DOI:
10.1039/C3PY00911D
(Paper)
Polym. Chem., 2014,
5, 132-143
Synthesis characterization and bulk-heterojunction photovoltaic applications of new naphtho[1,2-b:5,6-b′]dithiophene–quinoxaline containing narrow band gap D–A conjugated polymers†
Received
10th July 2013
, Accepted 30th July 2013
First published on 31st July 2013
Abstract
Alternating donor-acceptor (D–A) π-conjugated copolymers, poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX-I) and poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(thiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX2-II), were designed and synthesized based on the same thiophene-bridged naphtho[1,2-b:5,6-b′]dithiophene donor moiety, differing only at the quinoxaline acceptor counterpart by either additional electron-donating alkyl chain substitution in the thienyl ring attached to the quinoxaline base (in PTNDTT-QX-I) or a change in the location of the outward alkoxy side chain substituent of the phenyl rings (to the meta-position) adjoining the quinoxaline base (in PTNDTT-QX-II). The effect of alkyl chain positioning on the thermal, optical, and electrochemical properties, as well as field effect transistors and solar cell performances of the copolymers, were investigated and the results were compared with a previously published copolymer, PTNDTT-QX, which features a similar quinoxaline unit but is alkoxy substituted at the position para to its peripheral phenyl rings. Both polymers exhibited excellent thermal stability, with thermal decomposition temperatures over 400 °C. They absorbed light in the 300–700 nm range and exhibited optical band gaps of about 1.70 and 1.73 eV for PTNDTT-QX-I and PTNDTT-QX-II, respectively. Precise control of the alkyl/alkoxy chain positioning has made it possible to tune the HOMO energy levels between −5.14 and −5.29 eV and the LUMO energy levels between −3.44 and −3.55 eV. Bulk heterojunction photovoltaic devices of the structure ITO/PEDOT:PSS/polymer:PC71BM/LiF/Al were fabricated by using the polymers as the donors and [6,6]-phenyl C71-butyric acid methyl ester (PC71BM) as the acceptor. Power conversion efficiencies (PCEs) of 1.28% and 1.61% respectively were achieved for the photovoltaic devices based on PTNDTT-QX-I/PC71BM and PTNDTT-QX-II/PC71BM under AM 1.5 G simulated 1-sun solar illumination.
Introduction
Because of their many promising merits such as low cost, light weight, easy fabrication, and ability to form flexible devices, polymer solar cells (PSCs) that include electron-donating conjugated polymers and electron-accepting fullerene derivatives have drawn substantial academic and industrial research attention over the past decades.1–9 The bulk-heterojunction (BHJ) device has been the most successful PSC device architecture, utilizing a blend of donor (D) and acceptor (A) materials with maximized interfacial area for effective charge separation and transport of charge carriers.10–12 The use of regioregular poly(3-hexylthiophene) (P3HT) as the electron-donor and [6,6]-phenyl C61 butyric acid methyl ester as the electron-acceptor has been studied extensively in the last few years, advancing the progress of PSCs immensely.13–17 Despite the great potential of P3HT as a photoactive donor material, enabling power conversion efficiencies (PCEs) of up to 4–5%, the relatively large band gap (1.9 eV) and small energy difference between the highest occupied molecular orbital (HOMO) energy level of P3HT and the lowest unoccupied molecular orbital (LUMO) energy level of the fullerene acceptors make further PCE enhancement impossible in PSCs that use P3HT. The need to pursue materials other than P3HT has led to much interest in the development of alternating donor–acceptor (D–A) conjugated polymers, whose structure of repeated conjugated donor and acceptor units offers the opportunity to tune the band gaps and energy levels by using donor and acceptor units of varying strengths.18–24 Following tremendous efforts in the development of new donor materials and in the optimization of devices, PSCs have finally overcome the drawbacks that originated with P3HT, and device efficiency has successfully passed 8%.25–30
