 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding the scope of the crystallization-driven self-assembly of polylactide-containing polymers†
Anaïs
Pitto-Barry
a,
Nigel
Kirby
b,
Andrew P.
Dove
*a and
Rachel K.
O'Reilly
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk; a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 3236
bAustralian Synchrotron, 800 Blackburn Road, Clayton, Victoria 3168, Australia
First published on 29th November 2013
Abstract
We report the crystallization-driven self-assembly of diblock copolymers bearing a poly(L-lactide) block into cylindrical micelles. Three different hydrophilic corona-forming blocks have been employed: poly(4-acryloyl morpholine) (P4AM), poly(ethylene oxide) (PEO) and poly(N,N-dimethylacrylamide) (PDMA). Optimization of the experimental conditions to improve the dispersities of the resultant cylinders through variation of the solvent ratio, the polymer concentration, and the addition speed of the selective solvent is reported. The last parameter has been shown to play a crucial role in the homogeneity of the initial solution, which leads to a pure cylindrical phase with a narrow distribution of length. The hydrophilic characters of the polymers have been shown to direct the length of the resultant cylinders, with the most hydrophilic corona block leading to the shortest cylinders.
Introduction
The self-assembly of amphiphilic diblock copolymers in a selective solvent is governed by the lengths of the polymer blocks, their affinity for each other, for the solvent (the Flory–Huggins parameter) and the temperature. Using such parameters, a wide range of morphologies such as spheres, cylinders or vesicles have been targeted.1,2 Notably however, obtaining pure cylindrical micelle phases is challenging as the packing parameter, which dictates the self-assembly behavior, is required to be in a very narrow window between ⅓ and ½.An alternative method for the synthesis of cylindrical polymeric micelles is to use semi-crystalline polymers.3 Here, the formation of cylindrical micelles is driven by the crystallization of the core as long as the temperature is maintained below the Tm of the crystalline block.4 This crystallization-driven self-assembly (CDSA) process has been extensively studied by Winnik and Manners with a polyferrocenyldimethylsilane (PFS)-containing organometallic–organic diblock copolymer in alkane solvents. The length of the cylinders has been controlled via epitaxial growth, in a process similar to living polymerizations, leading to a narrow length dispersity of the resultant micelles.5 The active chain ends can be used to incorporate different unimers, still bearing the PFS core, which has led to the formation of cylindrical micelles6,7 and co-micelles with electronic8 or fluorescent properties9 amongst others.10 More complex morphologies have also been obtained which demonstrates the potential power of this approach. Variation of the core block, but still keeping analogous crystalline structures11 has produced scarf-like architectures; interference of the corona-forming block can lead to the creation of defects in the crystal growth fronts, which can produce lenticular platelets.12 Furthermore, multi-armed micelles and block co-micelles have been produced from colloidal nanoparticles of the organometallic homopolymer.13 Organic semi-crystalline blocks have also led to the formation of cylindrical micelles in organic solvents, such as poly(ethylene oxide) in n-heptane,14,15 or poly(acrylonitrile) in chloroform.16 The application of biodegradable and biocompatible materials for the preparation of cylindrical micelles has also been studied. Poly(ε-caprolactone)-b-poly(ethylene oxide) assembled in water has been shown to form cylindrical micelles alongside other morphologies depending on the ratio between the two blocks.17
Poly(lactide) fulfills the requirements of biomedical materials.18,19 It can be synthesized by ring-opening polymerization with a metal-free catalyst,20,21 with good molecular weight dispersity and precise architecture of polymer chains. The micellization phenomena of semi-crystalline poly(lactide)-containing polymers have been studied only recently.22–24 We have recently exploited its semi-crystalline property to promote CDSA in poly(acrylic acid)-b-poly(L-lactide) diblock copolymers (PAA-b-P-L-LA) at a temperature above the glass transition temperature, Tg, of the core.25,26 A sphere-to-cylinder transition has been observed, wherein the initial amorphous spherical micelles transform into crystalline seeds and subsequently cylinders. The use of a pro-hydrophilic monomer (tetrahydropyran acrylate) has however some drawbacks as the addition of acid is required to enable deprotection during the assembly step. Moreover, the assembly starts from a non-homogeneous solution, which often results in cylindrical micelles with relatively large length dispersities.
Herein we report the synthesis of amphiphilic diblock copolymers through a combination of ROP and RAFT polymerizations. The length of the hydrophobic P-L-LA block, has been targeted to be around 17.5 weight%, which is known to allow for the formation of cylindrical micelles via the crystallization-driven self-assembly process. Poly(4-acryloyl morpholine) (P4AM), poly(ethylene oxide) (PEO) and poly(N,N-dimethylacrylamide) (PDMA), have been used as hydrophilic blocks; the relative hydrophilic character of this block has been shown to result in control over the length of the cylinders.
Experimental section
Materials
Dry solvents were obtained by passing over a column of activated alumina using an Innovative Technologies solvent purification system. L-Lactide was purified using the following procedure: L-Lactide was dissolved in dichloromethane, passed through a silica plug and dried over magnesium sulfate, after which the solvent was removed. The L-lactide was dissolved in hot toluene which was removed before being transferred into a Schlenk flask under inert conditions. The procedure was repeated using dry toluene before further drying over 3 Å molecular sieves in dichloromethane twice. L-Lactide was then stored in a nitrogen-filled glove box. AIBN (2,2′-azo-bis(isobutyronitrile)) was recrystallized twice from methanol and stored in the dark at 4 °C. (−)-Sparteine was dried over CaH2, distilled, degassed, and stored under a nitrogen atmosphere. 1-(3,5-Bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea was prepared as previously reported.21 Recrystallized thiourea was dissolved in dry THF and dried over CaH2 before being stored in a nitrogen-filled glove box. 4-Acryloyl morpholine (4AM) and N,N-dimethylacrylamide (DMA) were purified using basic alumina columns prior to use. All other chemicals were used as received from Aldrich, Fluka, Acros and Iris Biotech.General considerations
All polymerizations were performed under moisture- and oxygen-free conditions either in a nitrogen-filled glove box or by standard Schlenk techniques. 1H NMR and 13C NMR spectra were recorded on a 250, 300, 400 or 500 MHz Bruker DPX FT-NMR spectrometer using deuterated solvents. Chemical shifts are reported as δ in parts per million using the residual protonated solvent as internal standard.27 Size exclusion chromatography (SEC) measurements in THF were obtained using a Varian 390-LC-Multi detector suite fitted with differential refractive index (DRI) and photodiode array (PDA) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μm (50 × 7.5 mm)) and two mixed-D columns (Varian Polymer Laboratories PLGel 5 μm (300 × 7.5 mm)). The mobile phase was tetrahydrofuran with 2% triethylamine eluent at a flow rate of 1.0 mL min−1 and samples were calibrated against narrow molecular weight PMMA standards (200–1.0 × 106 g mol−1) using Cirrus v3.3 software. SEC measurements in DMF were performed on a Varian 390-LC-Multi detector suite system equipped with a PLGel 3 μm (50 × 7.5 mm) guard column, two PLGel 5 μm (300 × 7.5 mm) mixed-D columns using DMF with 0.1% LiBr at 50 °C as the eluent at a flow rate of 1.0 mL min−1. An ultraviolet (UV) (set at 309 nm) and DRI were used as detectors. Narrow molecular weight PMMA standards (200–1.0 × 106 g mol−1) were used for calibration. SEC measurements in CHCl3 were performed on a Agilent 390-LC-Multi detector suite system equipped with a PLGel 3 μm (50 × 7.5 mm) guard column, two PLGel 5 μm (300 × 7.5 mm) mixed-D columns using CHCl3 with 5% TEA as the eluent at a flow rate of 1.0 mL min−1. A differential refractive index was used as detector. Narrow molecular weight PMMA standards (200–1.0 × 106 g mol−1) were used for calibration. Hydrodynamic diameters (Dh) and size distributions of particles were determined by dynamic light scattering (DLS) on a Malvern Zetasizer Nano ZS instrument operating at 25 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering) and the data was analyzed using Malvern DTS 6.20 software. The hydrodynamic radius was calculated from the Stokes–Einstein equation where particles are assumed to be spherical. In the case of cylindrical micelles, such an equation is not valid, however it is still possible to detect multiple populations in solution and obtain polydispersity information. Transmission electron microscopy (TEM) measurements were conducted on formvar/carbon grids, which were prepared by oxygen plasma treatment to make the surface hydrophilic. A drop of the solution (after freeze-drying and re-dispersion in nanopure water) was deposited onto a grid, blotted away after 2 min, left to air dry for 5 min and stained with phosphotungstic acid (PTA) (2% weight solution with pH adjusted to 7) for 2 min. Grids were examined with a transmission electron microscope (JEOL TEM-2000), operating at 200 kV. Small-angle X-ray scattering (SAXS) measurements were carried out on the SAXS/WAXS beamline at the Australian Synchrotron facility at photon energy of 8.2 keV. The samples in solution (0.5 mg mL−1) were run using 1.5 mm diameter quartz capillaries. The measurements were collected at sample to detector distance of 7.160 m to give a q range of 0.002 to 0.08 Å−1, where q is the scattering vector and is related to the scattering angle (2θ) and the photon wavelength (λ) by the following equation: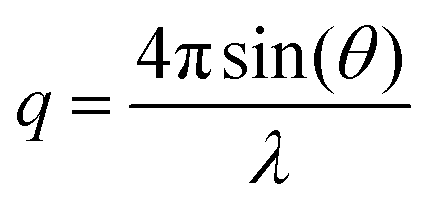
The scattering from a blank (H2O) was measured in the same location as sample collection and was subtracted for each measurement. The two-dimensional SAXS images were converted in one-dimensional SAXS profiles (I(q) versus q) by circular averaging, where I(q) is the scattering intensity. The number-average lengths of cylinders obtained by TEM were used to fit the SAXS raw data. The function used for the fitting from NCNR package was “cylinder – polydisperse radius” (“cylinder”, “sphere” and “cylinder – polydisperse length” as well as linear combination of these functions were also used but with no good fit).28 The scattering length density of the solvent (H2O) and the monomers were calculated using the “Scattering Length Density Calculator” provided by NIST Center for Neutron Research (SLD H2O = 9.46 × 10−6 Å−2).29 Limits for q range were applied for the fitting from 0.004 to 0.03 Å−1.
