid copolymer degradation.
Polymer characterization
PLA–PDMA and PLA–POEGMA block copolymers were assessed using a GPC system purchased from Polymer Laboratories at 30 °C. The system comprised two PL Gel 5 μm (7.5 × 300 mm) Mixed-C columns in series with a guard column. These were connected with a WellChrom K-2301 refractive index (RI) detector operating at 950 ± 30 nm. THF containing 2% v/v triethylamine and 0.05 wt/v % of butylhydroxytoluene (BHT) (flow rate 1 mL min−1) was used as an eluent.PLA–PMPC block copolymers were assessed using a Hewlett Packard HP1090 Liquid Chromatograph. Two PL Gel 5 μm Mixed-C columns in series with a guard column at 40 °C and a Gilson Model 131 RI detector were used. The eluent was a 3:1 v/v chloroform–methanol mixture with 2 mM lithium bromide (LiBr, flow rate of 1.0 mL min−1). Toluene (2 μL) was added to the sample as a flow rate marker.
Ten near-monodisperse linear poly(methyl methacrylate) (PMMA) standards (ranging from 1280 g mol−1 to 330000 g mol−1) were purchased from Polymer Laboratories (UK). These were used to calibrate the above two RI detectors. For both GPC systems, data analysis was carried out using Cirrus™ GPC Software supplied by the manufacturer.
The MWDs of the PLA–PGMA block copolymer, the branched PLA30–P(OEGMA30-stat-DSDMA1) block copolymer, the disulfide cleaved PLA30–P(OEGMA30-stat-TEMA2) linear block copolymer and the functionalized linear block copolymers PLA30–P(OEGMA30-stat-VSTEMA2) and PLA30–P(OEGMA30-stat-GluVSTEMA2) were assessed by DMF GPC. This system comprised two Polymer Laboratories PL gel 5 μm mixed C columns and one PL polar gel 5 μm guard column. These were arranged in series and maintained at 60 °C, followed by a Varian 390 LC RI detector. The DMF eluent containing 10 mM LiBr was kept at a flow rate of 1.0 mL min−1. Ten near monodisperse PMMA standards with MWs ranging from 625 g mol−1 to 618000 g mol−1 were used for calibration.
1H (Fig. 1a) and 13C NMR studies of the ROP–RAFT dual agent were performed in CDCl3. 1H NMR spectra of the PLA macroCTAs and the copolymers were acquired in the appropriate solvent for each block copolymer (Fig. 1b–f). A 250 or 400 MHz Bruker spectrometer was used for NMR analysis. The experimental DP values (DPNMR) of the PLA polymer block were obtained by comparing the integrated signal intensities of the aromatic RAFT end-group at 7.10–7.80 ppm with those of the –CH– protons of PLA at 5.14–5.24 ppm in d6-DMSO. The experimental DP values (DPNMR) of the RAFT-synthesized block were observed by comparing the integrated intensities of its characteristic protons with those of the methine protons from PLA. In more detail, for the calculations, the methyl protons of PDMA at 2.22–2.33 ppm in CDCl3 (Fig. 1c), the –COOCH2 protons of POEGMA block at 4.04 ppm in CDCl3 (Fig. 1d), the –CH2OH protons of PGMA at 3.38 ppm in d6-DMSO (Fig. 1e), and the methylene protons of the PMPC polymer block (Fig. 1f, protons e, f and g) at 3.65–4.14 ppm were used for the RAFT-synthesized block. For the PLA block, the methine protons of PLA at 5.07–5.24 ppm in CDCl3, at 5.13–5.21 ppm in d6-DMSO or 4.84–5.01 ppm in 1:1 CDCl3:CD3OD were taken into account. All the results are presented in Table 1.
DLS experiments were performed using a Zetasizer Nano-ZS (Malvern Instruments, UK). Aqueous copolymer solutions (0.1% w/v) were analyzed using disposable cuvettes and data were averaged over three consecutive runs.
TEM imaging of copolymer samples was performed using a FEI Tecnai G2 Spirit TEM 120 kV instrument equipped with an Orius SC1000 camera. The TEM samples were prepared using in-house carbon-coated copper grids. Grids were plasma glow-discharged for 45 s to create a hydrophilic surface. Each aqueous copolymer solution (0.1% w/w) was placed onto a freshly glow-discharged grid for 30 s. The grid was then blotted with a filter paper to remove excess solution. For positive staining of the deposited nanoparticles, a uranyl formate solution (0.75 w/v %) was used. This was placed using a micropipette on the sample-loaded grid for 20 s. The grid was then blotted with filter paper and dried using a vacuum hose.
Results and discussion
