 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An enzymatic biomimetic system: enhancement of catalytic efficiency with new polymeric chiral ionic liquids synthesised by controlled radical polymerisation†
Erno
Karjalainen
a,
Diana F.
Izquierdo
b,
Vicente
Martí-Centelles
b,
Santiago V.
Luis
b,
Heikki
Tenhu
*a and
Eduardo
García-Verdugo
*b
aUniversidad Jaume I, Departamento de Química Inorgánica y Orgánica, Campus del Riu Sec, E-12071 Castellón, Spain. E-mail: cepeda@uji.es
bLaboratory of Polymer Chemistry, Department of Chemistry, University of Helsinki, Finland. E-mail: heikki.tenhu@helsinki.fi
First published on 2nd December 2013
Abstract
The controlled radical polymerization of monomeric units containing chiral ionic liquids (CILs) allows the synthesis of intrinsically chiral polymers through a bottom-up design. These polymeric chiral ionic liquids (PCILs) show a well-defined three dimensional structure organized as the result of a complex non-covalent network of hydrogen-bonding contacts driven by the C2 hydrogen atoms of the imidazolium aromatic rings and the OH moieties present in the side functionalities of the main polymeric chain. The exchange of the chloride counter ion by the L-prolinate anion leads to new types of polymeric catalysts, which behave as efficient artificial aldolase biomimetic systems, being highly active and selective for the aldol reaction in water. These new polymeric catalysts are significantly more active than the corresponding monomeric counterpart when the reaction is performed either in water or in the presence of water. The increase in catalytic efficiency can be related to their 3D structure, displaying helical chirality in the polymeric chain as a function of their preparation methodology. Under suitable experimental conditions, these polymers are able to catalyse the consecutive aldol-dehydration process, behaving as synthetic mimics of the aldolase-dehydrogenase enzymatic system. Moderate enantioselectivities can be achieved under suitable conditions.
Introduction
Enzymes are increasingly used as biocatalysts for the production of fine and speciality chemicals.1 An important feature of enzymes is that they can act as bifunctional or even multifunctional catalysts, being able to catalyse cascade reactions with tremendously high catalytic efficiency and selectivity.2 Hence, enzymes have been seen as inspiration for the development of new synthetic catalytic systems.3 For this purpose, it should be borne in mind that although only certain groups, which are present in the catalytic site, are responsible for the catalysis, it is the three dimensional structure of these natural macromolecules which defines and explains their unique chemical reactivity. Different polymeric systems have been used to mimic the enzymatic catalytic behaviour.4 However, the preparation of a polymeric analogue for an enzyme is not a simple task. The structure of the polymer should provide a suitable chain architecture for locating the functional groups in such a way that the adopted three-dimensional structures pre-organize the functional groups for an efficient catalysis.5A suitable design of linear polymeric systems, prepared by controlled polymerization, especially those involving free radical polymerizations, seems to be a good biomimetic strategy for allowing large synthetic fragments to be located in close proximity and with the appropriate geometry and structure of the corresponding functional groups. Indeed, it is well known that different backbone structures can give rise to well-defined secondary and tertiary structures based on supramolecular interactions thus leading to organized systems such as micelles, rods, etc.6 The nature of the monomeric unit used as a structure-directing agent is a key parameter for their design. When the monomeric units are chiral molecules, it is possible to develop functional self-assembled systems that not only mimic nature, but also provide a cheap and simple route to complex multifunctional structures.7 In this regard, different examples of highly organized chiral polymers have been reported.8
A very specific element for the organisation of the final polymeric structure is the use of monomers based on ionic liquids (ILs), leading to polymers containing all beneficial properties of ILs (PILs).9 Thus, the preparation of functional nanostructured materials or composites based on ILs represents an exciting and significant area of opportunity for developing multifunctional, hierarchically structured materials.10 As in the case of ILs, one important advantage of PILs is their potential for displaying a large level of chemical and structural diversity and, hence, physicochemical characteristics. These can be readily tuned by selecting the adequate combination of the organic cation and the inorganic or organic anion.9 Thus, the large potential structural diversity of IL-units plays an important role in defining the final properties of PILs.11 Among them, chirality can be considered as one of the most interesting elements. Accordingly, a great deal of effort has been expended in the last few years to design and synthesize chiral ionic liquids (CILs).12 We have reported a simple and robust modular synthetic strategy that leads to a large variety of configurationally and structurally diverse CILs.13 The in-depth study of these CILs has allowed us to analyse in detail the structure–property relationships present in these compounds.13