Particle characterization
Dimensions of spheres and cylinders were determined by analyzing the TEM micrographs. At least 100 individual spheres or cylinders from different regions of the grid were carefully traced by hand to determine the contour length for the statistical length analysis. From these data Ln and Lw of each sample of cylindrical micelles were calculated by the equations shown below (L: length of cylinder, N: number):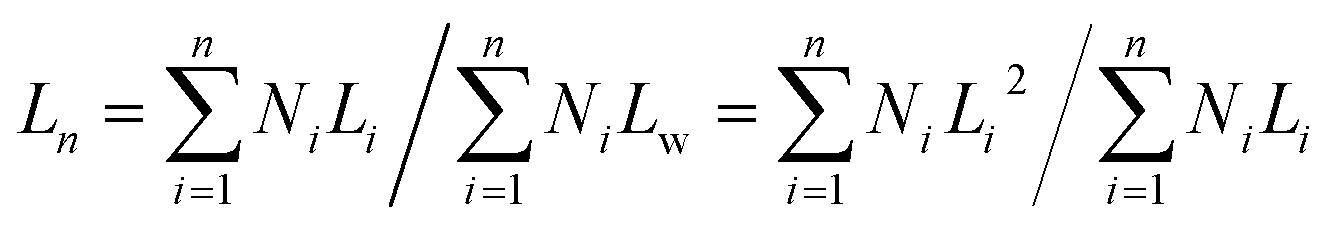
Polymerization procedures
Three freeze–pump–thaw cycles were performed on the reaction vessel for the removal of oxygen for all RAFT polymerizations. The ampoules were sealed under nitrogen before being immersed in an oil bath at the required temperature. ROP polymerizations were carried out in a glove box under an inert nitrogen atmosphere at ambient temperature. Molecular weights and dispersities were measured by SEC analysis, with 1H NMR spectroscopy used for the determination of the end group functionality and molecular weight by integration of the polymer backbone to the end group signals.Synthesis of poly(L-lactide) (1 (DP = 36) and 2 (DP = 38))
In a 20 mL scintillation vial equipped with a stirrer bar, a solution of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea organocatalyst (81.3 mg, 0.219 mmol), (−)-sparteine (25.7 mg, 0.110 mmol) and 4-(hydroxymethyl)benzyl carbonotrithioate CTA (50.0 mg, 0.125 mmol) in dry CH2Cl2 (1 mL) was added to a solution of L-lactide (632.6 mg, 4.389 mmol) in dry CH2Cl2 (4 mL). The reaction mixture was stirred for 2 h 45 min, before the polymer was precipitated from CH2Cl2 into cold diethyl ether. 1H NMR (300 MHz, CDCl3): δ 7.35–7.26 (m, 4H, Harom), 5.20–5.13 (q, 1H, 3JH–H = 7.0 Hz, CHbackbone), 4.61 (s, 2H, SCH2-Carom), 4.37–4.35 (q, 1H, 3JH–H = 6.8 Hz, last CHbackbone–OH), 3.39–3.34 (m, 2H, SCH2–Caliphatic), 2.66–2.64 (m, 1H, OH), 1.85–1.42 (d, 6H, 3JH–H = 7.0 Hz, CH3 backbone), 1.38–1.24 (m, CH2 endgroup), 0.90–0.86 (t, 3H, 3JH–H = 6.8 Hz, CH3 endgroup). 13C NMR (500 MHz, CDCl3): δ 169.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 135.9 (Carom), 134.7 (Carom), 129.6 (CHarom), 128.7 (CHarom), 69.1 (CH), 41.2 (SCH2–Carom), 37.3 (SCH2CH2), 32.0–22.8 (CHaliphatic), 20.7 (CH3–CHOH), 15.4 (CH3 backbone), 13.5 (CH3 end group). Mn,NMR (1) = 5.8 kDa, Mn,SEC,THF (1) = 11.7 kDa, Mn,NMR (2) = 5.9 kDa, Mn,SEC,THF (2) = 10.4 kDa.
O), 135.9 (Carom), 134.7 (Carom), 129.6 (CHarom), 128.7 (CHarom), 69.1 (CH), 41.2 (SCH2–Carom), 37.3 (SCH2CH2), 32.0–22.8 (CHaliphatic), 20.7 (CH3–CHOH), 15.4 (CH3 backbone), 13.5 (CH3 end group). Mn,NMR (1) = 5.8 kDa, Mn,SEC,THF (1) = 11.7 kDa, Mn,NMR (2) = 5.9 kDa, Mn,SEC,THF (2) = 10.4 kDa.
Synthesis of PEO454-P-L-LA28 (3)
In a 20 mL scintillation vial equipped with a stirrer bar, a solution of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea organocatalyst (8.6 mg, 0.023 mmol), (−)-sparteine (2.7 mg, 0.012 mmol) and PEO (20 kDa monomethyl ether (300 mg, 0.015 mmol)) in dry CH2Cl2 (1.6 mL) was added to a solution of L-lactide (67.0 mg, 0.465 mmol) in dry CH2Cl2 (0.3 mL). The reaction mixture was stirred for 40 h, before the polymer was precipitated from CH2Cl2 into cold diethyl ether. 1H NMR (400 MHz, CDCl3): δ 5.19–5.13 (q, 1H, 3JH–H = 7.0 Hz, CHP-L-LA backbone), 3.64 (s, 4H, CH2 PEO backbone), 3.38 (s, 3H, CH3 PEO), 2.66–2.64 (m, 1H, OH), 1.59–1.57 (d, 6H, 3JH–H = 7.2 Hz, CH3 P-L-LA). 13C NMR (400 MHz, CDCl3): δ 169.7 (CO), 70.1 (CH2), 69.1 (CH), 16.8 (CH3 PEO end group). Mn,NMR (3) = 24.0 kDa, Mn,SEC,CHCl3 (3) = 47.7 kDa.