In this work, amphiphilic diblock copolymers were synthesized consisting of a hydrolyzable aliphatic polyester block (PLA) and a methacrylic polymer block consisting of PDMA, POEGMA, PGMA or PMPC. The preparation of these copolymers is illustrated in Scheme 1. A combined ROP–RAFT agent 1 containing a hydroxyl group as the initiating site for ROP and a dithiobenzoate functionality for the RAFT polymerization was utilized. This CTA is the monoester product of the reaction between CADB and 1,6-hexanediol (Scheme 1a). The esterification was conducted in chloroform under reflux for 48 h using DCC (1.1 eq.) and DMAP (0.1 eq.) as catalyst. To ensure a monofunctional product, excess diol (5.0 eq.) was used. The ROP–RAFT dual agent 1 was obtained in 45% yield. Its chemical structure and high purity were confirmed by 1H NMR spectroscopy (Fig. 1a), 13C NMR and electrospray ionization mass spectroscopy (ESI-MS) analysis.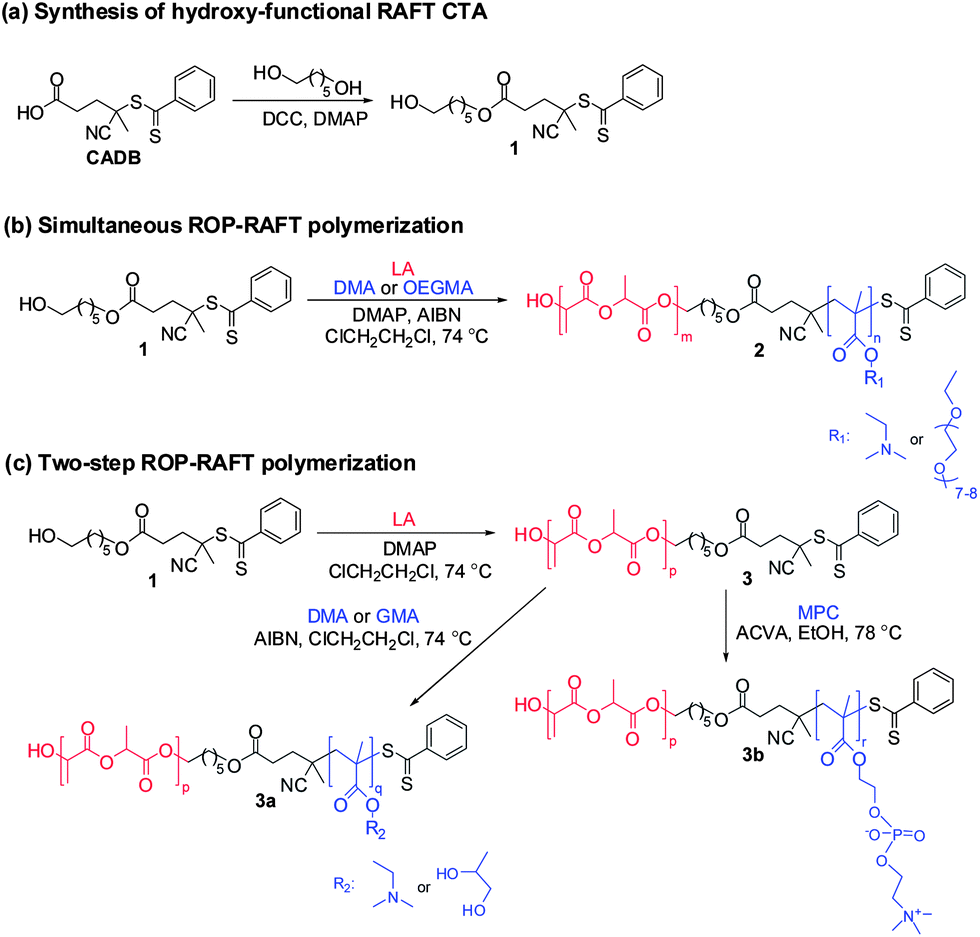 | ||
| Scheme 1 (a) Synthesis of hydroxyl-functional RAFT CTA for use as a ROP–RAFT dual agent in polymerizations. (b) Simultaneous ROP–RAFT copolymerization of (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide, LA) with 2-(dimethylamino)ethyl methacrylate (DMA) or oligo(ethylene glycol) methacrylate (OEGMA) at 74 °C in 1,2-dichloroethane to produce amphiphilic polylactide–methacrylic block copolymers 2. (c) Two-step ROP–RAFT polymerization; ROP of LA at 74 °C in 1,2-dichloroethane to synthesize PLA macro-CTAs that are subsequently used in the RAFT polymerization of DMA and glycerol monomethacrylate (GMA) at 74 °C in 1,2-dichloroethane or 2-(methacryloyloxy)ethyl phosphorylcholine (PMPC) at 78 °C in anhydrous ethanol for the preparation of amphiphilic polylactide–methacrylic block copolymers 3a and 3b, respectively. | ||
Subsequently, the ROP–RAFT dual agent 1 was used in the synthesis of block copolymers using both simultaneous one-step (Scheme 1b) and two-step (Scheme 1c) protocols. LA was used as the ROP monomer for the formation of all block copolymers (2, 3a and 3b). The resulting PLA homopolymer 3 was used as a macro-CTA in the second polymerization step (for the preparation of 3a or 3b). Various LA/1 molar ratios were used for the preparation of a PLAx–PDMAy block copolymer 2via simultaneous polymerization (Scheme 1b). An LA/1 molar ratio of 30 was also utilized for the synthesis of a PLA–POEGMA block copolymer 2 by the same simultaneous polymerization protocol (Scheme 1b). The same molar ratio was also used for PLA–PDMA 3a, PLA–PGMA 3a and PLA–PMPC 3b block copolymer syntheses by two-step polymerization (Scheme 1c). In all cases, the molar ratio of the RAFT monomer (DMA, OEGMA, GMA or PMPC) to 1 was kept constant at 30.
More specifically, for the synthesis of diblock copolymers by simultaneous ROP–RAFT polymerization (see Scheme 1b) either DMA or OEGMA was used as the RAFT monomer and 1,2-dichloroethane (bp 84 °C) as a solvent (55% w/w solids). A two-step process was required for the preparation of the diblock copolymer containing glycerol monomethacrylate (GMA) i.e. PLA–PGMA 3a. This was because the hydroxyl groups in the GMA monomer structure are capable of initiating unwanted ROP polymerization. Following the same two-step protocol (Scheme 1c), a PLA–PDMA block copolymer 3a was synthesized for comparison with its one-step synthetic process (Scheme 1b). Both steps were performed in 1,2-dichloroethane (36.0% and 55% w/w solids for first and second step, respectively). A two-step protocol was also required for the synthesis of the PLA–PMPC block copolymers. Such copolymers cannot be synthesized by simultaneous ROP–RAFT polymerization due to the insolubility of the MPC monomer in any suitable aprotic solvent for ROP. First, ROP of LA produced a PLA homopolymer 3 ([LA]0:[1]0 = 30 or 200) in 1,2-dichloroethane (36.0% w/w solids). Subsequently, MPC was employed as the RAFT monomer in the second step. Anhydrous ethanol (b.p. 78 °C, 20% w/w solids) was used for the synthesis of the PLA–PMPC block copolymers 3b (second step, Scheme 1c). Ethanol is a good solvent for both MPC monomer and PMPC. DMAP was selected as the catalyst for the ROP of LA instead of the more commonly-used catalyst stannous octoate (Sn(Oct)2) for this polymerization technique.6,7,16,17,46 DMAP can be used at lower temperatures,46 making it compatible with the RAFT polymerization conditions.24 2,2′-Azobis(isobutyronitrile) (AIBN) was used as the thermal initiator for RAFT polymerization. This initiator was utilized in both the one-step process 2 and the two-step process 3a ([1]0:[AIBN]0 = 1:0.20). ACVA was the initiator in the two-step process 3b ([1]0:[ACVA]0 = 1:0.25).