Here, we reported the development of self-organized complex multifunctional materials based on some CILs that are able to imitate, at least to some extent, natural complex systems such as the enzymatic ones. The presence of chiral IL-like units, along with the use of controlled radical polymerization (CRP) techniques, leads to polymeric macromolecules with a well-defined 3D structure. A simple exchange of the initial chloride anion by the L-proline anion yields new types of architectures, being able to behave as efficient artificial biomimetic systems.
Results and discussion
Synthesis of PCILs by different strategies
We have recently developed a general chemoenzymatic methodology to produce optically active trans-2-imidazoyl-cycloalkanols, by the nucleophilic opening of cycloalkene oxides (1) with imidazole (2) followed by the enzymatic kinetic resolution of the corresponding racemic alcohols (Scheme 1).13 In a similar way to that reported for the synthesis of N-benzylic imidazolium salts 9 and 10, the alkylation of either the racemic ((±)-3) or the enantiopure alcohol or ((+)-(S,S)-4) with 4-cholorovinylbenzene (6) leads to the corresponding monomeric imidazolium salts with good yields (Scheme 1, 84% for (±)-7 and 85% for (+)-(S,S)-8).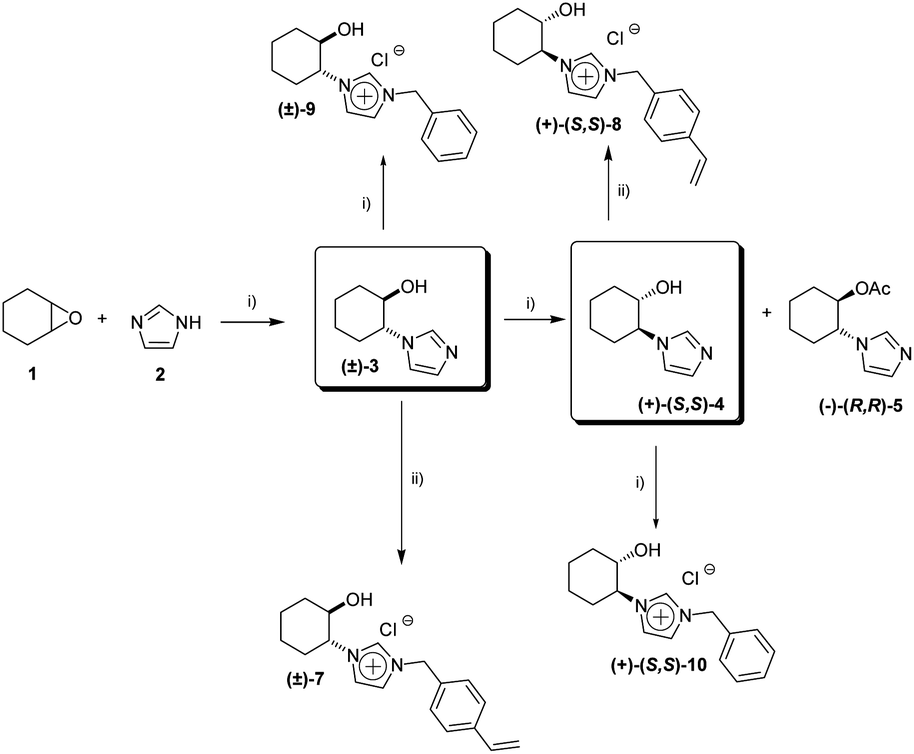 | ||
| Scheme 1 Synthesis of CIL monomeric species. (i) ref. 13. (ii) p-chlorovinylbenzene (6), ACN, reflux, 24 hours. | ||
Initially, the corresponding racemic and enantiopure monomers ((±)-7 and (+)-(S,S)-8) were polymerized by atom transfer radical polymerization (ARGET ATRP, Scheme 2).14 High levels of monomer conversion were observed by 1H-NMR after 14–19 hours. However, a discrepancy between the expected and the obtained molecular weight was found, especially for the chiral monomer (+)-(S,S)-8 (entries 1 and 2, Table 1). One possible explanation may be the involvement of the imidazolium monomer as a ligand for the copper species during the polymerisation process. Indeed, a change of colour, from blue to yellow, was observed when the copper solution was added to the monomer solution, which can be associated with the formation of a Cu–carbene complex. ILs have recently been used as solvents and ligands for the AGET ATRP of acrylonitrile, showing the active role of imidazolium salts for this type of polymerization.15
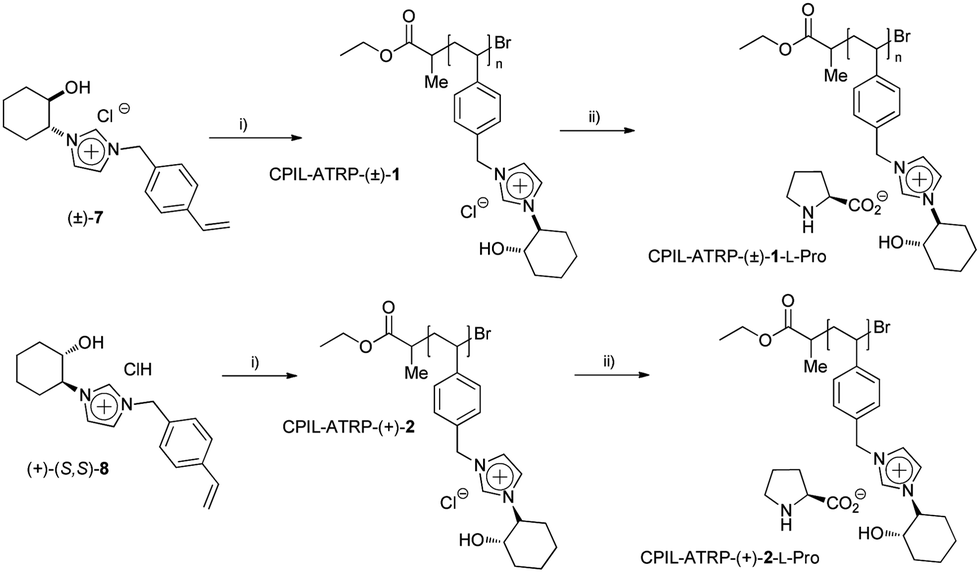 | ||
Scheme 2 ARGET-ATRP-polymerizations of CIL-monomers ((±)-7 and (+)-(S,S)-8). (i) 60 °C, Et2PrBr, Cu0, CuBr2/tris(2-dimethylaminoethyl)amine (Me6TREN), [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[CuBr2]:[L] 50:1:1:2. (ii) Amberlyst-OH, L-proline. :[I]:[CuBr2]:[L] 50:1:1:2. (ii) Amberlyst-OH, L-proline. | ||
| Entry | Polymer | Precursor | Time (h) | Conv.b (%) | M n(theor) (kg mol−1) | M n(exp) (kg mol−1) | PDI | [α]Df |
|---|---|---|---|---|---|---|---|---|
|
a ARGET-ATRP-polymerizations of CIL-monomer; 50:1:1:2 [M]:[I]:[CuBr2]:[L] molar ratio, I = ethyl 2-bromopropanoate (Et2PrBr), L = tris(2-dimethylaminoethyl)amine (Me6TREN), 60 °C in DMF.
b Calculated by 1H-NMR (MeOD).
c RAFT-polymerizations of CIL-monomer; 50:1:0.2 [M]:[CTA]:[I] molar ratio, 100 °C in 1:1 DMSO–water; CTA = (4-cyanopentanoic acid)-4-dithiobenzoate (CPA), I = azobiscyanopentanoic acid (ACPA).
d Determined by MALDI-TOF.
e Prepared by grafting from the polymer PClSt-1 and the imidazole (±)-3 or (+)-4 in a 1:1.2 ratio in DMF–H2O 2:1 at 80 °C.
f
c = 20 mg mL−1, MeOH, 25 °C.
|
||||||||
| 1 | PCIL-ATRP-(±)-1a | (±)-(S,S)-7 | 14 | 98 | 16.0 | 8.7 | 1.05 | — |
| 2 | PCIL-ATRP-(+)-2a | (+)-(S,S)-8 | 19 | 94 | 15.7 | 55.0 | 1.23 | 15.0 |
| 3 | PCIL-RAFT-(±)-3c | (±)-(S,S)-7 | 24 | 87 | 15.1 | 17.5 | 1.03 | — |
| 4 | PCIL-RAFT-(+)-4c | (+)-(S,S)-8 | 22 | 59 | 12.1 | 14.6 | 1.03 | 25.3 |
| 5 | PCIL-RAFT-graft-(±)-5e | (±)-(S,S)-3 | 27 | 99 | 8.0 | 5.4 | 1.13 | — |
| 6 | PCIL-RAFT-graft-(+)-6e | (+)-(S,S)-4 | 24 | 99 | 8.0 | 5.5 | 1.13 | 20.7 |
Alternatively, the application of reversible addition–fragmentation chain transfer (RAFT) polymerization was also evaluated (Scheme 3). A 1:1 DMSO–water mixture was used as the solvent in order to solubilise simultaneously both the monomer and the polymer formed (entries 3 and 4, Table 1).16 Although a lower conversion was observed for the enantiopure monomer than that obtained for the racemic one, a good control of the polymerization process was achieved, the polydispersity indices (PDI < 1.1) being low.
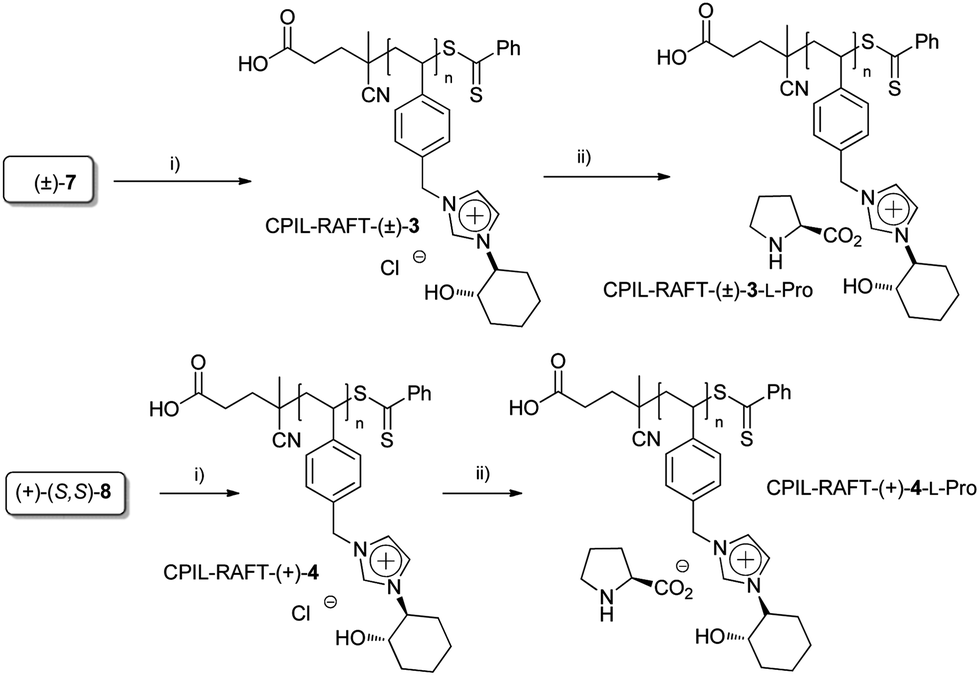 | ||
| Scheme 3 Synthesis of PCILs by RAFT polymerization. (i) Molar ratio [M]:[CTA]:[I] 50:1:0.2, 100 °C in 1:1 DMSO–water; CTA = (4-cyanopentanoic acid)-4-dithiobenzoate (CPA), I = azobiscyanopentanoic acid (ACPA). (ii) Amberlyst-OH, L-proline. | ||
The homopolymerization characteristics of the chiral monomer (+)-(S,S)-8 were also investigated. Fig. 1a depicts the monomer conversion calculated by 1H-NMR vs. reaction times. As could be expected, at short reaction times the polymerization shows a fast reaction rate, which is slowed down as the reaction time increases. Thus, initially a not well controlled polymerization reaction is observed with high concentrations of primary radicals being formed. However, when the reaction time increases, the polymerization control is established and the reaction proceeds with predictable molecular weights and low polydispersity indices (PDI < 1.1, Fig. 1b).