One of the current developments in the effort to enhance the performance of photoactive semiconductor materials in PSCs is the incorporation of π-conjugated, rigidly fused thiophene rings on a conjugated polymer backbone, since fused thiophene rings can make the molecular backbone more rigid and coplanar, thereby enhancing effective π-conjugation, extending absorption, lowering the band gap, and facilitating charge transport.31–34 Charge transport mobility is one of the most important factors determining overall device performance. Recently, various rigidly fused π-conjugated thiophene monomers and polymers have been synthesized and used in polymer solar cells, in efforts to improve the photovoltaic performances of PSC devices.35–44 Among the large number of fused-ring structures, naphthodithiophene derivatives with two thiophene rings fused at the end of a naphthalene unit have attracted particular scientific interest in recent times due to their symmetrical, planar structure and their potential to afford high charge carrier mobility.45–47 The solar cell performance of naphtho[2,1-b:3,4-b′]dithiophene units have been evaluated extensively by You's group.48–50 Marks and co-workers have used the 5,10-bis(2-ethylhexyloxy)naphtho[2,3-b:6,7-b′]dithiophene building block in small molecular organic solar cells, with encouraging PCE results.51 In addition, we recently developed the first small molecules based on naphtho[1,2-b:5,6-b′]dithiophene, combining it with benzothiadiazole conjugated units.52 Efficiencies of up to 2.2% were achieved in a solution-processed organic solar cell. Furthermore, we also employed this new naphtho[1,2-b:5,6-b′]dithiophene unit to synthesize a D–A conjugated polymer, PTNDTT-QX (shown in Chart 1), by combining it with a quinoxaline acceptor for PSCs that demonstrated a PCE of up to about ∼1.5% in elementary device characterization.53 These preliminary results indicate that, in addition to naphtho[2,1-b:3,4-b′]dithiophene and naphtho[2,3-b:6,7-b′]dithiophene moieties, naphtho[1,2-b:5,6-b′]dithiophene also has potential as a building block for designing new conjugated small molecules/polymers for organic solar cell applications.
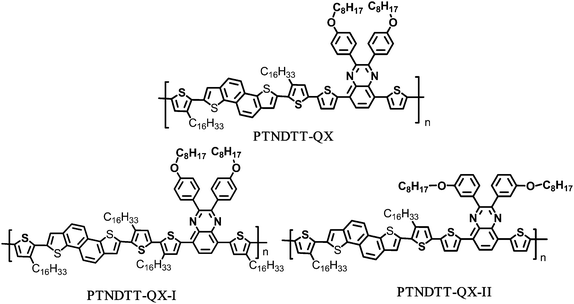 |
| | Chart 1 Chemical structure of the polymers. | |
In the present work, we developed two quinoxaline derivatives that differ in molecular structure with respect to the location of the alkyl and alkoxy groups. These quinoxaline acceptors were combined with the thiophene-bridged naphtho[1,2-b:5,6-b′]dithiophene unit to investigate the effects of the difference in alkyl chain positioning on the optoelectronic and photovoltaic properties of the resulting naphtho[1,2-b:5,6-b′]dithiophene–quinoxaline system. The new copolymers, poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(4-hexadecylthiophen-2-yl)-2,3-bis(4-octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX-I) and poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(thiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX-II) (see Chart 1 for structures), were synthesized by Stille polycondensation and studied to determine their thermal, optical, electrochemical, thin-film transistor and organic photovoltaic properties.
Experimental section
Materials
All chemicals and solvents were of reagent grade and were purchased from Aldrich, Fluka, TCI, or Lancaster Chemical Company. Toluene, chlorobenzene, tetrahydrofuran, and diethyl ether were distilled over sodium/benzophenone to keep them anhydrous before use. Chloroform was purified by refluxing with calcium hydride and then distilled. 2-Bromo-3-hexadecylthiophene, naphtho[1,2-b:5,6-b′]dithiophene (NDT) (1),54 2,3-bis-(4-octyloxyphenyl)-5,8-dibromo-quinoxaline (6),55 and 1-bromo-3-(octyloxy)benzene (10)56 were prepared according to the procedures described in the literature.