All reactions conducted in 1,2-dichloroethane (Scheme 1b and c polymers 3 and 3a) were performed at 74 °C for 24 h. The PLA–PMPC block copolymer synthesis 3b (Scheme 1c) was conducted in ethanol at 78 °C for 24 h. The monomer conversions were determined by 1H NMR analysis. Conversions were calculated based on the resonance intensities of the remaining unreacted monomers: LA at 5.03–5.10 ppm in CDCl3 (methine proton). DMA at 5.55 and 6.09 ppm, OEGMA at 5.52 ppm and 6.08 ppm in CDCl3, GMA at 5.66 and 6.05 ppm in d6-DMSO and MPC at 5.38 and 5.90 ppm in CDCl3/CD3OD (for four methacrylic protons for each monomer). As shown in Table 1, relatively high conversions were achieved for all monomers, ranging from 77% to 96% for the LA monomer and between 74% and 99% for the methacrylic monomers (DMA, OEGMA, GMA and MPC). The one-step synthesis of the PLA–POEGMA block copolymer gave the highest monomer conversions; 96% for LA and 99% for OEGMA (PLA29–POEGMA29, Table 1, entry 7). The corresponding monomer conversions for other block copolymers prepared by the one-step protocol using the same feed ratios ([LA]0:[RAFT monomer]0:[1]0 = 30:30:1) were 95% and 74% for PLA27–PDMA21 (Table 1, entry 8) and, 95% and >99% for PLA27–PGMA29 (Table 1, entry 9). For the PLA–PMPC block copolymer prepared by two-step polymerization (PLA25–PMPC25, Table 1, entry 11) the resulting conversions were 93% for LA (first step) and 85% for MPC (second step). In the one-step synthesis of PLAx–PDMAy block copolymers (Table 1, entries 1–6), the conversions of both LA and DMA monomers were not significantly affected by changing the feed ratio of LA monomer to 1 ([LA]0:[1]0 from 30 for entry 6 to 250 for entry 1). These values ranged from 77% to 93% for the LA monomer and between 79% and 97% for the DMA monomer.
All diblock copolymers were purified by dialysis against acetone. This was followed by methanol dialysis in the case of PLA–PGMA and PLA–PMPC block copolymers. This second dialysis step was essential for removal of any unreacted GMA or MPC monomer respectively, which are insoluble in acetone. After solvent evaporation, the dried block copolymers were characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC). The results are presented in Table 1, Fig. 1, 2 and 3.
The degrees of polymerization (DPs) calculated by 1H NMR (DPNMR) were found to be in agreement with the theoretical values (DPCalcd) calculated from the monomer feed ratios. These results suggest the well-controlled formation of block copolymers both for the one- and two-step protocols, except in the case where MPC was used as a monomer due to the partial insolubility of the PLA macro-CTA in ethanol, which was the polymerization solvent used for this RAFT synthesis. The molecular weight distributions (MWDs) of these copolymers were relatively narrow (Fig. 2 and 3): polydispersities ranged between 1.30 and 1.43 (see Table 1). The two PLA30–PDMA30 diblock copolymers had similar Mn (NMR) values of 8.1 kDa and 7.6 kDa when prepared by either the one-step (Table 1, entry 6) or two-step (Table 1, entry 8) protocols, respectively.
The MWD for the PLA30–PDMA30 diblock copolymer prepared by one-step polymerization was slightly narrower than that obtained via the two-step protocol (Fig. 3d). Polydispersities of 1.37 and 1.41 were obtained for the one-step and two-step protocols respectively (Table 1, entries 6 and 8). Thus using the more convenient protocol does not compromise the quality of the block copolymer. GPC analysis of PLA-based block copolymers using a refractive index detector is challenging because of the relatively low refractive index increment (dn/dc) of the aliphatic polyester component in common GPC eluents. For example, the dn/dc of PLA is 0.048 mL g−1 (ref. 70) in THF. This value is significantly lower than that for PMMA calibration standards (0.087 mL g−1 (ref. 71) in THF), which leads to inaccuracies in the GPC characterization of PLA-based block copolymers, particularly in the low molecular weight range.
Simultaneous ROP–RAFT polymerization using 1 was also utilized for the synthesis of PLA-based branched block copolymers. Here, either OEGMA or DMA was used as the RAFT monomer and DSDMA was selected as the cleavable cross-linker. A molar ratio of [LA]0:[RAFT monomer]0:[DSDMA]0:[1]0 = 30:30:1:1 was used to form a block copolymer with a linear PLA block and a branched statistical methacrylic block comprising OEGMA (or DMA) and DSDMA.
This synthetic route is presented in Scheme 2a. As shown in Fig. 4a, DMF GPC analysis (vs. PMMA calibration standards) of this disulfide-containing block copolymer indicated a relatively broad MWD (Mw/Mn = 2.36). This confirmed that the DSDMA comonomer had reacted not only intramolecularly but also intermolecularly,63,65 resulting in branching. Cleavage of the disulfide bonds (Scheme 2b) using excess Bu3P led to a much narrower MWD (Fig. 4a). The Mw/Mn was reduced from 2.36 to 1.37 for the resulting thiol-functionalized linear block copolymer PLA30–P(OEGMA30-stat-TEMA2) (where TEMA denotes 2-thioethyl methacrylate). Vinyl sulfone was reacted with the TEMA residues to afford 2-(2-(vinylsulfonyl)ethylthio)ethyl methacrylate (VSTEMA) units (Scheme 3a). For this thiol–ene reaction, a large excess of DVS (15 eq. relative to the thiol –SH group of TEMA) was used to ensure that only one of the DVS double bonds reacts with the thiol group. Thus possible inter- or intra-molecular cross-linking was avoided. The second double bond of the DVS thus remained unreacted and was utilized for further conjugation with thiol-containing molecules.72 The success of the reaction for the synthesis of the vinyl sulfone-functionalized amphiphilic block copolymer, PLA30–P(OEGMA30-stat-VSTEMA2), was confirmed by 1H NMR studies (see Fig. 4b and Fig. S1, ESI†). Specifically, in the 1H NMR (CDCl3) spectrum (Fig. S1†) the signal at 6.0–7.0 ppm is due to the pendant vinylsulfone protons. The signal at 2.75 ppm is assigned to the –OCH2CH2SCH2– protons. DMF GPC analysis of this copolymer indicates an Mw/Mn of 1.40, compared to 1.37 for the PLA30–P(OEGMA30-stat-TEMA2). This suggests that minimal branching occurred during RAFT copolymerization in this case. In principle, PLA30–P(OEGMA30-stat-VSTEMA2) can be reacted with thiol-functional oligopeptides, which can be useful for biological applications. This concept was tested in a model reaction using Glu (Scheme 3b). The conjugation reaction was performed under mild conditions (20 °C, 4 h, water). A Glu/VSTEMA molar ratio of 1.05 and a small amount of TCEP catalyst ([TCEP]0:[thiol]0 = 0.10) were used. The resulting PLA30–P(OEGMA30-stat-GluVSTEMA2) block copolymer had a similar Mn/Mw value (1.45 vs. 1.40) with the PLA30–P(OEGMA30-stat-VSTEMA2) precursor. However, the former copolymer had a much lower Mn (23600 g mol−1vs. 37800 g mol−1) (DMF GPC, Fig. 4a) than the latter copolymer. This is attributed to partial hydrolysis of the PLA block in water during the conjugation reaction and purification by dialysis. Successful Glu conjugation was confirmed by 1H NMR (CDCl3) since the new signal at 3.51 ppm is assigned to the –CH2CH(NH2)COOH proton. Also, in Fig. S2b (ESI†) the same peak was obtained at 3.48 ppm when D2O was used as an NMR solvent.