 | ||
| Fig. 1 Kinetic plots for the RAFT polymerization of ((+)-(S,S)-8). 50:1:0.2 [M]:[I]:[CTA] in 1:1 DMSO–H2O at 100 °C. (a) Conversion vs. time; (b) ln[M0]/[M] vs. time. | ||
As can be seen in Fig. 2, the number-average molecular weights (Mn(exp) (g mol−1)) are in good agreement with the theoretical values (Mn(theo) (g mol−1)) for polymerization degrees higher than 40%. The slight experimental molecular weight discrepancy may be attributed to the termination of intermediate radicals in the early stages of the reaction, the presence of polymers terminated by combination or the efficiency of the CTA being less than 1.17
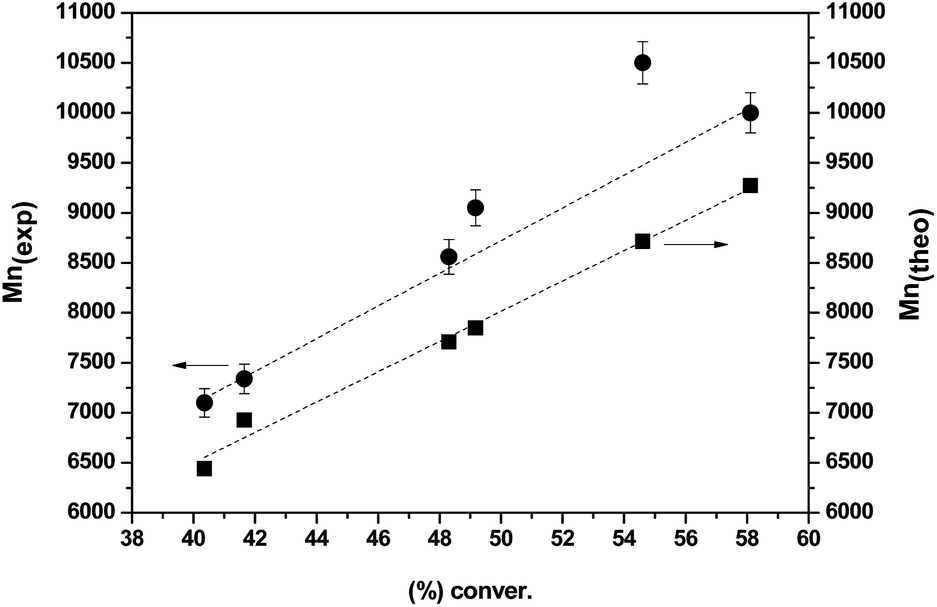 | ||
| Fig. 2 Experimental (circles) and theoretical (squares) molecular weights as a function of conversion for the RAFT-polymerization of (±)-7. 50:1:0.2 [M]:[I]:[CTA] molar ratio in 1:1 DMSO–H2O at 100 °C. | ||
Finally, the polymers PCIL-RAFT-graft-(±)-5 and PCIL-RAFT-graft-(+)-6 were prepared by chemical modification (grafting) of the corresponding poly-4-chloromethylstyrene (PClSt-1), prepared by RAFT polymerization. The synthesis of PClSt-1 was performed by bulk polymerization of p-chloromethylstyrene (6), at 100 °C for 24 h, using a 100:1:0.1 [M]:[CTA]:[I] molar ratio, with IBA as the chain transfer agent. The corresponding polymer was obtained with a 31% polymerization degree, 3.8 kg mol−1 being the molecular weight calculated by size exclusion chromatography and 1.13 its polydispersity.
The corresponding PCILs were prepared by substitution of the chloride groups by either the racemic ((±)-3) or the enantiopure ((+)-4) imidazoles. This chemical modification can be easily monitored by 1H-NMR. The 1H-NMR (DMSO-d6) of the polymer PCIL-RAFT-graft-(+)-6 isolated after the reaction of polymer PClSt-1 with the imidazole (+)-4 at 80 °C in 2:1 DMF–H2O shows a clear downfield shift of the benzylic signals (from 4.6 to 5.5 ppm) suggesting the substitution of the chloride group close to the benzylic protons by the more electronegative imidazolium fragment (see Fig. SI1 in the ESI†.). Furthermore, new proton signals assignable to the imidazolium units can also be observed at 3.6 ppm (CH–OH), 4.14 ppm (CH–N), 7.7 ppm and 10 ppm (imidazolium C4H/C5H and C2H respectively). The quantitative conversion of the CH2–Cl groups was also confirmed by ATR-FTIR and Raman spectroscopy.18 The comparison, for instance, of the Raman spectra of these polymers showed the disappearance of the bands at 1268 cm−1 and 678 cm−1 assignable to the C–Cl bond and the appearance of a new series of bands (e.g. 434.8, 488.8, 563.8, 801.8, 850.8, 1121.84, 1430.84, 1449.8 cm−1) associated with the presence of the imidazolium unit in the modified polymer CPIL-RAFT-graft-(+)-6 (see Fig. SI2 in the ESI†).18
The thermal stability of the new polymeric species was also studied by thermogravimetric analyses (see Fig. SI3†). In general, all polymers prepared showed good thermal stability, being stable up to 200 °C. The polymers decompose in two processes, the first being most likely the cleavage of the side chains at the benzylic position and the second one the depolymerization of the polystyrene-like main chain. It should be mentioned that polymers prepared by grafting start their decomposition at slightly higher temperatures than polymers prepared by direct polymerization.