General procedure for the RAFT polymerization from P-L-LA
In an ampoule equipped with a stirrer bar, a solution of P-L-LA, monomer and AIBN in dioxane was heated at 65 °C for 21 h, before the polymer was precipitated from dioxane into cold diethyl ether to afford a pure diblock copolymer.Synthesis of P4AM213-P-L-LA38 (4)
Use of the general procedure for RAFT polymerization with P-L-LA 2 (50 mg, 0.008 mmol), 4AM monomer (240 mg, 0.851 mmol) and AIBN (0.28 mg, 0.002 mmol) in dioxane (0.5 mL). 1H NMR (250 MHz, CDCl3): δ 5.21–5.13 (q, 1H, 3JH–H = 7.1 Hz, CHP-L-LA backbone), 3.82–3.19 (m, 2H, CH2 P4AM backbone), 2.78–1.67 (m, 5H, CHP4AM backbone and CH2 P4AM), 1.59–1.57 (d, 6H, 3JH–H = 7.1 Hz, CH3 P-L-LA). 13C NMR (400 MHz, CDCl3): δ 173.1 (COP4AM), 169.7 (COP-L-LA), 69.1 (CHP-L-LA), 66.9 (CH2OP4AM), 46.1 (CH2NP4AM), 42.5 (CH2NP4AM), 35.9–35.6 (CHP4AM and CH2 P4AM), 16.8 (CH3 P-L-LA). Mn,NMR (4) = 35.9 kDa, Mn,SEC,DMF (4) = 29.9 kDa.
Synthesis of P4AM166-P-L-LA36 (5)
Use of the general procedure for RAFT polymerization with P-L-LA 1 (100 mg, 0.017 mmol), 4AM monomer (408 mg, 2.886 mmol) and AIBN (0.57 mg, 0.003 mmol) in dioxane (1 mL). 1H NMR (400 MHz, CDCl3): δ 5.19–5.13 (q, 1H, 3JH–H = 7.0 Hz, CHP-L-LA backbone), 3.78–3.12 (m, 2H, CH2 P4AM backbone), 2.78–1.58 (m, 5H, CHP4AM backbone and CH2 P4AM), 1.58–1.57 (d, 6H, 3JH–H = 7.0 Hz, CH3 P-L-LA), 0.90–0.86 (t, 3H, 3JH–H = 6.8 Hz, CH3 endgroup). 13C NMR (400 MHz, CDCl3): δ 172.7 (COP4AM), 169.4 (COP-L-LA), 68.8 (CHP-L-LA), 66.8 (CH2OP4AM), 45.8 (CH2NP4AM), 42.1 (CH2NP4AM), 35.6–35.2 (CHP4AM and CH2 P4AM), 16.4 (CH3 P-L-LA). Mn,NMR (5) = 29.2 kDa, Mn,SEC,DMF (5) = 30.7 kDa.
Synthesis of PDMA248-P-L-LA36 (6)
Use of the general procedure for RAFT polymerization with P-L-LA 1 (100 mg, 0.017 mmol), DMA monomer (408 mg, 4.112 mmol) and AIBN (0.28 mg, 0.002 mmol) in dioxane (1 mL). 1H NMR (400 MHz, CDCl3): δ 5.18–5.13 (q, 1H, 3JH–H = 7.0 Hz, CHP-L-LA backbone), 3.12–3.2.21 (m, 7H, CHPDMA backbone), 1.85–1.58 (m, 2H, CHPDMA backbone), 1.58–1.56 (d, 6H, 3JH–H = 7.0 Hz, CH3 P-L-LA), 0.89–0.86 (t, 3H, 3JH–H = 6.8 Hz, CH3 endgroup). 13C NMR (400 MHz, CDCl3): δ 174.7 (COPDMA), 169.6 (COP-L-LA), 69.1 (CHP-L-LA), 37.1–35.8 (CHaliph PDMA), 16.6 (CH3 P-L-LA). Mn,NMR (6) = 30.4 kDa, Mn,SEC,DMF (6) = 39.0 kDa.
Results and discussion
Synthesis of polymers
Ring-opening polymerization (ROP) and reversible-addition fragmentation chain transfer (RAFT) polymerization were combined to synthesize poly(L-lactide)-containing diblock copolymers according to a method previously reported.25,26 Three different hydrophilic blocks were targeted: poly(4-acryloyl morpholine), poly(N,N-dimethylacrylamide), and poly(ethylene oxide) (commercially available with a monomethylether protected end) (see Scheme 1) with a targeted hydrophilic weight fraction of ca. 80% in each case. Such a hydrophilic/hydrophobic ratio would usually be expected to afford spherical micelles.1,2 however the semi-crystalline nature of the isotactic poly(lactide) block has been shown to promote crystallization-driven self-assembly and therefore under conditions that promote crystallization, the formation of cylindrical micelles is preferred.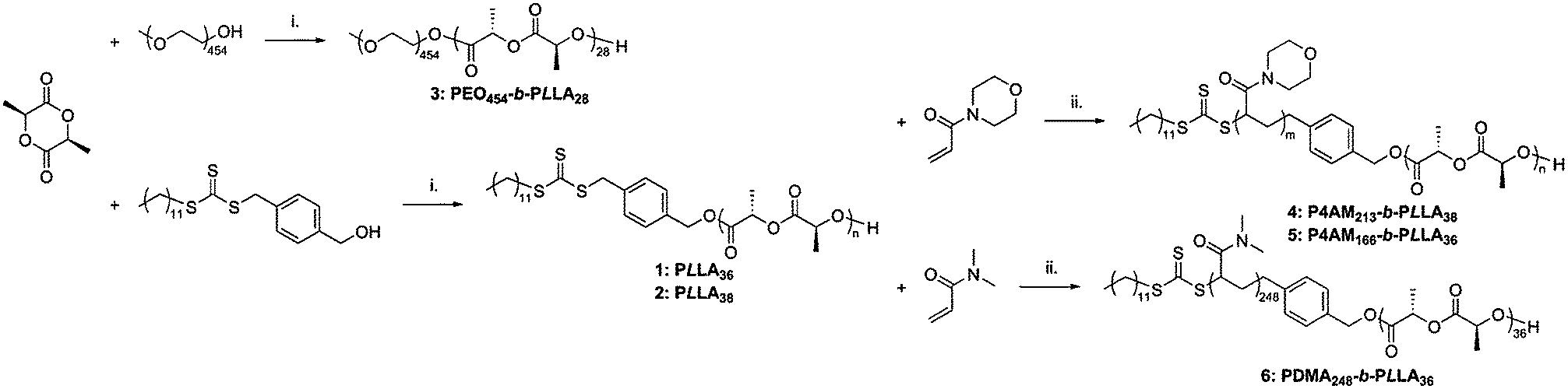 | ||
| Scheme 1 Preparation of block copolymers 3–6. Conditions: (i) 1-(3,5-bis(trifluoromethyl) phenyl)-3-cyclohexyl-thiourea, (−)-sparteine, CH2Cl2; (ii) AIBN, dioxane, 65 °C. | ||
The ROP of L-lactide was achieved in dichloromethane at ambient temperature, using (1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea) with (−)-sparteine.21 This organocatalytic system has a high selectivity towards ROP over (hydroxymethyl)benzyl carbonotrithioate25 as a dual-headed initiator/chain transfer agent.30
Monomer conversion was monitored by 1H NMR spectroscopy by comparing the reduction of the quadruplet resonance from the L-lactide monomer at δ = 5.03 ppm with the appearance of a quartet at δ = 5.17 ppm that is attributed to the proton located on the asymmetric carbon of both the monomer and polymer (see ESI, Fig. S1 and S2†). Low dispersity values, ĐM (ca. 1.06), and monomodal size exclusion chromatography (SEC) traces also indicated the ROP was well controlled (Table 1).