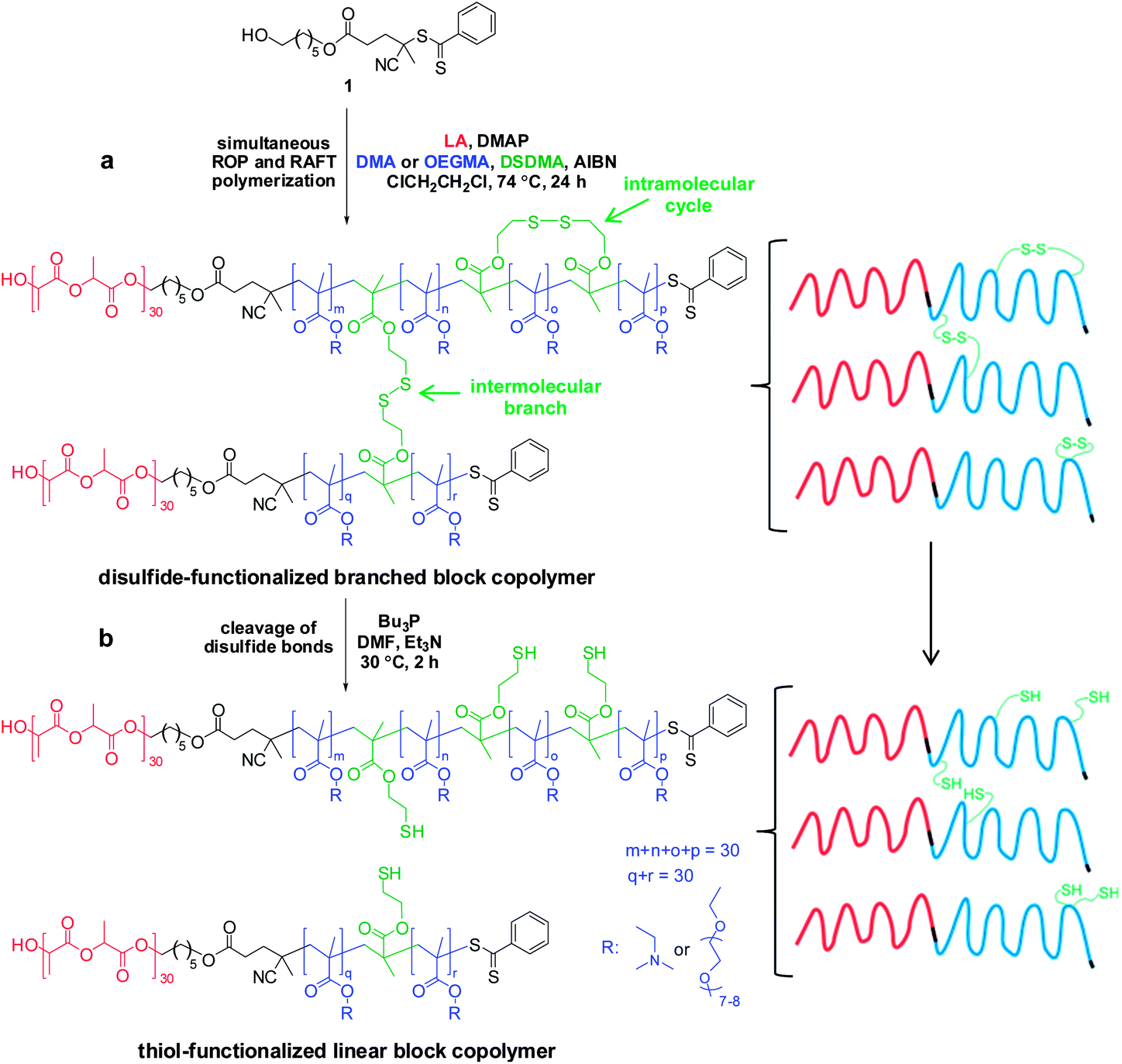 | ||
| Scheme 2 Preparation of thiol-functionalized block copolymers. (a) Synthesis of a disulfide-functionalized branched block copolymer by simultaneous ROP of LA and RAFT statistical copolymerization of OEGMA or DMA with DSDMA using a dual ROP–RAFT reagent 1 at 55% w/w solids, at 74 °C for 24 h. Polymerization conditions: [LA]0:[R]0:[DSDMA]0:[ROP–RAFT reagent]0 relative molar ratios 30:30:1:1. (b) Reductive cleavage of the disulfide bonds in the methacrylic block using Bu3P in DMF at 30 °C for 2 h, results in the formation of linear statistical block copolymer containing thiol groups that can be used for further functionalization. Reaction conditions: [Bu3P]0:[Et3N]0:[disulfide bond]0 relative molar ratios 3.0:2.1:1. | ||
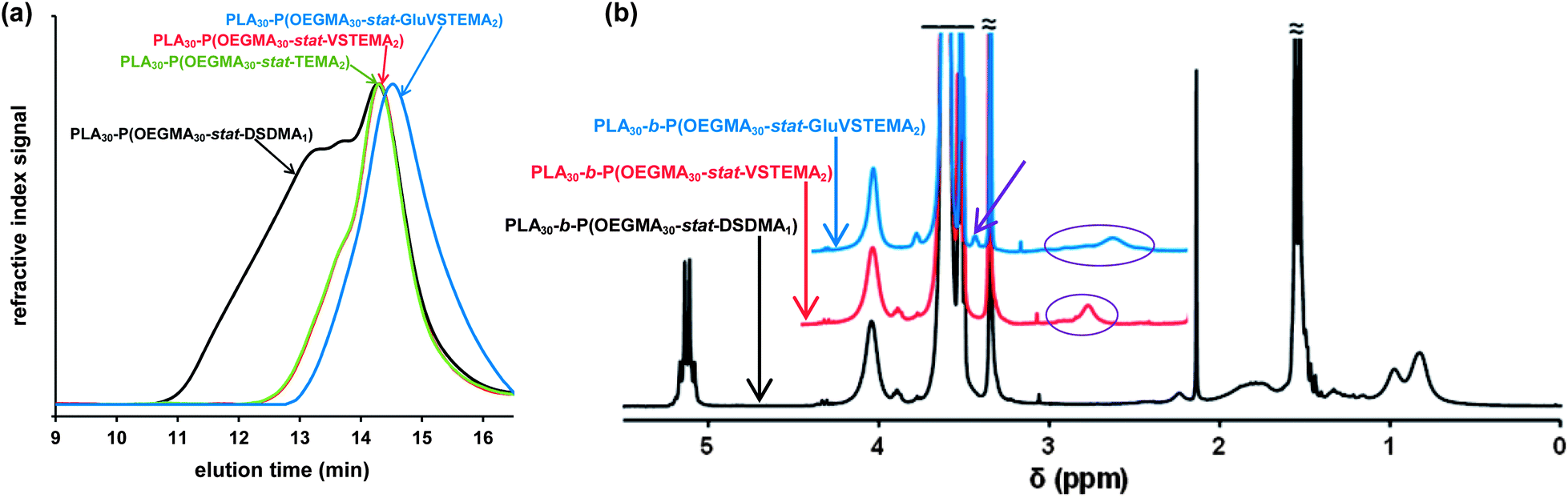 | ||
| Fig. 4 (a) DMF GPC curves vs. poly(methyl methacrylate) standards and (b) 1H NMR spectra (right; recorded in CDCl3) obtained for the PLA30–P(OEGMA30-stat-DSDMA1) branched block copolymer, the PLA30-b-P(OEGMA30-stat-TEMA2) linear block copolymer obtained after disulfide cleavage using tributyl phosphine, the PLA30–P(OEGMA30-stat-VSTEMA2) linear block copolymer after functionalization with divinyl sulfone and the PLA30-b-P(OEGMA30-stat-GluVSTEMA2) after L-glutathione (Glu) conjugation (LA = lactide; OEGMA = oligo(ethylene glycol) methacrylate); DSDMA = disulfide-based dimethacrylate; TEMA = 2-thioethylmethacrylate; VSTEMA = 2-(2-(vinylsulfonyl)ethylthio)ethyl methacrylate, GluVSTEMA = glutathione conjugated 2-(2-(vinylsulfonyl)ethylthio)ethyl methacrylate). | ||
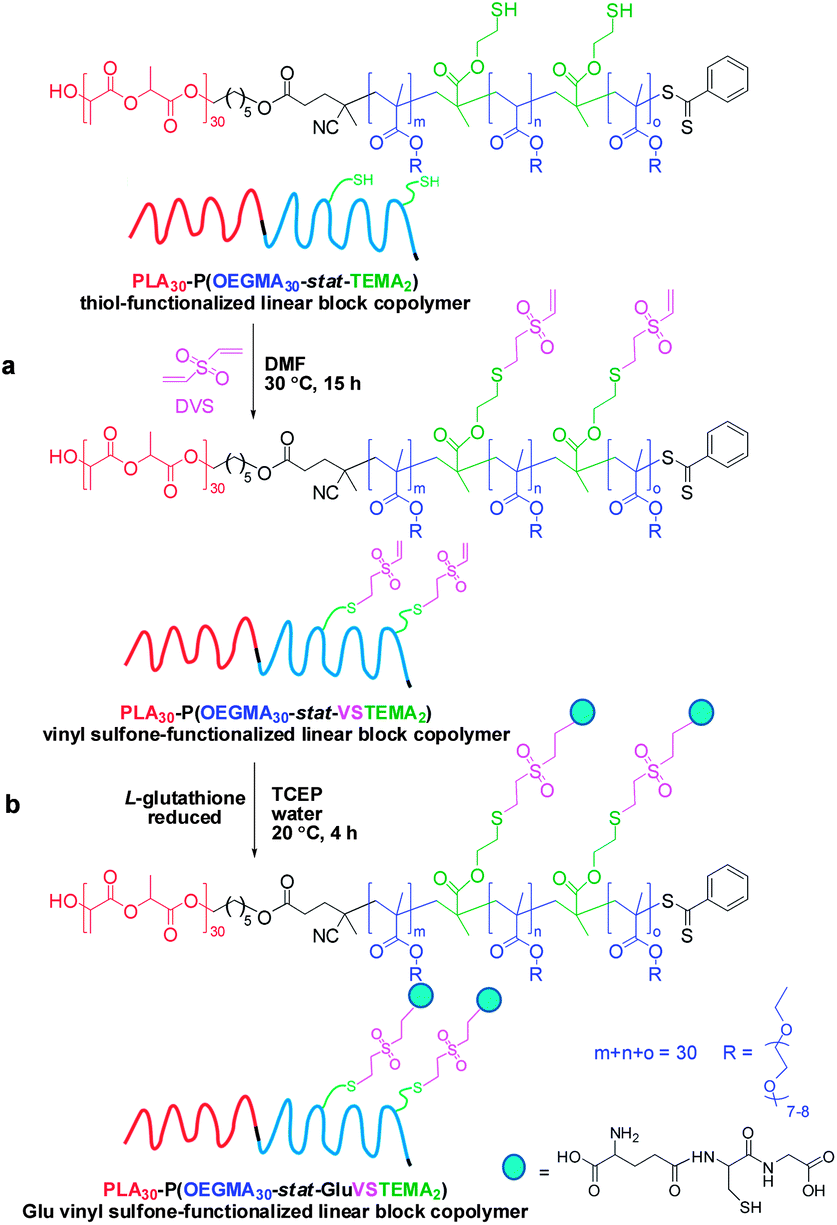 | ||
| Scheme 3 Preparation of a L-glutathione-conjugated linear block copolymer, PLA30–P(OEGMA30-stat-GluVSTEMA2). (a) Synthesis of a vinyl sulfone-functionalized linear block copolymer PLA30–P(OEGMA30-stat-VSTEMA2) by in situ conjugation of excess divinyl sulfone to the thiol-functionalized linear block copolymer, PLA30–P(OEGMA30-stat-TEMA2). Reaction conditions: DMF, 30 °C, 15 h, [DVS]0:[thiol]0 relative molar ratio 15:1. (b) Conjugation of glutathione to the vinyl sulfone-functionalized linear block copolymer PLA30–P(OEGMA30-stat-VSTEMA2). Reaction conditions: water, room temperature, 4 h, [TCEP]0:[thiol]0 molar ratio = 0.1. | ||