Chiroptical properties and the secondary structure of chiral poly(ionic liquids)
The chiroptical properties of the polymers prepared using enantiopure imidazoles were also studied. It is expected that the presence of the chiral imidazolium units in the para-substituted styrene along with the polymerization method employed may influence both the chirality and the stereoregularity of the macromolecules formed. Indeed, the specific rotation values obtained in methanol (see Table 1) for the polymers were always higher than those of the chiral momoner ([α]D = 10.1°, c = 20 mg mL−1 in MeOH at 25 °C). The specific rotation value of the polymer PCIL-RAFT-(+)-4 prepared by RAFT polymerization of the chiral monomer is significantly higher ([α]D = 25.3°, c = 20 mg mL−1 in MeOH at 25 °C) than the one for PCIL-ATRP-(+)-2 obtained by ATRP polymerization ([α]D = 15.0°, c = 20 mg mL−1 in MeOH at 25 °C). It is noteworthy that the PCILs synthesized by chemical modification of a preformed polymer (PCIL-RAFT-graft-(+)-6) afforded a lower specific rotation value ([α]D = 20.0°, c = 20 mg mL−1 in MeOH at 25 °C) than the analogous PCILs prepared by polymerisation of the chiral monomer. The circular dichroism (CD) spectra of these polymers were obtained to further explore the effect of the chiral imidazolium units on the secondary structures of the main polymeric chain. Fig. 3 depicts the CD spectra of the PILs prepared either by polymerization of the chiral monomer ((+)-(S,S)-8) or by grafting of the enantiopure imidazole ((+)-4) on an already preformed poly(p-chlorostyrene) (PClSt-1). All these enantiopure PCILs presented CD signals that exhibit a negative maximum at ca. 200 nm followed by a positive one at ca. 225 nm (positive Cotton effect). The bands of the UV-vis spectra in this region correspond to the benzene chromophore of the polystyrene backbone. It must be noted that neither the enantiopure chiral monomer ((+)-(S,S)-8) nor, as it is logic, the unmodified poly(p-chloromethylstyrene) shows any noticeable CD signals. The fact that all the PCILs present a clear Cotton effect evidences that the presence of the hydrogen-bonding interaction involving the side-chain groups is arranging the main polymeric chain in a chiral orientation. The relative intensities of the CD-signals observed for the different polymers studied can be used as a probe to monitor the different degree of polymer organisation and the main-chain chirality induced by the type of methodology used for their synthesis.8 Hence, the polymer prepared by RAFT polymerization presents the highest degree of helical chain conformation. Most likely, the RAFT polymerisation leads to a more stereoregular polymer with a better defined secondary structure. The polymer prepared by chemical modification, attending its CD-spectra, shows clearly a much lower degree of helical conformation, suggesting that a certain degree of chirality is transferred to the main-chain during the polymerization process, although the ATRP polymerization seems not to be very efficient in this regard. | ||
| Fig. 3 CD spectra of PCIL-ATRP-(+)-2 (black dots), PCIL-RAFT-(+)-4 (red line) and PCIL-RAFT-graft-(+)-6 (blue dashed line) in MeOH (0.032 mg mL−1). | ||
The crystallographic data obtained for the related imidazolium salts ((+)-(S,S)-10) have shown that the protons of the cationic component tend to form strong H-bonds with the anionic component.13 Similar interactions between side groups in the polymer, mainly involving the –OH group, the imidazolium hydrogen atoms and the anions, are responsible for providing a well-defined secondary structure. In good agreement with the former results, some differences can be noted for the 1H-NMR of the polymers prepared either by grafting or by polymerisation. Thus, the signals corresponding to the OH group showed a slight downfield shift (to ca. 5.8 ppm) as well as those of the imidazole protons (C4–H and C5–H at ca. 7.8 ppm and C2–H at ca. 10 ppm) for the polymer PCIL-RAFT-(+)-4 in comparison with PCIL-RAFT-graft-(+)-6 suggesting a stronger network of interactions for the polymer prepared by polymerisation (see Fig. SI5†).
We have also performed theoretical molecular modelling calculations (MMFF) in order to try to understand the driving interactions responsible for the secondary structure of the polymeric chain.19 Both, experimental data and calculations, strongly support the stabilization effect of the OH⋯anion hydrogen-bonding interactions, although secondary interactions between the adjacent imidazolium units (such as additional H bonding, mainly through C2–H and C4–H; π–π or C–H⋯ π interactions involving aromatic rings) may also contribute to the organisation of the polymeric chain. The interactions between the side chain groups are likely to fix the orientation of the chain giving place to the emergence of the observed Cotton effect ascribed to the exciton-coupling of the side-chain neighbouring groups arranged in a mutual chiral orientation in a well-defined polar environment (see Fig. 4).
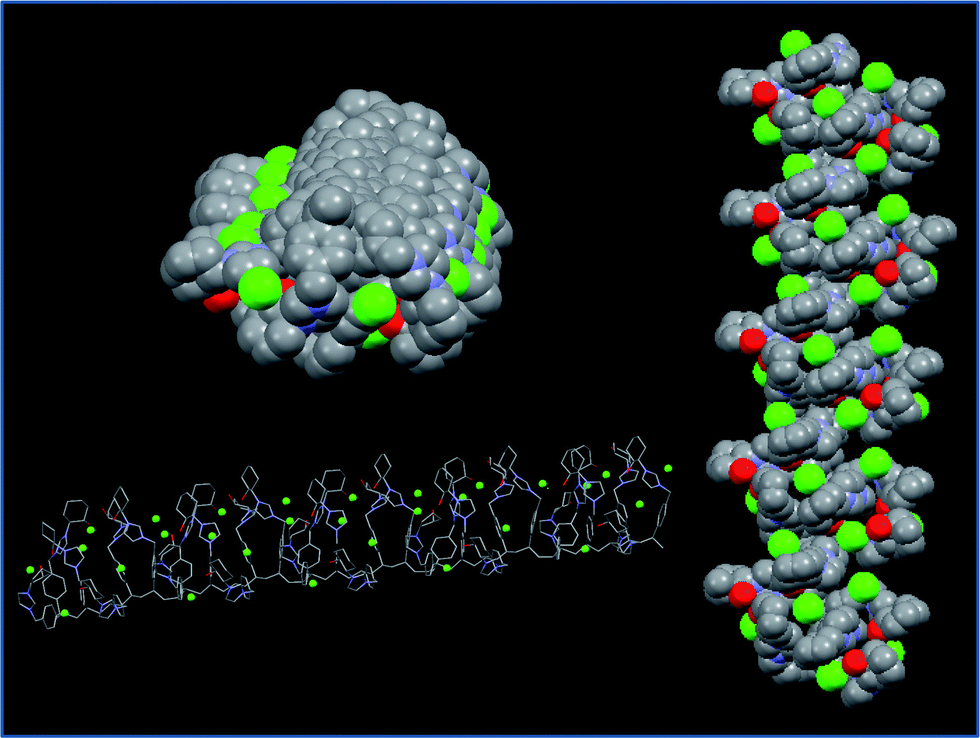 | ||
| Fig. 4 Optimised structure obtained from computational calculations for the polymer prepared from the enantiopure monomer (+)-(S,S)-8. | ||
Organocatalytic properties of the PCIL-L-proline polymers
The aldol reaction is an effective method for the formation of carbon–carbon bonds in organic synthesis. Both, stoichiometric and catalytic methods, are known for the synthesis of aldol products with controlled stereochemistry.20 Biological methodologies, such as aldolase enzymes21 and catalytic antibodies,22 have also been extensively developed for accomplishing aldol reactions with high efficiency and selectivity. Alternatively, several small organic molecule catalysts, based on L-proline and related derivatives, have also been shown to act as “aldolase” mimics under mild conditions.23 Different efforts have been devoted to develop an efficient strategy for the immobilization of organocatalysts onto different supports.24,25 In general, the immobilization methodology is based on the development of side-chain or end-functionalized polymeric catalysts. Good results have been obtained when L-proline is bonded to crosslinking PS-DVB resins by click chemistry.26 Alternatively, well-defined copolymers of styrene and L-proline-functionalized styrene, being able to self-assemble into well-defined aggregates, have also been reported.27,28 Fewer efforts have been made, however, to develop homopolymeric L-proline organocatalysts, which may adopt a 3D structure leading to more efficient catalysts than the non-supported catalysts. Depending on the 3D structure adopted by the linear polymer, one may anticipate a synergistic effect on the outcome of the catalytic reaction. Furthermore, the possibility of using water in place of organic solvent(s) is also an important feature to be considered in order to add another element of “greenness” to the process, resembling more closely enzymatic systems.29 Taking into account all these considerations, the PILs previously prepared could be easily converted into L-proline derivatives by simple counterion exchange by the treatment of the aqueous solution of the corresponding polymer with a polymer supported base (Amberlite-OH) followed, after filtration of the insoluble resins, by neutralization with a slight excess of L-proline. This procedure afforded, after dialysis, different PILs/PCILs having the amino acid carboxylate as the counter ion.30 The related non-polymeric analogues were also prepared according to the same procedure. The L-proline PCILs were analysed by 1H-NMR, ATR-IR and Raman spectroscopy. These analyses indicated the complete exchange of the chloride ions by L-prolinate.Catalysis in water as the solvent
The L-proline-PCIL derivatives were tested as catalysts for the reaction between p-nitrobenzaldehyde (11) and acetone (12) in aqueous media without the addition of organic solvents. This process is often used in the literature as an aldolic organocatalytic benchmark reaction (Scheme 4).23 The reaction was performed in a 0.1 M solution of aldehyde in nanopure water with ten equivalents of acetone and 40 mol% loading of the corresponding polymeric catalyst at room temperature for 20 h. The results obtained are summarised in Table 2. | ||
| Scheme 4 Synthesis of PCILs by RAFT polymerization and grafting. (i) Molar ratio [M]:[CTA]:[I] 1000:10:1, 100 °C; CTA = 2-cyano-2-propyl dodecyl trithiocarbonate, I = azobis(isobutyronitrile) (AIBN). (ii) 0.34 M of (±)-3 in DMF–MeOH 2:1, 80 °C for 25 hours. (iii) 0.34 M of (+)-(S,S)-4 in DMF–MeOH 2:1, 80 °C for 25 hours. (iv) Methyl imidazole in DMF–MeOH 2:1, 80 °C for 25 hours. | ||
| Entry | Catalyst | Conv.b (%) | (13)b (%) | (14)b (%) | eec (%) |
|---|---|---|---|---|---|
|
a 1:10:0.4 RCHO–acetone–catalyst molar ratio, room temperature, 20 hours; 0.1 M of aldehyde in H2O–acetone 13:1 (v/v), 15 mg mL−1.