General instrumentation
1H and 13C NMR spectra were recorded on a JEOL FT-NMR (400 MHz) spectrophotometer using CDCl3 or CDCl2CDCl2 as the solvent. Chemical shifts were reported as δ values (ppm) relative to the internal standard tetramethylsilane (TMS). Elemental analyses were carried out using a CE Instruments Flash EA 1112 series. Number-average (Mn) and weight-average (Mw) molecular weights of the polymers were determined via gel permeation chromatography (GPC) with a Viscotec GPC system (Model 350 HTGPC). The calibration curve was constructed with a series of monodisperse polystyrene standards (Shodex). GPC analysis was performed with polymer–chloroform (HPLC grade) solutions at a flow rate of 1 mL min−1 at 25 °C. The UV-vis absorption spectra were obtained using a Shimadzu UV-2550 spectrophotometer. Photoluminescence (PL) spectra of films or solutions in chlorobenzene were obtained using a FP-6500 (JASCO). Thermogravimetric analysis (TGA) was carried out with a TA Instrument Q-50 at a scanning rate of 20 °C min−1 under a nitrogen atmosphere. The temperature of degradation (Td) corresponded to a 5% weight loss. Differential scanning calorimetry (DSC) experiments were performed on a TA Instrument (DSC 2910) at a heating rate of 15 °C min−1 under a nitrogen atmosphere. Cyclic voltammetry (CV) measurements for polymer films deposited on ITO glass were performed on a VersaSTAT3 (METEK), using a solution of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) (0.10 M) in chloroform, under argon, at a scan rate of 50 mV s−1 at room temperature. A Pt wire and Ag/AgCl were used as the counter and reference electrodes, respectively. The surface morphology was measured using a Digital Instruments Multimode atomic force microscope (AFM) controlled by a Nanoscope IIIa scanning probe microscope 20 controller.
OFET device fabrication and characterization
Thin-film transistor devices were fabricated on silicon wafers with a bottom-contact geometry (channel length, L = 12 μm, width, W = 120 μm). A heavily n-doped silicon wafer with a 300 nm thermal silicon dioxide (SiO2) layer was used as the substrate/gate electrode, with the top SiO2 layer serving as the gate dielectric. The SiO2 surface was cleaned, dried, and pretreated with a solution of 10 mM octyltrichlorosilane (OTS-8) in toluene at room temperature for 2 h under nitrogen to produce nonpolar and smooth surfaces, onto which the polymers were spin-coated. The semiconductor layer was spin-coated at 2000 rpm from a 0.5 wt% o-dichlorobenzene solution, to a thickness of 70 nm. Subsequently, a series of gold source–drain electrode pairs were deposited by vacuum evaporation through a shadow mask. Silicon oxide on the backside of the silicon wafer of the TFT device was removed with HF to provide a conductive gate contact. All device fabrication procedures and measurements were carried out in air at room temperature.
Photovoltaic device fabrication and characterization
Organic photovoltaic cells, with the structure glass/ITO/PEDOT:PSS/polymer:PC71BM/LiF/Al, were prepared on commercial ITO-coated glass substrates with a sheet resistance of 7 Ω □−1. Prior to use, the substrates were cleaned with deionized water. Sonication of the substrate in acetone followed by isopropanol was done to obtain clean substrates. A thin film of PEDOT:PSS (35 nm) (AI 4083, H. C. Starck) was spin-coated (3000 rpm, 40 s) onto the substrate and was dried at 140 °C for 20 min. The active layer was prepared with a thickness of about 70–100 nm on the surface of the PEDOT:PSS, using spin-coating with different polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC71BM (Nano-C, USA) ratios (from 1:1 to 1:4 w/w) in o-dichlorobenzene, and was dried at RT for 3 h in glove box under nitrogen. The devices were completed by deposition of a 0.5 nm layer of LiF and a 150 nm layer of Al. These layers were thermally evaporated at a pressure of 1 × 10−6 Torr at room temperature. The active area was 0.09 cm2. The current–voltage (I–V) characteristics of the photovoltaic devices in the dark and under white light illumination were measured at AM 1.5 G solar simulation (Newport) and 100 mW cm−2 conditions, adjusted with a standard PV reference cell (2 cm × 2 cm), a monocrystalline silicon solar cell, (calibrated at NREL, Colorado, USA) with a Keithley 2400 source-measure unit. The external quantum efficiency (EQE) was determined using a Polaronix K3100 spectrometer.