A gel network was obtained when DMA was used as a comonomer for the synthesis of a disulfide-containing branched amphiphilic block copolymer, PLA30–P(DMA30-stat-DSDMA1) (Scheme 2a). Thus characterization of this precursor copolymer using GPC or 1H NMR was not feasible. Gel formation indicates a significantly higher degree of branching than for the analogous reaction using OEGMA instead of DMA. A viscous solution rather than a gel was obtained from the synthesis of the PLA30–P(OEGMA30-stat-DSDMA1) branched block copolymer. Presumably, the sterically congested nature of the OEGMA residues (vs. DMA residues) hinders intermolecular cross-linking. Subsequently, the disulfide bonds of the PLA30–P(DMA30-stat-DSDMA1) branched block copolymer were reduced using the same protocol as described above for the OEGMA-based branched block copolymer (Scheme 2b). This resulted in the formation of a thiol-functionalized PLA30–P(DMA30-stat-TEMA2) block copolymer, which was functionalized with a DOTA ligand via thiol–ene chemistry. More specifically, excess maleimido-monoamide-DOTA (2.1 eq. relative to disulfide bonds) was used to convert the TEMA residues to DOTATEMA residues (Scheme 4a) to produce a PLA30–P(DMA30-stat-DOTATEMA2) block copolymer. The DOTA is a well-studied macrocyclic that can be used for metal conjugation. It is known to form very stable metal complexes.73–75 The high electron density of heavy metals enables high resolution imaging of the block polymer in transmission electron microscopy (TEM) studies, as discussed below. Indium was used to form a complex with the DOTA-functionalized polymer. This was achieved by using excess InCl3 (2.0 eq. relative to disulfide bonds, Scheme 4b). Successful metal complexation was confirmed by inductively-coupled plasma atomic emission spectroscopy (ICP-AES). It was calculated that there were approximately 1.16 In atoms per PLA30–P(DMA30-stat-InDOTATEMA2) block copolymer chain. Self-assembly of the PLA27–PGMA29 block copolymer during its synthesis in 1,2-dichloroethane was examined by TEM. Due to the insolubility of the PGMA block in this polymerization solvent the block copolymer self-assembled in situ to form various nanoparticles. Micelles and vesicles with diameters ranging from 50 to 500 nm were observed (see Fig. S4†).
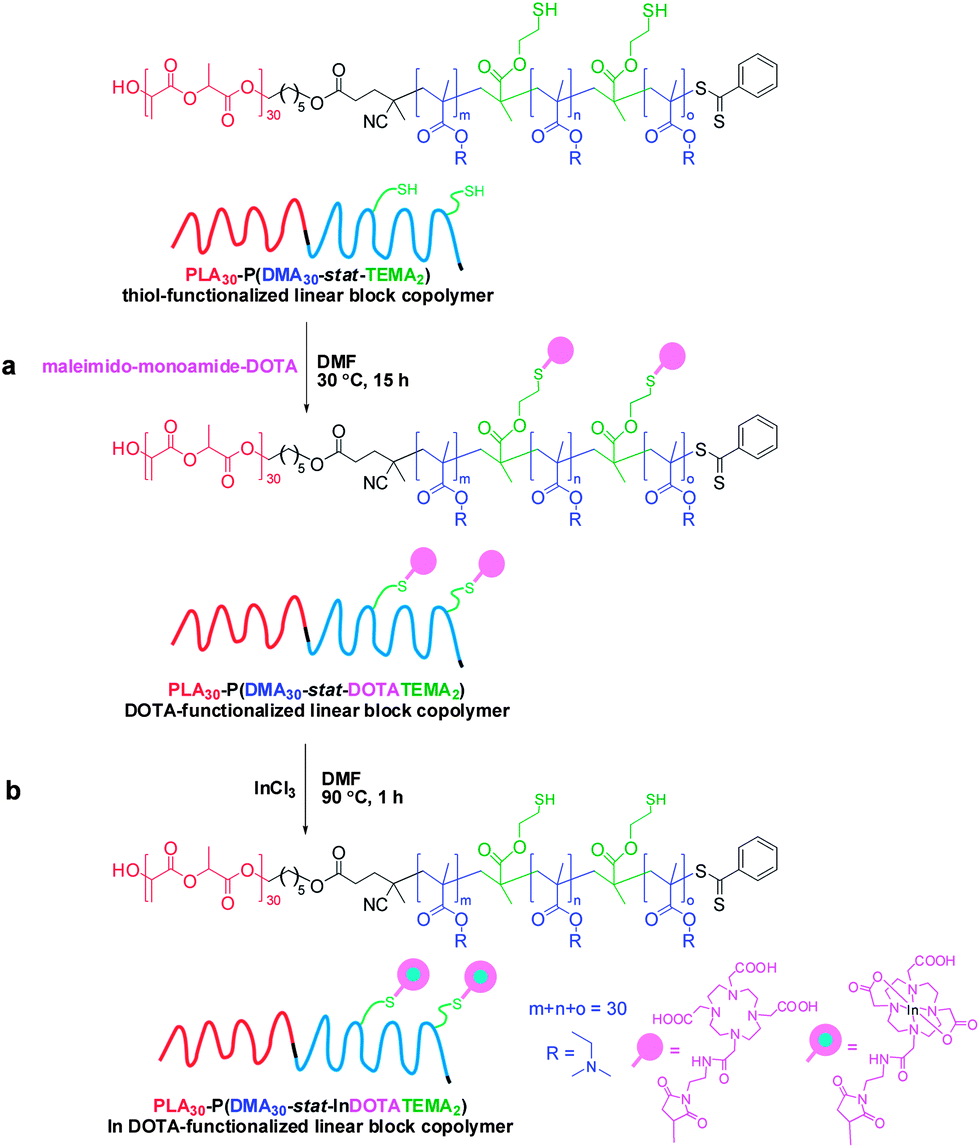 | ||