b Calculated by 1H-NMR from the crude of the reaction.
c Enantiomeric excess calculated by HPLC.
|
|||||
| 1 | L-Proline | <5% | — | — | — |
| 2 | (+)-10L-Pro | <5% | — | — | — |
| 3 | (±)-9 | <5% | — | — | — |
| 4 | PCIL-ATRP-(+)-2 | 41 | 38 | 3 | 0 |
| 5 | PCIL-ATRP-(±)-1-L-Pro | 96 | 45 | 51 | — |
| 6 | PCIL-ATRP-(+)-2-L-Pro | 96 | 26 | 70 | 0 |
| 7 | PCIL-RAFT-(+)-4 | 15 | 15 | 0 | 0 |
| 8 | PCIL-RAFT-(±)-3-L-Pro | 97 | 76 | 21 | — |
| 9 | PCIL-RAFT-(+)-4-L-Pro | 96 | 57 | 39 | 0 |
As expected, the reaction catalysed by L-proline, under these conditions, produced less than 5% of aldehyde conversion. Aldolase-type organocatalytic reactions are typically performed in organic solvents, such as DMSO, DMF, or chloroform, where the addition of a small amount of water often accelerates reactions and/or improves enantioselectivities.31 However, those organocatalytic reactions generally result in very poor yield and stereoselectivity when performed in bulk water.32 Both the L-prolinate imidazolium salt ((+)-10-L-Pro) and the related chloride imidazolium salt ((±)-9) did not show any significant catalytic activity.33 The polymers PCIL-ATRP-(+)-2 and PCIL-RAFT-(+)-4 (not bearing proline units, entries 4 and 7 in Table 2) were also then tested as potential catalysts for this benchmark reaction. Both polymers showed some catalytic activity (40 and 15% conversion respectively). It is noteworthy that the polymeric derivatives bearing L-proline units were finally able to consume almost quantitatively the aldehyde, showing, thus, better activity than the related monomeric species. In all the cases, different amounts of the dehydrated product (14) were observed. The degree of dehydration seems to be related to both the enantiopurity of the polymeric backbone (compare entries 5 and 6 or 8 and 9 in Table 2, for racemic vs. enantiopure) and the polymerisation method used in the polymer preparation (ATRP vs. RAFT, compare entries 6 and 9). Unfortunately, unlike native aldolases or aldolase antibodies, when the process was carried out in bulk water these aldolase-type organocatalytic reactions resulted in a very poor stereoselectivity, the aldol being obtained as a racemic mixture (Scheme 5).
 | ||
| Scheme 5 Aldol benchmark reaction. | ||
Our results also highlight the very remarkable importance of the nature of the polymeric backbone to enhance the catalytic activity. The monomeric unit (±)-9, without L-proline moieties, is not capable of catalysing the aldol reaction, while the related polymers (Table 2, entries 3, 4 and 7) showed moderate catalytic activity. In these cases, the reaction should occur through a base catalysed process, as in the absence of L-proline an enamine mechanism is ruled out. In order to assess the relative basicity of the polymers and the monomeric counterparts, a pH indicator dye (bromothymol blue), which allows a naked-eye analysis, was used. The non-polymeric imidazolium salt ((±)-9) does not produce any change in the colour of the aqueous solution (the yellow colour is indicative of a non-basic solution) while the polymer (PCIL-ATRP-(+)-2) turned the solution blue. Thus, the first reason for the improved catalytic behaviour of the polymers is that they are stronger bases than their monomeric analogues. The increase in basicity of the polymeric systems can be based on the cooperative action of adjacent basic residues located in proximity to the polymeric structure.34 A strong stabilization of the resulting conjugated acid through H-bonding interactions with anions or the hydroxyl groups, suitably orientated in the microenvironment of the base as a consequence or the organized 3D structure of the main polymeric chain, could then occur. This cooperative assistance can be considered similar to the proton relay systems observed in enzymatic catalysis.35 On the other hand, the organized polymeric structure can also implement the activation process of both the nucleophile and the electrophile when the prolinate anion is present. Most likely, adjacent prolinate groups can participate in the catalytic process, one of them activating the nucleophile via enamine formation, and the adjacent one activating the electrophile via hydrogen bond formation.36 The additional groups present in the main polymeric chain (imidazolium and –OH group) can also be involved in the activation process, forming hydrogen bonds with the aldehyde or with water molecules. These additional interactions, which are not possible in the monomeric counterpart, will reduce the energy of the transition state in the C–C bond formation step.
To analyse the system in more detail, the kinetic profiles of aldol appearance for (+)-10-L-Pro and PCIL-ATRP-(±)-1-L-Pro were obtained (Fig. 5). The amount of catalysts was reduced to 10 mol% and a ratio of 5:1 H2O–acetone was used. Once again, the non-polymeric catalyst presented a very slow reaction rate. Yields of aldol lower than 10% were achieved even for prolonged reaction times (>50 hours). In contrast, the polymeric catalyst showed a very efficient catalytic behaviour, yielding 89% aldol in only 30 min. This means that the polymeric catalyst provides a highly efficient process, highlighting the important contribution of the macromolecular structure to the catalytic activity.
 | ||
| Fig. 5 Aldol yield (%) vs. time for the aldol reaction catalysed by PCIL-ATRP-(±)-1-L-Pro (red dots) or (+)-10-L-Pro (black squares). 7:3 H2O–acetone; 1:10:0.4 RCHO–acetone–cat molar ratio; 0.357 M; 17 mg polymer per mL of H2O–acetone. Each point corresponds to the yield of a single reaction after isolation and analysis by 1H-NMR. | ||
The effect of catalyst concentration was also evaluated. The concentration of catalyst PCIL-ATRP-(±)-1-L-Pro was varied from 85 to 13 mg of polymer per mL of water, using the standard catalytic conditions, with the aldehyde–acetone–catalyst molar ratio being kept constant (1:10:0.4). The results obtained after 20 hours are summarized in Fig. 6.