:PC71BM (Nano-C, USA) ratios (from 1:1 to 1:4 w/w) in o-dichlorobenzene, and was dried at RT for 3 h in glove box under nitrogen. The devices were completed by deposition of a 0.5 nm layer of LiF and a 150 nm layer of Al. These layers were thermally evaporated at a pressure of 1 × 10−6 Torr at room temperature. The active area was 0.09 cm2. The current–voltage (I–V) characteristics of the photovoltaic devices in the dark and under white light illumination were measured at AM 1.5 G solar simulation (Newport) and 100 mW cm−2 conditions, adjusted with a standard PV reference cell (2 cm × 2 cm), a monocrystalline silicon solar cell, (calibrated at NREL, Colorado, USA) with a Keithley 2400 source-measure unit. The external quantum efficiency (EQE) was determined using a Polaronix K3100 spectrometer.
Syntheses of the monomers
2,7-bis(trimethylstannyl)naphtho[1,2-b:5,6-b′]dithiophene (2).
To a solution of 1 (0.5 g, 2.08 mmol) in THF (20 mL), TMEDA (0.68 mL, 4.57 mmol) was added at −50 °C under stirring; 1.6 M n-butyllithium in hexane (4.57 mmol, 2.85 mL) was then added dropwise at −78 °C. The solution was stirred at −78 °C for 30 min and subsequently refluxed for 2 h under an N2 atmosphere. The reaction mixture was cooled to −78 °C, and trimethyltin chloride in THF (1.04 mL, 5.0 mmol) was added in one portion. The reaction mixture was warmed to room temperature and was stirred for 6 h. The reaction was quenched with 20 mL of water and then extracted with dichloromethane. The organic layer was washed twice with 20 mL of brine and dried with anhydrous MgSO4. The solvent was removed, and the resulting residue was recrystallized from acetone to yield 0.82 g (70%) of 2 as a white solid. 1H NMR (400 MHz, CDCl3, ppm) δ: 7.56 (s, 2H, ArH), 7.92 (d, 2H, ArH), 8.05 (d, 2H, ArH), 0.47 (s, 18H, Sn(CH3)3). 13C NMR (100 MHz, CDCl3, ppm) δ: 143.3, 138.7, 138.5, 133.2, 125.4, 121.9, 121.4. Elemental analysis: calculated for C20H24S2Sn2: C, 42.44; H, 4.27; S, 11.33. Found: C, 42.51; H, 4.19; S, 11.39.
2,7-Bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene (4).