| Scheme 4 Preparation of an In-conjugated linear block copolymer, PLA30–P(DMA30-stat-InDOTATEMA2). (a) Synthesis of a DOTA-functionalized linear block copolymer, PLA30–P(DMA30-stat-DOTATEMA2), by in situ conjugation of maleimido-monoamide-DOTA to the thiol-functionalized linear block copolymer, PLA30–P(DMA30-stat-TEMA2). Reaction conditions: DMF, 30 °C, 15 h, maleimido-monoamide-DOTA (2.1 eq. relative to disulfide bonds). (b) Conjugation of indium to the PLA30–P(DMA30-stat-DOTATEMA2) block copolymer. Reaction conditions: DMF, 90 °C, 1 h, InCl3 (2.0 eq. relative to disulfide bonds). | ||
Self-assembly of all PLA-based amphiphilic block copolymers to form various nanostructures in water was obtained by the solvent switch method. Table 1 summarizes DLS hydrodynamic diameters observed for aqueous copolymer dispersions after removal of the organic solvent. TEM images for representative block copolymer samples are displayed in Fig. 5. Addition of water to an acetone solution of copolymer was used for the self-assembly of PLAx–PDMAy and PLA29–POEGMA29 block copolymers. A solvent switch from acetone/methanol to water was used for the PLA27–PGMA29 and from chloroform/methanol to water for the PLAx–PMPCy block copolymers. In the case of the PLAx–DMAy block copolymers, varying the PLA block DP led to different morphologies (Fig. 5a–f). Hydrodynamic diameters of these nanoparticles ranged between 113 nm and 685 nm (Table 1, entries 1 to 6). In the case of the highly asymmetric PLA173–PDMA24, vesicles make up the majority of nanoparticles (214 nm, Table 1 entry 1), but a small number of micelles were also formed (Fig. 5a). The micelle fraction increased as the DP of the PLA was reduced (from Fig. 5a–f). For the PLA26–PDMA25 block copolymer, micelles are the major population (Fig. 5f). Various nanostructures (vesicles, worm-like micelles and spherical micelles) were observed for PLA29–POEGMA29 block copolymer (Fig. 5g). The TEM image of this sample also suggested partial crystallization of the PLA block, similar to previous studies on PLA–PEG block copolymers and blends.76–78 The same sample had a hydrodynamic diameter of 550 nm (Table 1 entry 7) and a high polydispersity. The solvent switch protocol for the PLA27–PGMA29 block copolymer resulted in a mixture of large vesicles (338 nm, Table 1 entry 9) and some micellar structures. The highly asymmetric PLA184–PMPC23 block copolymer self-assembled to produce a mixture of vesicles (1430 nm, Table 1 entry 10), worm-like micelles and spherical micelles as judged by TEM (Fig. 5i).