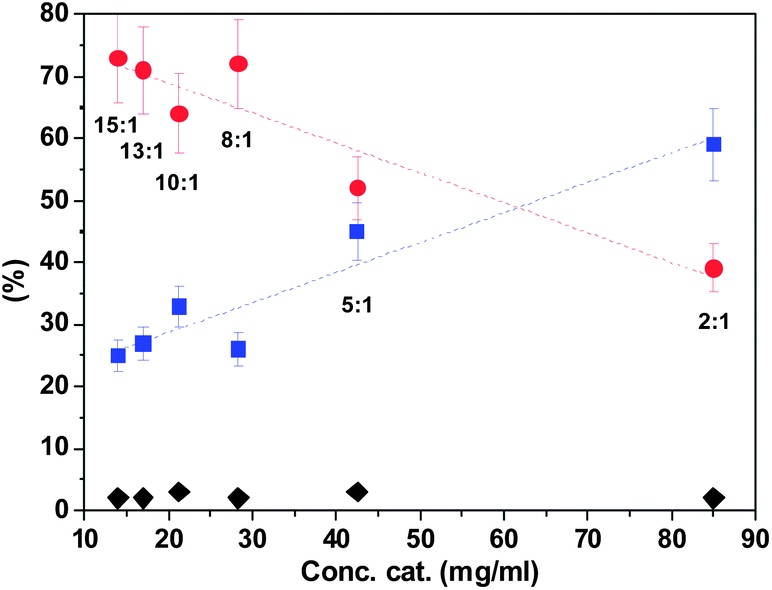 | ||
| Fig. 6 Effect of catalyst concentration on the product distribution. 1:10:0.4 RCHO–acetone–cat molar ratio, ratio H2O–acetone (v/v) is indicated in the graph. 24 hours, 25 °C. Catalyst: PCIL-ATRP-(±)-1-L-Pro. Black diamonds: aldehyde 11; red dots: aldol 13; blue squares: dehydrated compound 14. Each point corresponds to the yield of a single reaction after isolation and analysis by 1H-NMR. | ||
For all the cases assayed, the conversion of the aldehyde was almost quantitative and different amounts of the dehydration product were found, along with the aldol product, depending on the concentration of the catalyst. Consistently, an increase in the catalyst concentration led to a higher dehydration degree. It is noteworthy that the enantioselectivity of the aldol compound varied with the catalyst concentration antiparallel to the formation of the dehydrated compound. The lower catalyst concentration (15 mg polymer per mL) provided the higher enantioselectivity, reaching a ca. 65% ee for the (S) enantiomer, along with the lower amount of dehydrated compound (25%). This could be indicative of an enantioselective dehydration of the R-aldol obtained in the former condensation step. When a reaction was performed with PCIL-ATRP-(+)-2-L-Pro (15 mg mL−1) the reaction led to 60% yield of the aldol with a 76% ee (S) and 40% of the dehydration product. These results were partly in contradiction with our previous experiences, where the aldol 13 was obtained as a racemic mixture. Thus, a control experiment was carried out using the same polymer (15 mg mL−1) but for a shorter time (two hours). A 98% yield for the racemic aldol was found with no trace of dehydration product. Then, the aldol was extracted from the aqueous reaction media with dimethylether and the polymer dried and re-suspended in an aqueous solution of the same concentration of the aldol in the absence of additional acetone. After 20 hours, the organic products were extracted from the aqueous phase and analysed. The mixture contained 56% of aldol with 86% ee (S enantiomer) and 44% of dehydrated product. Thus, these results reveal that the C–C bond formation is extremely fast and not enantioselective, while the dehydration process is much slower and takes place enantioselectively. Both consecutive processes are catalysed by the polymers, as only minor amounts of the dehydrated product were found, under the same experimental conditions, in the absence of any polymer. In this regard, our polymeric systems mimic the tandem aldol condensation/dehydration. Such a tandem process has already been reported for D-aminoacylase in the presence of different N-heterocycles.37
Catalysis in acetone in the presence of water
Under the experimental reaction conditions reported above, in water, the catalyst is soluble, but not the aldehyde nor the corresponding aldol. However, by increasing the amount of acetone (from acetone–H2O 1:13 to 4:1 v/v), all components form a single phase allowing to monitor the reaction evolution by 1H-NMR. This allows obtaining more easily the concentration profiles vs. time for the different catalytic systems and calculating the reaction rate constants for the aldol reaction (pseudo-first order kinetics). The obtained results are summarised in Table 3. All the polymers show a significant improvement in catalytic efficiency when compared with their monomeric counterpart. The chiral enantiopure polymeric catalyst prepared by RAFT polymerisation (PCIL-RAFT-(+)-4-L-Pro) was the most effective one with a ca. 6.6 × 103 fold acceleration over the relative monomeric organocatalyst (entries 1 and 5, Table 3). The importance of the chiral organisation of the polymeric backbone is highlighted by the decrease of the relative activity (1.4 × 103vs. 6.6 × 103) observed for the polymer obtained by RAFT polymerisation of the racemic monomer (±)-7 (entries 4 and 5, Table 3). The lower relative efficiency of the polymers obtained by chemical modification of a preformed chloropolystyrene confirmed the importance of the polymerisation process in the bottom-up approach to generate 3D highly organised and efficient catalysts. The polymers prepared by ATRP showed a lower activity than those synthesised by RAFT, although similar trends were also present. Thus, the catalyst with an enantiopure polymeric backbone was more active than the racemic one (entries 2 and 3, Table 3). In general, our catalytic results and the CD-studies previously discussed suggest that, during the polymerisation process, a certain degree of chiral transference to the main polymeric chain can occur, leading to a more organised polymeric structure and to a more efficient catalyst. Hence, the importance of how the macromolecule is built-up from the related monomeric units is clearly highlighted. The right self-assembling of the monomeric units generates a highly ordered structure with a complex hierarchical architecture able to mimic the behaviour of an aldose in terms of high catalytic efficiency in comparison with the corresponding monomeric building blocks, although unfortunately no enantiopreference was observed for the different polymers assayed. It should be mentioned that the monomer analogue (+)-10-L-Pro only led to a modest 35% ee (R).
| Entry | Catalyst | k | k rel. |
|---|---|---|---|
|
a 1:30:0.1 RCHO–acetone–catalyst molar ratio, 0.38 M solution, 24 mg polymer per mL, r.t., acetone-d6–H2O 4:1.
|
|||
| 1 | (+)-10-L-Pro | 0.00883 | 1 |
| 2 | PCIL-ATRP-(±)-1-L-Pro | 2.4 | 289 |
| 3 | PCIL-ATRP-(+)-2-L-Pro | 13.2 | 1590 |
| 4 | PCIL-RAFT-(±)-3-L-Pro | 11.28 | 1359 |
| 5 | PCIL-RAFT-(+)-4-L-Pro | 55.26 | 6658 |
| 6 | PCIL-RAFT-graft-(±)-5-L-Pro | 7.2 | 867 |
| 7 | PCIL-RAFT-graft-(+)-6-L-Pro | 5.7 | 687 |
| 8 | PCIL-RAFT-graft-(+)-7-L-Pro | 9.6 | 1108 |
According to the important effect observed for the chiral polymeric backbone on the reaction rates, the behaviour of D-prolinate as the counter anion was studied to investigate any match/mismatch effect between the chiral components. Thus, the kinetic profiles for the model reaction catalysed by 5 molar% of either PCIL-ATRP-(+)-2-L-Pro or PCIL-ATRP-(+)-2-D-Pro were obtained. The aldehyde was converted to the corresponding racemic aldol even when low molar loadings of the catalyst were used. Nevertheless, both catalysts led to similar reaction rates suggesting the absence of any match/mismatch effect between the polymeric cation and the chiral anion. Taking into account this result, no further analysis of the presence of match/mismatch effects was carried out with the other systems.
The activity enhancement is most likely to be attributed to the non-covalent network of hydrogen-bonding contacts leading to a well-defined three dimensional polymeric structure. Fig. 7 shows the structures obtained by molecular mechanics calculations for the polymers obtained from (+)-(S,S)-8 and (+)-(S,S)-8-L-Pro. It can be seen how the optimised models present a significant variation in their conformations after the exchange of chloride by L-prolinate. It can be observed how the structure with L-prolinate has a tighter helical conformation than the polymer with chloride that displays an almost linear structure. Besides, the imidazolium units in CILP-(+)-(S,S)-8-L-Pro are located around the main polymeric chain also in an helical arrangement, while in the structure calculated for the polymer derived from (+)-(S,S)-8 these units are located only in one side of the main polymeric chain (Fig. 7). Finally, it can be observed how pyrrolidine fragments can be located in close proximity (ca. 4.5–6 Å for N–N distances). This arrangement of the proline residues could support their cooperative effect.