| M n,NMR (kDa) | M n,SEC (kDa) | Đ M | DPa (P-L-LA) | DPa (2nd block) | Hydro-phobic wt fraction (%) | |
|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by SEC analysis in THF. c Determined by SEC analysis in DMF. d Determined by SEC analysis in CHCl3. | ||||||
| P-L-LA, 1 | 5.8 | 11.7 | 1.06 | 36 | — | 100 |
| P-L-LA, 2 | 5.9 | 10.4 | 1.06 | 38 | — | 100 |
| PEO-b-P-L-LA, 3 | 24.0 | 47.7d | 1.02d | 28 | 454 | 16.8 |
| P4AM-b-P-L-LA, 4 | 35.9 | 33.1c | 1.09c | 38 | 213 | 15.4 |
| P4AM-b-P-L-LA, 5 | 29.2 | 31.0c | 1.08c | 36 | 166 | 18.1 |
| PDMA-b-P-L-LA, 6 | 30.4 | 41.5c | 1.09c | 36 | 248 | 17.4 |
The P-L-LA homopolymers with the trithiocarbonate end group 1 and 2 were chain-extended by RAFT polymerization in dioxane ([macro-CTA]0 = 100 mg mL−1) at 65 °C with 10 or 20 mol% of AIBN. The monomer conversion of both 4-acryloylmorpholine (4-AM) and N,N-dimethylacrylamide (DMA), was monitored by 1H NMR spectroscopy. After 4 h, a monomer conversion of about 80% was obtained in both cases, with full conversions being obtained after 21 h (see ESI, Fig. S4–S6†). SEC analysis of the diblock copolymers revealed that the ĐM values of polymers P4AM213-b-P-L-LA38 (4), P4AM166-b-P-L-LA36 (5) and PDMA248-b-P-L-LA36 (6) were low (≤1.14) (Fig. 1 and Table 1). Moreover, analysis of the RI- and UV-detected (λ = 309 nm) SEC traces indicated that no loss of the trithiocarbonate end group had occurred (Fig. 1).
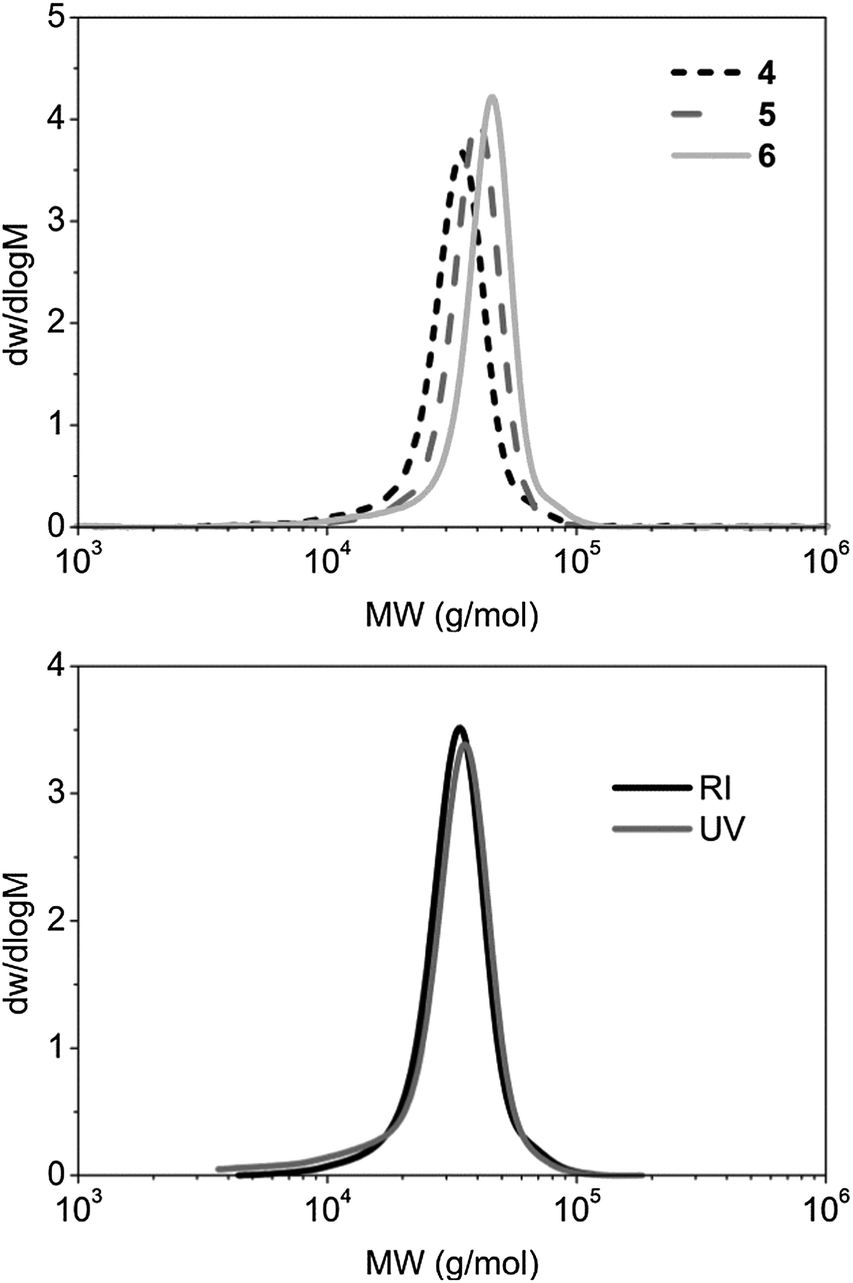 | ||
| Fig. 1 RI SEC traces of P4AM213-b-P-L-LA38 (4), P4AM166-b-P-L-LA36 (5) and PDMA248-b-P-L-LA36 (6) in DMF (top): RI and UV (λ = 309 nm) SEC traces of P4AM213-b-P-L-LA38 (4) (bottom). | ||
PEO454-b-P-L-LA28 (3) was synthesized from a commercially available PEO (20 Kg mol−1) monomethyl ether, the hydroxyl functionality acting as the initiator. The same catalytic system was used as for the preparation of homopolymers P-L-LA 1 and 2. As the PEO initiator required more solvent for its dissolution, the reaction was performed under more dilute conditions than the synthesis of 1 and 2. Such a dilution of the reagents combined with an increase of the viscosity led to an increase of the reaction time (from 3 to 40 h) in order to obtain a monomer conversion of more than 90% (see ESI, Fig. S3†). A linear relationship between the Neperian logarithmic of the consumption of monomer and the time of reaction confirmed the controlled nature of the ROP (Fig. 2). Low dispersity values, ĐM (ca. 1.02), and monomodal size exclusion chromatography (SEC) traces also indicated that the ROP was well controlled.