In a 50 mL flame-dried two-neck flask fitted with a condenser, 2,7-bis(trimethylstannyl)naphtho[1,2-b:5,6-b′]dithiophene (2) (2.0 g, 3.53 mmol), 2-bromo-3-hexadecylthiophene (3.41 g, 8.82 mmol, 2.5 eq.), and dichlorobis-(triphenylphosphine)palladium(II) (118 mg, 0.17 mmol) were added and subjected to three vacuum–argon fill cycles. Argon-degassed chlorobenzene (20 mL) was added, and the mixture was stirred for 20 min under argon. The reaction mixture was heated to reflux for 24 h and was monitored by TLC. After the reaction was complete, the reaction was quenched with 20 mL of water and the compound was extracted with dichloromethane. The organic layer was washed twice with 20 mL of brine and dried with anhydrous MgSO4. Dichloromethane was removed under reduced pressure. The resulting crude product was purified by column chromatography using hexane as the eluent to afford compound 4 (2.1 g, 70%). 1H NMR (400 MHz, CDCl3, ppm) δ: 8.01 (d, 2H, ArH), 7.88 (d, 2H, ArH), 7.47 (s, 2H, ArH), 7.26 (s, 2H, ArH), 7.08 (d, 2H, ArH), 2.92 (t, 4H), 1.74 (m, 4H), 1.44–1.24 (m, 52H), 0.88 (t, 6H). 13C NMR (CDCl3, 100 MHz, ppm) δ: 140.95, 138.60, 137.77, 135.95, 130.44, 125.81, 124.89, 123.67, 122.72, 121.54, 32.13, 31.13, 30.96, 29.85, 29.83, 29.75, 29.69, 29.56, 22.89, 14.32. Elemental analysis: calculated for C54H76S4, C, 76.00; H, 8.98; S, 15.03. Found: C, 76.06; H, 8.91; S, 15.07.
2,7-Bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene distannane (5).
2,7-Bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene distannane (5) was prepared using a method similar to that given above for compound 2. In brief, to a solution of 4 (2.0 g, 2.34 mmol) in THF (20 mL), TMEDA (0.77 mL, 5.15 mmol) was added at −50 °C under stirring, and then 1.6 M n-butyllithium in hexane (5.61 mmol, 3.15 mL) was added dropwise at −78 °C under an N2 atmosphere. After 30 min, the solution was refluxed for 2 h. The reaction mixture was cooled to −78 °C, and a 1.0 M solution of trimethyltin chloride (5.85 mmol, 5.85 mL) was added in one portion. The reaction mixture was warmed to room temperature and was stirred for 6 h. The reaction was quenched with 20 mL of water and the compound was extracted with dichloromethane. The solvent was removed and the residue was recrystallized from acetone to yield 2.0 g (72%) of 5 as a yellow solid. 1H NMR (400 MHz, CDCl3, ppm) δ: 7.96 (d, 2H, ArH), 7.86 (d, 2H, ArH), 7.45 (s, 2H, ArH), 7.06 (s, 2H, ArH), 2.93 (t, 4H), 1.77–1.67 (m, 4H), 1.43–1.23 (m, 52H), 0.89 (t, 6H), 0.50 (s, 18H, Sn(CH3)3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 141.77, 138.52, 138.26, 137.67, 137.58, 136.22, 135.98, 125.52, 123.40, 122.80, 122.42, 121.23, 31.91, 30.87, 30.73, 29.70, 29.62, 29.46, 29.36, 29.24, 29.69, 22.69, 14.12. Elemental analysis: calculated for C60H92S4Sn2, C, 61.12; H, 7.86; S, 10.88. Found: C, 61.23; H, 7.71; S, 10.80.
5,8-Bis(4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (8).
To a 50 mL flame-dried two-neck flask, 5,8-dibromo-2,3-bis(4-(octyloxy)phenyl)quinoxaline (6) (0.7 g, 1.0 mmol), 2-(4-hexadecylthiophen-2-yl)-4,4′,5,5′-tetramethyl-1,3,2-dioxaborolane (7) (1.04 g, 2.41 mmol), and Pd(PPh3)4 (116 mg, 10 mol%) were added and subjected to three vacuum–nitrogen fill cycles. Nitrogen-degassed toluene (18 mL) and 2 M aqueous solution of K2CO3 (6 mL) were subsequently added. The reaction mixture was heated to reflux for 24 h and was monitored by TLC. After the reaction was complete, the reaction was quenched with water and the organic layer was extracted with dichloromethane. Dichloromethane was removed under reduced pressure. The resulting crude product was purified by column chromatography, using dichloromethane as the eluent, to produce 8 (0.8 g, 69%). 1H NMR (300 MHz, CDCl3, ppm) δ: 8.03 (s, 2H), 7.73 (d, 6H), 7.08 (S, 2H), 6.89 (d, 4H), 3.99 (t, 4H), 2.68 (t, 4H), 1.81 (m, 4H), 1.70 (m, 4H), 1.34–1.25 (m, 72H), 0.87 (t, 12H). 13C NMR (CDCl3, 100 MHz, ppm) δ: 160.17, 151.23, 142.94, 138.88, 137.25, 132.07, 131.47, 131.21, 128.08, 126.60, 123.77, 114.40, 68.22, 32.14, 32.04, 30.87, 29.93, 29.88, 29.80, 29.66, 29.60, 29.57, 29.51, 29.46, 26.30, 22.90, 22.88, 14.31. Elemental analysis: calculated for C76H114N2O2S2, C, 79.25; H, 9.98; N, 2.43; S, 5.57; Found: C, 79.38; H, 10.05; N, 2.49; S, 5.41.