 | ||
| Fig. 5 TEM images obtained using a solvent switch protocol for the following diblock copolymer particles prepared by a combination of ROP and RAFT polymerizations: acetone to water solvent switch protocol was used for (a) PLA173–PDMA24; (b) PLA137–PDMA27; (c) PLA58–PDMA27; (d) PLA48–PDMA28; (e) PLA41–PDMA30; (f) PLA26–PDMA25; (g) PLA29–POEGMA29; (h) acetone/methanol (1/1 v/v) to water for PLA27–PGMA29; (i) chloroform/methanol (3/1 v/v) to water for PLA184–PMPC23; (j) acetone to water for PLA30–P(DMA30-stat-InDOTATEMA2). Uranyl formate was used as the positive staining agent for samples a–f, h and i. Phosphotungstic acid was the positive staining agent for sample g. No staining was used for sample j. | ||
Following the same solvent switch protocol, the self-assembly of the In-labelled PLA30–P(DMA30-stat-InDOTATEMA2) was examined. For this sample, nanostructures with a mean DLS diameter of 265 nm were observed. These morphologies could be imaged by TEM (Fig. 5j and Fig. S4a†) without requiring a staining agent. This was possible because of the high electron density of the heavy metal conjugated to the copolymer chains.
Conclusions
A combination of two “living” polymerization techniques via either one or two steps was achieved using a novel hydroxyl-functionalized dithiocarbonate-based ROP–RAFT dual agent. Simultaneous polymerization of an aliphatic ester and a methacrylic monomer for the one-pot synthesis of amphiphilic block copolymers has been established using a facile metal-free formulation. This was achieved by ROP of LA (catalyzed using DMAP) and RAFT polymerization of DMA or OEGMA. In contrast, a two-step approach was essential for the synthesis of PLA–GMA and PLA–PMPC block copolymers. The simultaneous ROP–RAFT polymerization protocol was also utilized for the facile synthesis of new branched amphiphilic block copolymers, which serve as precursors for thiol-functionalized linear block copolymers. For this latter synthesis, the disulfide cross-linking agent DSDMA was employed to incorporate latent thiol groups. Cleavage of the disulfide bonds was performed under reducing conditions, resulting in thiol-functionalized linear block copolymers. These biodegradable block copolymers can be conjugated with molecules such as peptides or ligands for heavy metals using facile thiol–ene chemistry, which suggests potential biomedical applications.Acknowledgements
EPSRC is gratefully acknowledged for funding a postdoctoral fellowship to support E.T. (EP/I001697/1). Biocompatibles UK Ltd. is also thanked for supplying the MPC monomer. Dr Jeppe Madsen is acknowledged for his helpful advice regarding copolymer functionalization.Notes and references
- O. W. Webster, Science, 1991, 251, 887–893 CrossRef CAS PubMed.
- M. Barz, F. K. Wolf, F. Canal, K. Koynov, M. J. Vicent, H. Frey and R. Zentel, Macromol. Rapid Commun., 2010, 31, 1492–1500 CrossRef CAS PubMed.
- J. Z. Du and S. P. Armes, Langmuir, 2008, 24, 13710–13716 CrossRef CAS PubMed.
- J. Z. Du and S. P. Armes, Langmuir, 2009, 25, 9564–9570 CrossRef CAS PubMed.
- J. Z. Du and S. P. Armes, Soft Matter, 2010, 6, 4851–4857 RSC.
- M. Hales, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Langmuir, 2004, 20, 10809–10817 CrossRef CAS PubMed.
- D.-H. Han and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 789–799 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6396–6408 CrossRef CAS.
- C. Lefay, D. Glé, M. Rollet, J. Mazzolini, D. Bertin, S. Viel, C. Schmid, C. Boisson, F. D'Agosto, D. Gigmes and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 803–813 CrossRef CAS.
- J. Li, J. Ren, Y. Cao and W. Yuan, Polymer, 2010, 51, 1301–1310 CrossRef CAS PubMed.
- A. K. Mishra, V. K. Patel, N. K. Vishwakarma, C. S. Biswas, M. Raula, A. Misra, T. K. Mandal and B. Ray, Macromolecules, 2011, 44, 2465–2473 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- A. O. Saeed, S. Dey, S. M. Howdle, K. J. Thurecht and C. Alexander, J. Mater. Chem., 2009, 19, 4529–4535 RSC.
- C. Schmid, J. Falkenhagen and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1–10 CrossRef CAS.
- P.-J. Shi, Y.-G. Li and C.-Y. Pan, Eur. Polym. J., 2004, 40, 1283–1290 CrossRef CAS PubMed.
- Y. You, C. Hong, W. Wang, W. Lu and C. Pan, Macromolecules, 2004, 37, 9761–9767 CrossRef CAS.