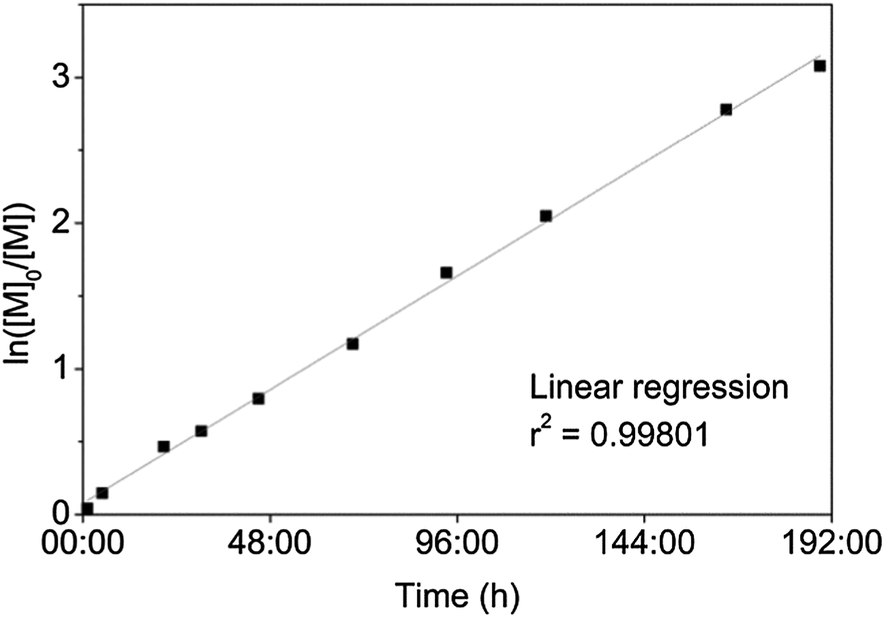 | ||
| Fig. 2 Plot of ln([M]0/[M]) against the time of reaction for the ROP of L-lactide with PEO(20 Kg mol−1) monomethyl ether as initiator. | ||
Crystallization-driven self-assembly of polymers
In order to demonstrate that the formation of cylindrical micelles is governed by the presence of crystallinity within the P-L-LA core domain regardless the hydrophilic corona domain, we have explored the CDSA of different block copolymers, all bearing a poly(L-lactide) core block. Poly(4-acryloyl morpholine), poly(ethylene oxide) and poly(N,N-dimethylacrylamide) have been used as the hydrophilic domains to mirror the acrylic acid polymers utilized in our previous work26 and demonstrate the hydrophilic block versatility for the CDSA process. The solvent ratio, concentration of polymer, and use of well-defined spherical micelles as a starting point have been studied to vary the dynamics of the formed cylindrical micelles.Effect of cosolvent additive for P4AM-P-L-LA
CDSA was conducted with a polymer concentration of 20 mg mL−1 in a mixture of THF and water at 65 °C in a closed vial equipped with a needle through the cap (see ESI† for detailed procedure). A range of initial THF contents (10, 20 and 30 vol%) for the assembly process was studied for the block copolymer P4AM213-b-P-L-LA38 (4) with the 4AM corona. Each sample was analyzed after 27 and 51 h, always following the same work-up procedure (freeze-drying of the sample then redispersion in nanopure water at a concentration less than 1 mg mL−1, see ESI† for more detailed procedure). Dynamic light scattering (DLS) and TEM analyses of the samples were used to characterize these assemblies. TEM analyses revealed an increase of cylinder length with time for experiments with 10 and 30 vol% THF while a slight decrease of length was observed for the experiment with 20 vol% THF content (Fig. 3 and ESI, S8–S10†). In all cases, a mixed phase of spheres and cylinders was observed by TEM analysis. Furthermore, the diameter of the smaller, spherical particles was not observed to change significantly (from ca. 18 nm) during the different assembly procedures (see ESI, Fig. S8–S10†). DLS and TEM analyses were the most consistent for the cylinders assembled in the 20 vol% THF solution (see ESI, Fig. S7†). The differences in population number between DLS and TEM are a consequence of the scattering of a particle being related to the radius by a factor power of 6, hence large objects scatter much more than small structures. Under these conditions, no trend in final cylinder length could be observed with changing solvent composition, which differs from the results reported for the PAA-P-L-LA block copolymer system.26 This can be explained by the permanently hydrophilic nature of the P4AM component and hence the requirement for adjusted assembly conditions.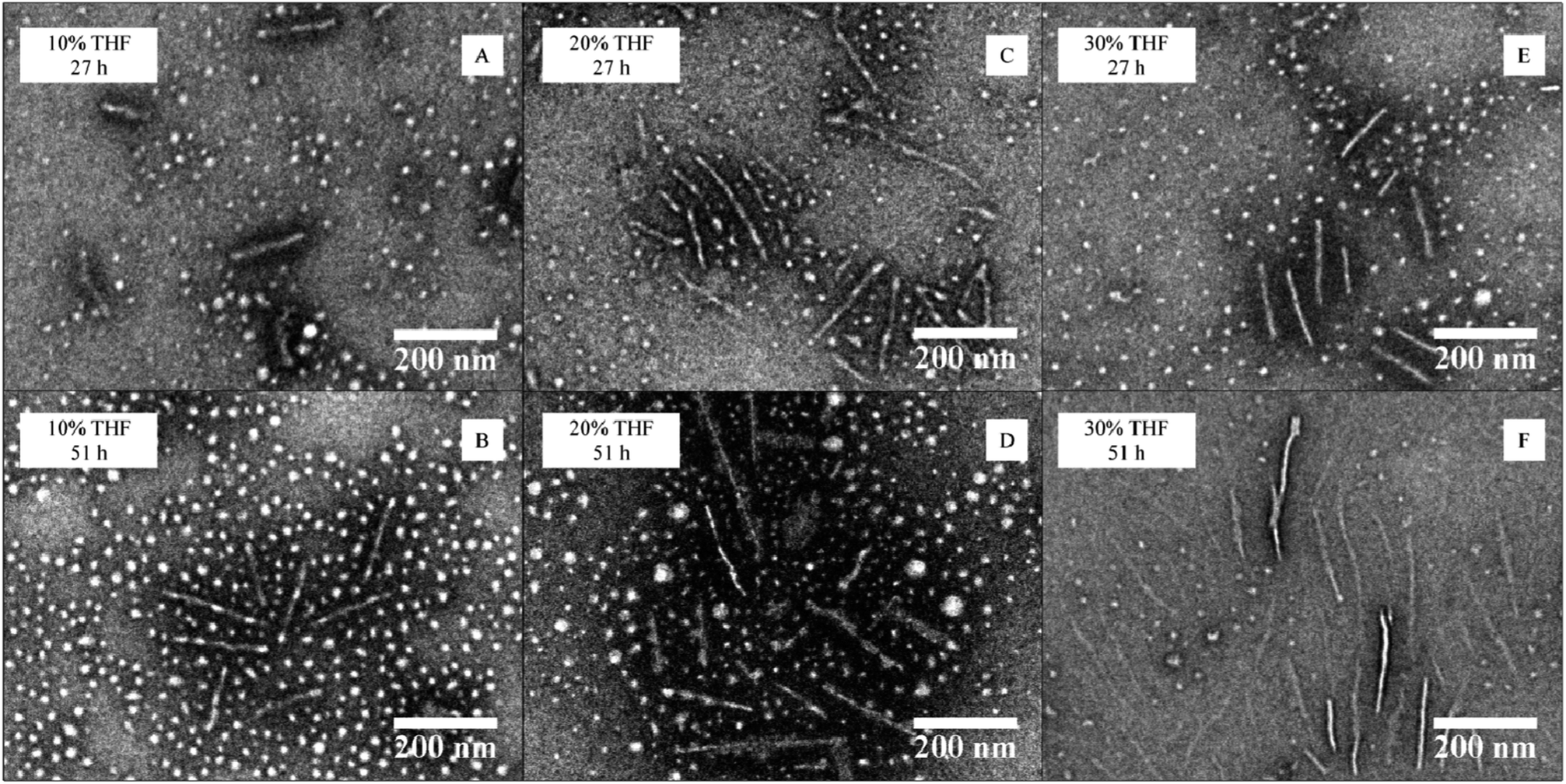 | ||
| Fig. 3 TEM data for the CDSA of P4AM213-b-P-L-LA38 (4) at 65 °C at different THF contents (10–30 vol%) and heating times (27 or 51 hours). | ||
Effect of polymer concentration
Maintaining the THF–water ratio at 20/80 v/v, the effect of the polymer concentration upon CDSA behavior was conducted at 10 and 20 mg mL−1, as described previously for polymer 4. Block copolymers in which the hydrophilic block was P4AM (5) and PDMA (6) respectively were studied under these conditions. Each sample was analyzed after 1 and 2 days, always following the same work-up procedure.A high concentration regime was chosen to favor the micelle core nucleation based on previous experimental observations for our PAA-b-P-L-LA system (more than 5 mg mL−1).26 DLS and TEM analyses of the samples were used to characterize these assemblies. TEM analyses revealed a mixed phase for the CDSA of block copolymer P4AM166-b-P-L-LA36 (5) regardless of the time or the conditions of the process. The diameter of spheres slightly increased during the assembly process (increase of around 1.5 nm) and interestingly this population was more dominant than the cylindrical phase. The cylinder lengths remained almost the same during the assembly process for the less concentrated experiment whereas a slight decrease of the length of cylinders was observed for the more concentrated system during the assembly process (Fig. 4 and ESI, S12 and S13†). In both cases, a slight decrease of the dispersity of the cylinder lengths was observed over time, although this dispersity was still rather broad, highlighting the ill-defined nature of these assemblies.
 | ||
| Fig. 4 TEM data for the CDSA of P4AM166-b-P-L-LA36 (5) at 65 °C at different polymer concentrations (10 and 20 mg mL−1) and heating times (1–2 days). | ||
Further study of the effects of block copolymer concentration on self-assembly behaviour was undertaken using PDMA248-b-P-L-LA36 (6). For this system a mixed morphology phase was again observed by TEM regardless of the time and concentration of the assembly procedure. A slight increase of the sphere diameter size occurred during the process, however the length of the observed cylinders remained the same regardless of the assembly time at 10 mg mL−1. When the assembly was performed at 20 mg mL−1, an increase in cylinder length was noted (Fig. 5 and ESI, S15 and S16†). Disappointingly, the polymer concentration was not shown to have an impact on the homogeneity of the assembly process as mixed morphology phases were obtained in all of the experiments.