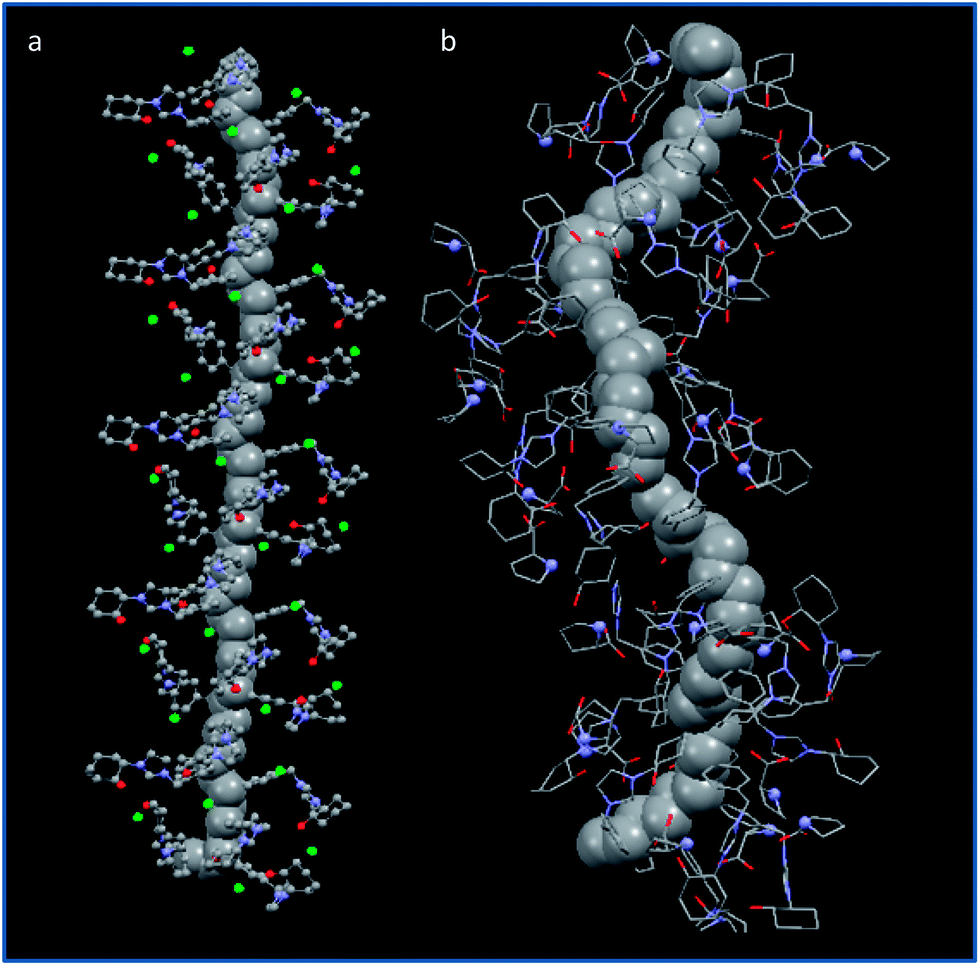 | ||
| Fig. 7 Optimised structures for the polymers built-up from the enantiopure monomers (a) (+)-(S,S)-8 and (b) (+)-(S,S)-8-L-Pro. The carbons of the main polymeric chain are represented in space filling for a better view. | ||
Catalysis in polar organic solvents in the absence of water
Finally, the use of polar anhydrous organic solvents was evaluated (Table 4). As expected, the solvent has a significant influence on the reaction outcome. Thus, in polar aprotic solvents (acetone and DMSO) both high yield and good enantioselectivity for the aldol were found (entries 1 and 2, Table 4). The use of an organic protic polar solvent like MeOH, able to disrupt, to a large extent, the ion-pairing and the hydrogen-bonding of the CIL ((+)-10-L-Pro), leads to a significant reduction in yield and enantioselectivity (entry 3, Table 4) in comparison with aprotic solvents. Regarding the polymeric catalysts, when aprotic polar solvents were assayed, low yields were obtained for PCIL-ATRP-(+)-2-L-Pro (entries 1 vs. 4 and 2 vs. 5, Table 4), while PCIL-RAFT-(+)-4-L-Pro yielded the aldol almost quantitatively. Both polymeric catalysts gave better yields than the homogeneous counterpart when the reaction was assayed in MeOH. This activity enhancement is in good agreement with the results found in water and in the presence of water. Regarding the enantioselectivity, the polymers led to lower chiral induction. The stronger basicity of the polymeric catalysts in comparison with their monomeric analogue may well account for the enantioselectivity reduction. The nature of the polymer is once again the determining factor. Thus, the polymer prepared by RAFT polymerisation (PCIL-RAFT-(+)-4-L-Pro) produced a moderate enantioselectivity (50% ee), while the one polymerised by ATRP (PCIL-ATRP-(+)-2-L-Pro) gave almost racemic aldol mixtures. Finally, the use of an organic polar protic solvent such as MeOH, which is able to disrupt the hydrogen-bonding and the corresponding 3D organisation of the polymers, leads, independently of the nature of the polymer used, to a significant reduction in the enantioselectivity (entries 6 and 9, Table 3). In general, these results highlight once again that during the polymerisation processes some degree of chirality transfer to the main polymeric chain can take place leading to a more properly organised polymeric structure and, therefore, a more efficient catalyst in terms of both activity and enantioselectivity.| Entry | Solvent | Catalyst | Yieldb (%) | eec (%) |
|---|---|---|---|---|
|
a 1:10:0.4 RCHO–acetone–catalyst ratio; 0.314 M solution, 24 mg polymer per mL of solvent, r.t., 24 hours.
b Determined by 1H-NMR.
c Determined by Chiral-HPLC (Chiracel OH).
|
||||
| 1 | DMSO | (+)-10-L-Pro | 94 | 78 |
| 2 | Acetonec | (+)-10-L-Pro | 97 | 77 |
| 3 | MeOH | (+)-10-L-Pro | 52 | 20 |
| 4 | DMSO | PCIL-ATRP-(+)-2-L-Pro | 72 | 17 |
| 5 | Acetone | PCIL-ATRP-(+)-2-L-Pro | 59 | 2 |
| 6 | MeOH | PCIL-ATRP-(+)-2-L-Pro | 93 | 10 |
| 7 | DMSO | PCIL-RAFT-(+)-4-L-Pro | 97 | 50 |
| 8 | Acetone | PCIL-RAFT-(+)-4-L-Pro | 92 | 55 |
| 9 | MeOH | PCIL-RAFT-(+)-4-L-Pro | 90 | 6 |
Conclusions
Our results demonstrate that chiral monomeric imidazolium salts can be used to build up polymers with a well organised secondary structure. The right self-assembling of the monomeric units generates a highly ordered macromolecular structure with a complex hierarchical architecture, which can provide an adequate microenvironment for an efficient catalytic activity, mimicking the one found in natural enzymes, whose three-dimensional structure defines and explains their unique reactivity. The main observations are that:(i) A highly active and selective system for the aldol reaction can be obtained for reactions both in water and in the presence of water. Thus, for instance, in acetone in the presence of water (H2O–acetone 1:4) the polymeric catalysts obtained can be up to ca. 6.6 × 103 fold more active than their corresponding monomeric counterpart, while in water the only active catalyst is the polymeric one. This enhancement in catalytic activity can be related to some degree of chirality transfer to the main polymeric chain, producing a more properly organised polymeric structure and, therefore, a more efficient catalyst.
(ii) The polymeric system is able to catalyse the consecutive aldol-dehydration process behaving as a synthetic mimic of the aldolase-dehydrogenase enzymatic system. Indeed, under the right conditions of concentration and acetone–water ratio an enantioselective dehydration of the R aldol is preferentially obtained.
(iii) A better transfer of the chirality is obtained for the polymers prepared by RAFT polymerization. The catalysts prepared by RAFT polymerization of the chiral monomer are more active and enantioselective than the analogous ones prepared by ATRP or grafting methods.