- K. V. Bernaerts and F. E. Du Prez, Prog. Polym. Sci., 2006, 31, 671–722 CrossRef CAS PubMed.
- S. M. Howdle, C. J. Duxbury, W. X. Wang, M. de Geus and A. Heise, J. Am. Chem. Soc., 2005, 127, 2384–2385 CrossRef PubMed.
- S. M. Howdle, J. X. Zhou, S. Villarroya, W. X. Wang, M. F. Wyatt, C. J. Duxbury and K. J. Thurecht, Macromolecules, 2006, 39, 5352–5358 CrossRef.
- H. U. Kang, Y. C. Yu, S. J. Shin, J. Kim and J. H. Youk, Macromolecules, 2013, 46, 1291–1295 CrossRef CAS.
- H. U. Kang, Y. C. Yu, S. J. Shin and J. H. Youk, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 774–779 CrossRef CAS.
- M. Le Hellaye, C. Lefay, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3058–3067 CrossRef CAS.
- Y. K. Li, E. Themistou, J. Zou, B. P. Das, M. Tsianou and C. Cheng, ACS Macro Lett., 2012, 1, 52–56 CrossRef CAS.
- K. Matyjaszewski and W. Jakubowski, Macromol. Symp., 2006, 240, 213–223 CrossRef.
- D. Mecerreyes, G. Moineau, P. Dubois, R. Jerome, J. L. Hedrick, C. J. Hawker, E. E. Malmstrom and M. Trollsas, Angew. Chem., Int. Ed., 1998, 37, 1274–1276 CrossRef CAS.
- M. Nasser-Eddine, C. Delaite, G. Hurtrez and P. Dumas, Eur. Polym. J., 2005, 41, 313–318 CrossRef CAS PubMed.
- T. Ozturk, M. Goktas and B. Hazer, J. Appl. Polym. Sci., 2010, 117, 1638–1645 Search PubMed.
- M. Seo, C. J. Murphy and M. A. Hillmyer, ACS Macro Lett., 2013, 2, 617–620 CrossRef CAS.
- K. J. Thurecht, A. M. Gregory, S. Villarroya, J. Zhou, A. Heise and S. M. Howdle, Chem. Commun., 2006, 42, 4383–4385 RSC.
- S. Villarroya, J. Zhou, K. J. Thurecht and S. M. Howdle, Macromolecules, 2006, 39, 9080–9086 CrossRef CAS.
- C. Cheng, E. Khoshdel and K. L. Wooley, Macromolecules, 2007, 40, 2289–2292 CrossRef CAS.
- Y. C. Yu, G. Li, H. U. Kang and J. H. Youk, Colloid Polym. Sci., 2012, 290, 1707–1712 CAS.
- T. Endo, General Mechanisms in Ring-Opening Polymerization, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier, J. -M. Raquez, Wiley-VCH, Weinheim, Germany, 2009, pp. 53–63 Search PubMed.
- D. Bourissou, O. Dechy-Cabaret and B. Martin-Vaca, Chem. Rev., 2004, 104, 6147–6176 CrossRef PubMed.
- K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Controlled Ring-Opening Polymerization: Polymers with Designed Macromolecular Architecture, in Adv. Polym. Sci.: Degradable Aliphatic Polyesters,Springer-Verlag, Berlin: Heidelberg, New York, 2002, 157, pp. 41–65 Search PubMed.
- G. Moad, J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- M. H. Stenzel, Complex Architecture Design via the RAFT Process: Scope, Strengths and Limitations, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008, pp. 315–372 Search PubMed.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- G. Battaglia and A. J. Ryan, J. Phys. Chem. B, 2006, 110, 10272–10279 CrossRef CAS PubMed.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- C. M. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510–3513 CrossRef CAS PubMed.
- J. Madsen, S. P. Armes, K. Bertal, H. Lomas, S. MacNeil and A. L. Lewis, Biomacromolecules, 2008, 9, 2265–2275 CrossRef CAS PubMed.
- L. Wang, C. M. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566–1570 CrossRef CAS.
- J. C. Gaulding, M. H. Smith, J. S. Hyatt, A. Fernandez-Nieves and L. A. Lyon, Macromolecules, 2012, 45, 39–45 CrossRef CAS PubMed.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS PubMed.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- N. J. Warren, C. Muise, A. Stephens, S. P. Armes and A. L. Lewis, Langmuir, 2012, 28, 2928–2936 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., Hoboken, New Jersey, 2004 Search PubMed.
- Y. T. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- J. Rosselgong and S. P. Armes, Macromolecules, 2012, 45, 2731–2737 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145–2156 CrossRef CAS.
- J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar, J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1, 1041–1045 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- J. Zou, C. C. Hew, E. Themistou, Y. K. Li, C. K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS.
- J. R. McKee, V. Ladmiral, J. Niskanen, H. Tenhu and S. P. Armes, Macromolecules, 2011, 44, 7692–7703 CrossRef CAS.
- S. Olivier and R. Walkenhorst, Matériaux, 2002, 1–5 Search PubMed.
- D. W. van Krevelan and K. te Nijenhuis, Properties of Polymers: Their Correlation with Chemical Structure; their Numerical Estimation and Prediction from Additive Group Contributions, Elsevier Science, Slovenia, 2009 Search PubMed.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS PubMed.
- C. Wu, F. Virzi and D. J. Hnatowich, Nucl. Med. Biol., 1992, 19, 239–244 CAS.
- J. Fichna and A. Janecka, Bioconjugate Chem., 2003, 14, 3–17 CrossRef CAS.
- S. Liu and D. S. Edwards, Bioconjugate Chem., 2000, 12, 7–34 CrossRef.
- P. Cerrai, M. Tricoli, L. Lelli, G. D. Guerra, R. S. Delguerra, M. G. Cascone and P. Giusti, J. Mater. Sci.: Mater. Med., 1994, 5, 308–313 CrossRef CAS.
- N. Kang, M. E. Perron, R. E. Prud'homme, Y. B. Zhang, G. Gaucher and J. C. Leroux, Nano Lett., 2005, 5, 315–319 CrossRef CAS PubMed.
- S. Y. Lee, I. J. Chin and J. S. Jung, Eur. Polym. J., 1999, 35, 2147–2153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional 1H NMR spectra and TEM images obtained for these amphiphilic block copolymers. See DOI: 10.1039/c3py01446k |
| ‡ Present address: School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK. E-mail: E-mail: e.themistou@qub.ac.uk |
| This journal is © The Royal Society of Chemistry 2014 |