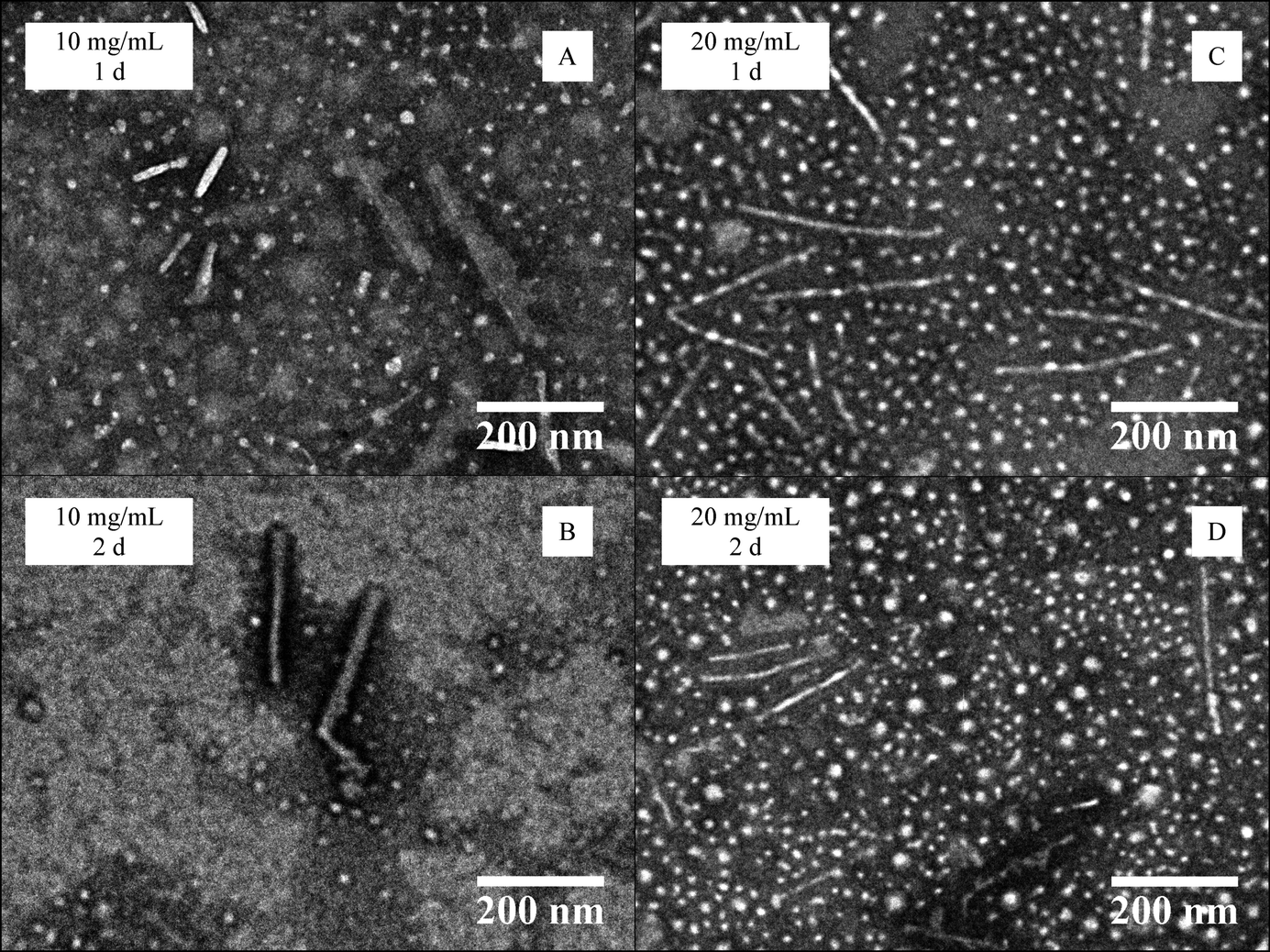 | ||
| Fig. 5 TEM data for the CDSA of PDMA248-b-P-L-LA36 (6) at 65 °C at different polymer concentrations (10–20 mg mL−1) and heating times (1–2 days). | ||
Effect of slow addition of non-solvent for P4AM-P-L-LA (5)
The high length dispersities (>1.10) obtained for the previous experiments suggested a growth of the cylinders from a large diameter distribution of spherical seeds. Such a large distribution may be explained by the low solubility of the unimer chains at the beginning of the self-assembly. Moreover, both spherical and cylindrical micelles have been observed by TEM analysis, which indicates that spherical micelles may be trapped in the solution and cannot evolve further through the proposed unimer exchange process which enables cylindrical micelle formation.In order to improve this solubility, a new experimental procedure was developed: water (2 mL) was added slowly using a peristaltic pump with a rate of 0.5 mL h−1 after the solubilization of copolymer in THF (0.5 mL) (see ESI† for detailed procedure). Such a slow addition of water ensured the polymer was soluble in the mixture of solvents as evidenced by the formation of clear solutions. The production of cylindrical micelles with more controlled lengths was expected with this experimental setup as more seeds should be created given an increase solubility of unimers. To explore this, the CDSA of block copolymer P4AM166-b-P-L-LA36 (5) was conducted at 20 mg mL−1 with 20 vol% THF using both slow and rapid addition of water (i.e. with and without a peristaltic pump). Shorter cylinders (measured by TEM analysis) were obtained from the slow water addition experiment after 3 days (see Fig. 6‡ and ESI, S17†). TEM analysis indicated a narrower length distribution was obtained when the slow addition of water was used and this enabled increased control of the cylinder growth during the 3 day self-assembly process (Table 2‡). It is proposed that the formation of a homogeneous solution and hence complete dissolution of the diblock polymer resulted in a more controlled initiation growth step and this leads in turn to the formation of well-defined cylindrical micelles. Moreover, almost no spherical micelles were observed by TEM analysis, which indicates that the kinetics of the unimer exchange is no longer blocked when complete dissolution of the polymer is achieved.