Acknowledgements
This work was supported by GV-PROMETEO/2012/020, CTQ-2011-28903 and the Santiago-Grisolia GV Program (GRIS48/10). Cooperation of the SCIC of the UJI for instrumental analyses is acknowledged. Financial support from the Finnish Funding Agency for Technology and Innovation TEKES (grant # 40207/09), Academy of Finland (grant # 264990) and ERA.Net RUS (Project name SILICAMPS) is gratefully acknowledged.Notes and references
- (a) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088 CrossRef CAS PubMed; (b) M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138 CrossRef CAS PubMed; (c) E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Rev., 2011, 111, 3998 CrossRef CAS PubMed; (d) E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2005, 105, 313 CrossRef PubMed.
- R. Xue and J. M. Woodley, Bioresour. Technol., 2012, 115, 183 CrossRef CAS PubMed.
- V. Sharma and M. Bachwani, Curr. Enzyme Inhib., 2011, 7, 178 CrossRef CAS.
- (a) Y. Yin, Z. Dong, Q. Luo and J. Liu, Prog. Polym. Sci., 2012, 37, 1476 CrossRef CAS PubMed; (b) R. Breslow, S. Bandyopadhyay, M. Levine and W. Zhou, ChemBioChem, 2006, 7, 1491 CrossRef CAS PubMed; (c) Artificial Enzymes, ed. R. Breslow, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- R. P. Mengens and G. Roelfes, Chem.–Eur. J., 2011, 17, 8514 CrossRef PubMed.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551 CrossRef CAS.
- R. K. O'Reilly, Polym. Int., 2010, 59, 568 CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629 CrossRef CAS PubMed; (b) J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469 CrossRef CAS PubMed.
- (a) K. Nakamura, K. Fukao and T. Inoue, Macromolecules, 2012, 45, 3850 CrossRef CAS; (b) V. Sans, N. Karbass, M. I. Burguete, V. Compañ, E. García-Verdugo, S. V. Luis and M. Pawlak, Chem.–Eur. J, 2011, 17, 1894 CrossRef CAS PubMed; (c) P. M. Carrasco, A. Ruiz De Luzuriaga, M. Constantinou, P. Georgopanos, S. Rangou, A. Avgeropoulos, N. E. Zafeiropoulos, H.-J. Grande, G. Cabañero, D. Mecerreyes and I. Garcia, Macromolecules, 2011, 44, 4936 CrossRef CAS.
- (a) K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235 CrossRef CAS; (b) A. Winkel, P. V. G. Reddy and R. Wilhelm, Synthesis, 2008, 999 CAS; (c) X. Chen, X. Li, A. Hu and F. Wang, Tetrahedron: Asymmetry, 2008, 19, 1 CrossRef CAS PubMed; (d) A. D. Headley and B. Ni, Aldrichimica Acta, 2007, 40, 107 Search PubMed; (e) C. Baudequin, D. Brjgeon, J. Levillain, F. Guillen, J.-C. Plaquevent and A. C. Gaumont, Tetrahedron: Asymmetry, 2005, 16, 3921 CrossRef CAS PubMed.
- (a) E. Busto, V. Gotor-Fernández, N. Ríos-Lombardía, E. García-Verdugo, I. Alfonso, S. García-Granda, A. Menéndez-Velázquez, M. I. Burguete, S. V. Luis and V. Gotor, Tetrahedron Lett., 2007, 48, 5251 CrossRef CAS PubMed; (b) N. Ríos-Lombardía, E. Busto, V. Gotor-Fernández, V. Gotor, R. Porcar, E. García-Verdugo, S. V. Luis, I. Alfonso, S. García-Granda and A. Menéndez-Velázquez, Chem.–Eur. J., 2010, 16, 836 CrossRef PubMed.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139 CrossRef CAS.
- H. Chen, D. Liu, L. Chen and R. Qu, Mater. Chem. Phys., 2011, 28, 331 CrossRef PubMed.
- E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef.
- A. Favier and M. Charreyre, Macromol. Rapid Commun., 2006, 9, 653 CrossRef.
- B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis and M. J. Vicent, Tetrahedron, 2001, 57, 8675 CrossRef CAS.
- B. J. Deppmeier, A. J. Driessen, T. S. Hehre, W. J. Hehre, J. A. Johnson, P. E. Klunzinger, J. M. Leonard, I. N. Pham, W. J. Pietro and J. Yu, Spartan '08, build 132 (March 27 2009), Wavefunction Inc., Irvine CA, 2009 Search PubMed.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed, 2000, 39, 1352 CrossRef CAS.
- For reviews on the use of natural aldolase enzymes in synthesis see: (a) H. J. M. Gijsen, L. Qiao, W. Fitz and C.-H. Wong, Chem. Rev., 1996, 96, 443 CrossRef CAS PubMed; (b) C.-H. Wong and G. M. Whitesides, Enzymes in Synthetic Organic Chemistry, Pergamon Press, Oxford, 1994 Search PubMed; (c) N. Wymer and E. J. Toone, Curr. Opin. Chem. Biol., 2000, 4, 110 CrossRef CAS.
- (a) J. Wagner, R. A. Lerner and C. F. Barbas, Science, 1995, 270, 1797 CAS; (b) T. Hoffmann, G. Zhong, B. List, D. Shabat, J. Anderson, S. Gramatikova, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 1998, 120, 2768 CrossRef CAS; (c) G. Zhong, R. A. Lerner and C. F. Barbas, Angew. Chem., Int. Ed., 1999, 38, 3738 CrossRef CAS.
- V. Bisai, A. Bisai and V. K. Singh, Tetrahedron, 2012, 68, 4541 CrossRef CAS PubMed.
- (a) Y. C. Yang and D. E. Bergbreiter, Pure Appl. Chem., 2013, 85, 493 CrossRef CAS; (b) F. Cozzi, Adv. Synth. Catal., 2006, 348, 1367 CrossRef CAS; (c) J. D. Revell, D. Gantenbein, P. Krattiger and H. Wennemers, Biopolymers, 2006, 84, 105 CrossRef CAS PubMed.
- (a) P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis and R. K. O'Reilly, Macromolecules, 2012, 45, 2377 CrossRef CAS; (b) A. Lu, P. Cotanda, J. P. Patterson, D. A. Longbottomb and R. K. O'Reilly, Chem. Commun., 2012, 48, 9699 RSC.
- D. Font, S. Sayalero, A. Bastero, C. Jimeno and M. A. Pericas, Org. Lett., 2008, 10, 337 CrossRef CAS PubMed.
- (a) A. Lu, D. Moatsou, D. A. Longbottom and R. K. O'Reilly, Chem. Sci., 2013, 4, 965 RSC; (b) A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233 CrossRef CAS PubMed; (c) A. C. Evans, A. Lu, C. Ondeck, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2010, 43, 6374 CrossRef CAS.
- (a) D. Zhang, C. Ren, W. Yang and J. Deng, Macromol. Rapid Commun., 2012, 33, 652 CrossRef CAS PubMed; (b) A. Ikeda, K. Terada, M. Shiotsuki and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3783 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed.
- W. Chen, Y. Zhang, L. Zhu, J. Lan, R. Xie and J. You, J. Am. Chem. Soc., 2007, 129, 13879 CrossRef CAS PubMed.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed, 2004, 43, 1983 CrossRef CAS PubMed; (b) A. I. Nyberg, A. Usanp and P. M. Pihko, Synlett, 2004, 1891 CAS.
- N. Mase and C. F. Barbas, III, Org. Biomol. Chem., 2010, 8, 4043 CAS.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem.–Eur. J., 2012, 18, 3362 CrossRef CAS PubMed.
- (a) B. Escuder, F. Rodrıguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC; (b) B. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478 CrossRef PubMed.
- P. Haake, G. Wallerberg and J. Boger, J. Am. Chem. Soc., 1971, 93, 4938 CrossRef CAS.
- T. Kehat, K. Goren and M. Portnoy, New J. Chem., 2012, 36, 394 RSC.
- X. Chen, B.-K. Liu, H. Kang and X.-F. Lin, J. Mol. Catal. B: Enzym., 2011, 68, 71 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py01364b |
| This journal is © The Royal Society of Chemistry 2014 |