5,8-bis(5-bromo-4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (9).
A solution of 5,8-bis(4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (8) (0.6 g, 0.52 mmol) in CHCl3 was cooled to 0 °C while protected from light. N-bromosuccinimide (0.185 g, 1.04 mmol) was added in small portions over 20 min, and the reaction was then stirred for 3 h. The mixture was poured into water (100 mL) and was extracted with CHCl3. The organic layer was dried over anhydrous MgSO4, and the solvent was removed by rotary evaporation. The crude product was purified by recrystallization from MeOH to yield compound 11 as a red-yellow solid (0.670 g, 94%). 1H NMR (CDCl3, 300 MHz, ppm) δ: 7.97 (s, 2H), 7.68 (d, 4H), 7.49 (s, 2H), 6.85 (d, 4H), 4.01 (t, 4H), 2.62 (t, 4H), 1.81 (m, 4H), 1.66 (m, 4H), 1.35–1.25 (m, 72H), 0.87 (t, 12H).13C NMR (CDCl3, 100 MHz, ppm) δ: 160.27, 151.64, 141.28, 137.96, 136.64, 132.11, 130.95, 130.31, 126.38, 125.16, 114.37, 114.15, 68.27, 32.14, 32.08, 30.05, 29.93, 29.88, 29.80, 29.75, 29.62, 29.58, 29.53, 29.47, 26.31, 22.90, 14.34. Elemental analysis: calculated for C76H112Br2N2O2S2, C, 69.70; H, 8.62; N, 2.14; S, 4.90; Found: C, 69.64; H, 8.57; N, 2.31; S, 4.82.
1,2-Bis(3-(octyloxy)-phenyl)-ethane-1,2-dione (13).
The intermediate 1,2-bis(3-(octyloxy)-phenyl)-ethane-1,2-dione (13) was synthesized using a procedure similar to that reported for 1,2-bis-[4-(2-ethyl-hexyloxy)-phenyl]-ethane-1,2-dione.57 A Grignard reagent was prepared by drop-wise addition of 1-bromo-3-(octyloxy)benzene (7.41 g, 26 mmol) in THF (9 mL) to a suspension of magnesium (0.74 g, 30.9 mmol) in THF (15 mL). In a separate flask, a solution of LiBr (4.52 g, 52 mmol) was added to a stirred suspension of CuBr (3.73 g, 26 mmol) in THF (19 mL). The mixture was stirred until it became homogeneous and was then cooled in an ice-water bath. The Grignard reagent was added to the LiBr–CuBr suspension. Oxalyl chloride (1.55 g, 12.22 mmol) was added to the cold solution, and after 20 min the reaction was quenched with saturated NH4Cl. The mixture was extracted with dichloromethane. The combined ethyl acetate extract was dried over anhydrous Na2SO4 and the solvent was removed to afford a yellow solid. This product was purified by column chromatography using hexane:dichloromethane (5:1) as the eluent. Compound 13 was obtained as a light-yellow solid (3.75 g, 31%). 1H NMR (CDCl3, 300 MHz, ppm) δ: 7.53 (s, 2H), 7.41 (d, 2H), 7.39 (dd, 2H), 7.21 (d, 2H), 3.90 (t, 4H), 1.74 (m, 4H), 1.57–1.30 (m, 20H), 0.90 (t, 6H).13C NMR (CDCl3, 100 MHz, ppm) δ: 194.56, 159.85, 134.19, 129.92, 122.94, 122.23, 113.54, 70.76, 39.38, 30.46, 29.04, 23.80, 23.01, 14.06, 11.09. Elemental analysis: calculated for C30H42O4, C, 77.21; H, 9.07; Found: C, 77.39; H, 9.12.