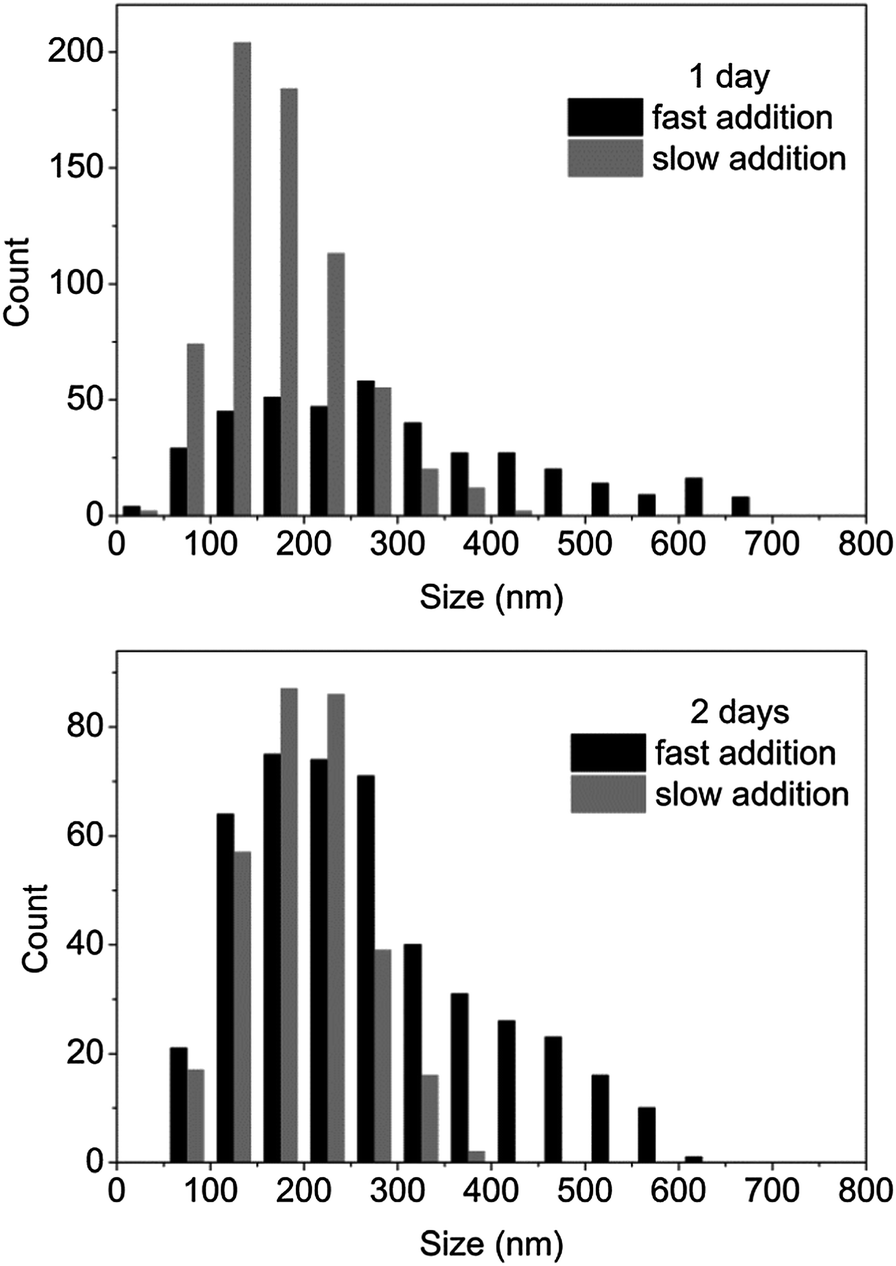 | ||
| Fig. 6 Histograms for cylindrical micelles prepared from the CDSA of P4AM166-b-P-L-LA36 (5) at 20 mg mL−1, 20 vol% THF at different times (1–2 days) and water speed additions (fast or slow). | ||
| 1 Day | 2 Days | 3 Days | ||
|---|---|---|---|---|
| Fast water addition | L n (nm) | 293 ± 155 | 264 ± 124 | 202 ± 112 |
| L w/Ln | 1.28 | 1.22 | 1.31 | |
| Slow water addition | L n (nm) | 173 ± 68 | 198 ± 62 | 214 ± 75 |
| L w/Ln | 1.16 | 1.10 | 1.12 | |
Variation of the corona
To further demonstrate the utility of these optimized conditions for the CDSA of P-L-LA-containing block copolymers, the variation of the hydrophilic corona was investigated. Poly(ethylene oxide) is one of the most commonly-applied hydrophilic polymers.31 Block copolymer PEO454-b-P-L-LA28 (3) was applied in the optimized self-assembly procedure (dissolution of the polymer in THF and slow addition of water via a peristaltic pump with a final concentration of polymer of 20 mg mL−1 and a THF content of 20 vol%, see ESI† for detailed procedure). In contrast with block copolymer P4AM166-b-P-L-LA36 (5), PEO454-b-P-L-LA28 (3) was not fully soluble in THF even after several hours of stirring. Notably however, only a few drops of water were necessary to obtain a clear solution and the self-assembly procedure was completed as described in the ESI.† Analysis of the TEM images indicated that the cylinders’ length increased during the 3 day assembly process (see Fig. 7 and ESI, Fig. S19†) and in comparison to the earlier assembly attempts, very few spheres were observed after 3 days of the process.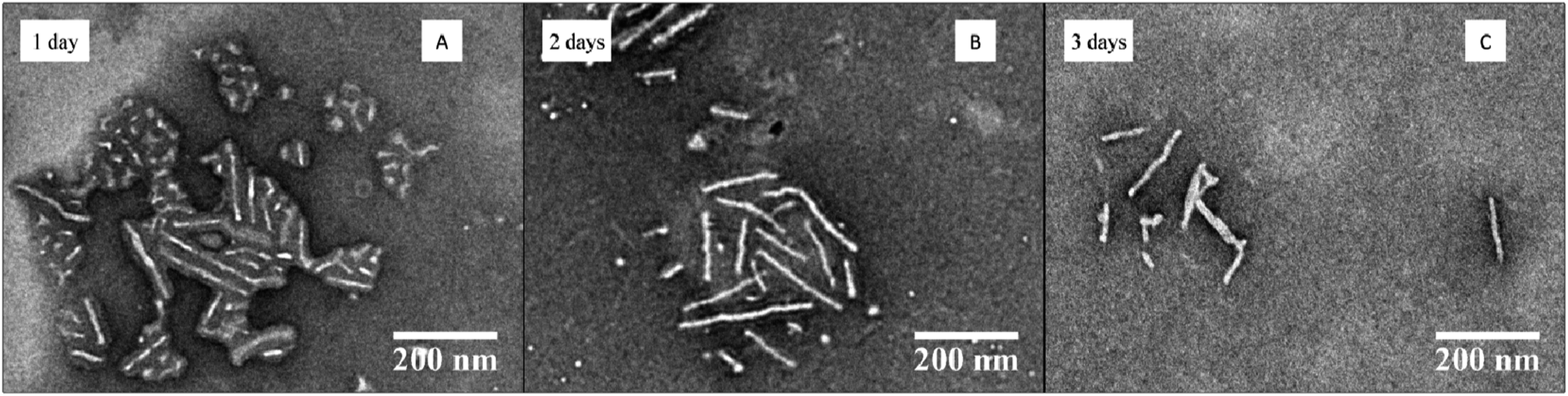 | ||
| Fig. 7 TEM data for the CDSA of PEO454-b-P-L-LA28 (3) at 65 °C, 20 mg mL−1 and 20 vol% THF with slow addition of water at different times (1–3) days. | ||
The influence of a more hydrophilic corona block was then investigated. Block copolymer PDMA248-b-P-L-LA36 (6) was self-assembled under the same conditions as the previous sample (at 20 mg mL−1, 20 vol% THF with the slow addition of water) (see ESI† for detailed procedure). This copolymer was fully soluble in THF after a few minutes of stirring. Following slow water addition and heating, the solution became slightly cloudy after 20 h. DLS measurements on the resultant solution revealed the formation of well-defined nanostructures and a transition from the initial micelles to bigger particles was observed over the 3 day assembly process (see ESI, Fig. S20†). TEM analysis also indicated that the cylinders’ length increased during assembly (Fig. 8 and ESI, Fig. S21†). However, a bimodal distribution of nanostructures was observed after 3 days by DLS and TEM analysis which indicates the formation of long cylindrical micelles alongside shorter cylinders. This can be explained as the length of the cylinders is governed by the rate of propagation which in turn depends on the rate of unimer exchange. Different lengths of cylinders can sometimes be obtained if this process is fast compared to the initiation process.
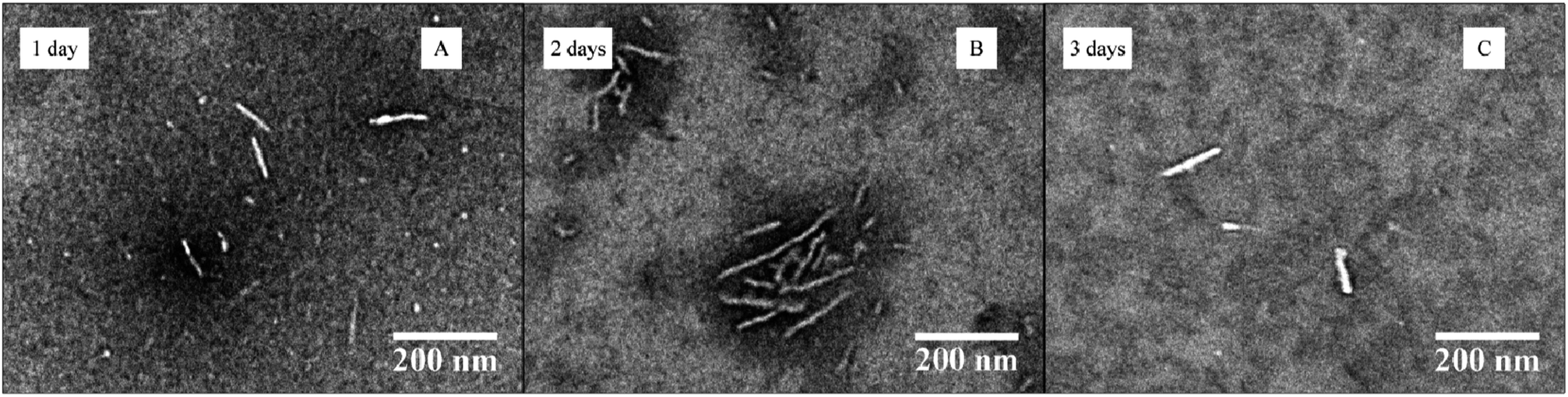 | ||
| Fig. 8 TEM data for the CDSA of PDMA248-b-P-L-LA36 (6) at 65 °C, 20 mg mL−1 and 20 vol% THF with slow addition of water at different times (1–3 days). | ||
Small-Angle X-Ray Scattering (SAXS) analyses were performed on the resultant assemblies from the CDSA of polymers (PEO block (3), P4AM block (5) and PDMA (6)) with the slow addition of water. Analysis of the scattering raw data strongly indicated the presence of cylindrical micelles (see ESI, Fig. S22†). The main curvature of the three curves is different for each diblock copolymer (and varies between 0.01 and 0.02 Å−1), which suggests different radius of nanostructure in all 3 systems. Moreover, the slope of the curve between 0.004 Å−1 and the main curvature is not the same in all the three samples therefore it indicates different lengths of cylindrical micelles are formed in assemblies using the 3 different polymers. A range of different models were used to fit the data and obtain further insight into the resultant micelle morphologies. The “sphere” model did not fit well at all and the best results were instead obtained for the “cylindrical polydisperse radius” model (see ESI, Fig, S22†). Indeed, this model provided a better fit than a linear combination with the “sphere” model, which confirms observations made by TEM analysis. The widths of the cylindrical micelles in solution were calculated to be 15 nm (3), 22 nm (5) and 13 nm (6). The values of fitted widths are in accordance with results obtained for the self-assembly of PAA-b-P-L-LA blocks under similar conditions.30 Fitted cylinder widths by SAXS were smaller than the widths from the dry state TEM as the coronas of the micelles are hydrated in solution and hence they are less visible in solution during SAXS analysis than in dry state TEM analysis. Widths determined by SAXS are more representative of the core dimensions, which is composed of semi-crystalline P-L-LA. This hypothesis was confirmed by the values obtained for the scattering length density (SLD) of the micelles (1.10 × 10−5 Å−2), which are very close to the value for polylactide alone (1.12 × 10−5 Å−2) and significantly different from the values for the different coronas (1.03 × 10−5 Å−2 for PEO and P4AM and 8.92 × 10−5 Å−2 for PDMA).
Interestingly, the length of the cylinders determined by TEM analysis was shorter for block copolymers PEO454-b-P-L-LA28 (3) and PDMA248-b-P-L-LA36 (6) than the lengths obtained for block copolymer P4AM166-b-P-L-LA36 (5) under the same conditions. Indeed, the equilibrium cylinder length correlates well to the hydrophilicity of the corona, given that the hydrophobic/hydrophilic weight ratio was similar for all three blocks. The hydrophilic characters of P4AM, PDMA and PEO were estimated for different degrees of polymerization by using the software Molinspiration.31 As the software is not suitable for large molecules/polymers, only short oligomeric molecules with degrees of polymerization (DPs) from 5 to between 25 and 40, depending of the monomer, were used to predict the hydrophilic tendency of the polymers (see ESI, Fig. S23†). These analyses reveal that PDMA and PEO have a similar hydrophilic character with P4AM being less hydrophilic. Interestingly, under the optimized assembly conditions, the longest cylinders were obtained for the least hydrophilic corona block (P4AM). These results suggest that it may be possible to vary the length of the resultant cylinders by varying the hydrophilic character of the corona. This provides a further method for tuning the dimensions of the cylindrical micelles formed from the CDSA of poly(lactide) semi-crystalline polymers.
Conclusions
Three different diblock copolymers bearing a hydrophobic P-L-LA block have been synthesized from a unique combination of ROP and RAFT polymerizations. P4AM, PDMA and PEO have been used as the hydrophilic part of the polymer. Narrow dispersities determined by SEC have shown the excellent control of these polymerizations. A targeted hydrophobic weight fraction ratio of around 17.9% has been obtained for each polymer, in order to mimic the previously synthesized PAA-b-P-L-LA. Cylindrical micelles have been obtained through the CDSA process as a consequence of the semi-crystallinity of P-L-LA. Furthermore, the experimental conditions have been improved to obtain a better dispersity of the final length of the cylindrical micelles. The control over the cylinders' length has been related to the hydrophilicity of the corona. The more hydrophilic corona (PDMA) has led to the shortest cylinders. The rates of both nucleation and growth processes can explain this trend, as the solubility of the unimers changes with the solubility of the corona-forming block. Varying the size of cylinders can thus be governed by many parameters from the polymer itself: the hydrophilic weight fraction ratio, the length of the hydrophilic block and the hydrophilic character of the corona.Acknowledgements
The authors wish to thank the EPSRC and the University of Warwick for funding this research, the Swiss National Science Foundation for an Early Postdoc Mobility fellowship (Grant no PBNEP2-142949 to A.P.B.). The Warwick Research Development Fund is also thanked for funding. Some items of equipment used in this research were funded by Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Corbion-Purac are gratefully acknowledged for the kind donation of L-Lactide monomer.Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- W.-N. He and J.-T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef CAS PubMed.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- H. Wang, M. A. Winnik and I. Manners, Macromolecules, 2007, 40, 3784–3789 CrossRef CAS.
- I. Korczagin, M. A. Hempenius, R. G. Fokkink, M. A. Cohen Stuart, M. Al-Hussein, P. H. H. Bomans, P. M. Frederik and G. J. Vancso, Macromolecules, 2006, 39, 2306–2315 CrossRef CAS.
- H. Wang, A. J. Patil, K. Liu, S. Petrov, S. Mann, M. A. Winnik and I. Manners, Adv. Mater., 2009, 21, 1805–1808 CrossRef CAS.
- F. He, T. Gädt, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2011, 133, 9095–9103 CrossRef CAS PubMed.
- J. Schmelz, F. H. Schacher and H. Schmalz, Soft Matter, 2013, 9, 2101–2107 RSC.
- T. Gadt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- S. F. Mohd Yusoff, M.-S. Hsiao, F. H. Schacher, M. A. Winnik and I. Manners, Macromolecules, 2012, 45, 3883–3891 CrossRef CAS.
- H. Qiu, G. Cambridge, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2013, 135, 12180–12183 CrossRef CAS PubMed.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842–8845 CrossRef CAS PubMed.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163–3173 RSC.
- M. Lazzari, D. Scalarone, C. E. Hoppe, C. Vazquez-Vazquez and M. A. Lòpez-Quintela, Chem. Mater., 2007, 19, 5818–5820 CrossRef CAS.
- Z.-X. Du, J.-T. Xu and Z.-Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- J. Zhang, L.-Q. Wang, H. Wang and K. Tu, Biomacromolecules, 2006, 7, 2492–2500 CrossRef CAS PubMed.
- T. Fujiwara, M. Miyamoto, Y. Kimura, T. Iwata and Y. Doi, Macromolecules, 2001, 34, 4043–4050 CrossRef CAS.
- D. Portinha, F. Boué, L. Bouteiller, G. Carrot, C. Chassenieux, S. Pensec and G. Reiter, Macromolecules, 2007, 40, 4037–4042 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- NIST SLD calculator, http://www.ncnr.nist.gov/resources/sldcalc.html.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013 DOI:10.1021/ma401634s.
- S. Grob, Molinspiration Property Calculation Service, http://www.molinspiration.com.
Footnotes |
| † Electronic supplementary information (ESI) available: Further polymer and nanostructure characterization. See DOI: 10.1039/c3py01048a |
| ‡ Lengths indicated in Table 2 and Fig. 6 are only representative of the cylindrical morphology. Spheres coexist in these systems, and are predominant in the assemblies where the fast water addition is used. For assemblies formed by the slow addition of water, spheres are still visible as well as very small cylinders, both of which have been counted in the calculation of the smaller dimension material. |
| This journal is © The Royal Society of Chemistry 2014 |