5,8-bis(5-bromothiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline (19).
To a solution of 4,7-bis(5-bromothiophen-2-yl)benzo[1,2,5]thiadiazole (17) (1.41 g, 3.09 mmol) in acetic acid (48 mL) was added zinc dust (2.4 g, 38.4 mmol). The reaction mixture was stirred at 80° C over a period of 3 h and then filtered. The pH of the solution was increased to over 7 by drop-wise addition of 2 M NaOH. The aqueous solution was subsequently extracted with dichloromethane. The organic phase was dried over Na2SO4 and the solvent was evaporated in vacuo. The orange solid thus obtained was dissolved in acetic acid (48 mL). 13 (942 mg, 2.02 mmol) was added to the solution and the reaction mixture was stirred overnight at 60° C. The precipitate obtained after the reaction was collected, washed with water, and purified with column chromatography using hexane–dichloromethane (4:1) as the eluent to afford 5,8-bis(5-bromothiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline (19) as orange crystals (1.1 g, 60%). 1H NMR (CDCl3, 300 MHz, δ): 8.09 (s, 2H), 7.57 (d, 2H), 7.55 (s, 2H), 7.22 (t, 2H), 7.12 (d, 2H), 7.10 (d, 2H), 6.98 (d, 2H), 4.04 (t, 4H), 1.82 (m, 4H), 1.54 (m, 4H), 1.34 (m, 16H) 0.90 (t, 6H). 13C NMR (CDCl3, 100 MHz, δ): 159.36, 151.92, 139.47, 139.37, 136.42, 130.53, 129.03, 125.62, 125.43, 122.90, 117.17, 115.17, 68.32, 31.85, 29.41, 29.33, 29.3, 26.17, 22.68, 14.11. Elemental analysis: calculated for C44H48Br2N2O2S2, C, 61.39; H, 5.62; N, 3.25; S, 7.45; Found: C, 61.14; H, 5.79; N, 3.19; S, 7.51.
Syntheses of the polymers
Poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX-I).
2,7-Bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene distannane (5) (515 mg, 0.44 mmol; 1.1 eq.) and 5,8-bis(5-bromo-4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (9) (520 mg, 0.39 mmol; 1.0 eq.) were dissolved in anhydrous chlorobenzene (20 mL). The solution was deoxygenated by flushing with argon for 30 min. Pd2(dba)3 (18 mg, 5 mol% with respect to the monomer) and P(o-tolyl)3 (12 mg, 10 mol%) were added to the reaction mixture under an argon atmosphere. The reaction mixture was stirred at reflux for 24 h under an argon atmosphere. After the reaction mixture was cooled to room temperature, the resulting polymer was slowly poured into 200 mL of well stirred methanol containing 20 mL of hydrochloric acid. The precipitate was collected on a membrane filter and was subjected to sequential Soxhlet extraction with methanol, acetone, and hexane to remove the low molecular weight fractions. The desired polymer was then collected through extraction with chloroform and reprecipitated into methanol to afford the polymer (74%). 1H NMR (300 MHz, CDCl3, ppm) δ: 8.2–7.1 (m, br, 20H), 4.2 (br, 4H), 3.1 (m, br, 8H), 1.9–0.8 (m, br, 154H). GPC (THF) Mn = 13.57 kD; Mw = 32.25 kD; PDI = 2.4.
Poly[2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene-5,5′-diyl-alt-5,8-bis(thiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline-5,5′-diyl] (PTNDTT-QX-II).
A similar copolymerization as that given above for PTNDTT-QX-I was carried out between monomers 5 and 19 to afford the polymer PTNDTT-QX-II. In brief, compound 5 (801 mg, 0.68 mmol; 1.1 eq.), compound 19 (530 mg, 0.61 mmol; 1.0 eq.), Pd2(dba)3 (28 mg, 5 mol% with respect to the monomer) and P(o-tolyl)3 (18 mg, 10 mol%) were refluxed in anhydrous chlorobenzene (20 mL) for 24 h under an argon atmosphere. The reaction mixture was cooled to room temperature, precipitated into methanol, and purified following the procedure described above to yield polymer PTNDTT-MQX (68%). 1H NMR (300 MHz, CDCl3, ppm) δ: 8.2–7.1 (m, br, 22H), 4.1 (br, 4H), 2.9 (m, br, 4H), 1.9–0.8 (m, br, 92H). GPC (THF) Mn = 8.3 kD; Mw = 68.1 kD; PDI = 8.2.
Results and discussion
Synthesis and thermal stability
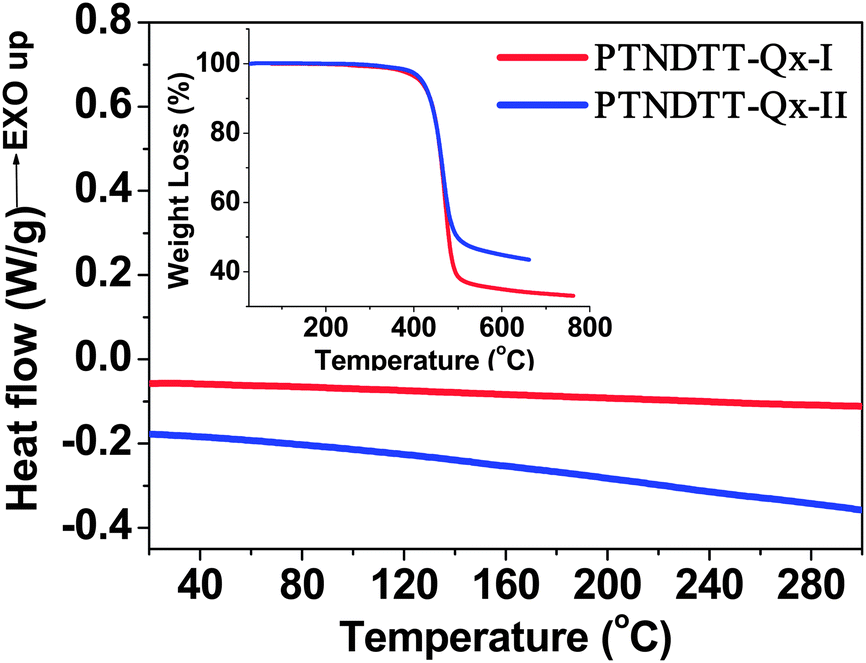
The general synthetic routes for the monomers and polymers are presented in Scheme 1. Monomer 2,7-bis(3-hexadecylthiophene- 2-yl)naphtho[1,2 b:5,6 b′]dithiophene (5) was synthesized starting from naphtho[1,2 b:5,6 b′]dithiophene (1) following our previously reported method.52 A Pd(PPh3)4-catalyzed Suzuki coupling reaction between 5,8-dibromo-2,3-bis(4-(octyloxy)phenyl)quinoxaline (6) and 2-(4-hexadecylthiophen-2-yl)-4,4′,5,5′-tetramethyl-1,3,2-dioxaborolane (7) produced 5,8-bis(4-hexadecylthiophen-2-yl)-2,3-bis(4 (octyloxy)phenyl)quinoxaline (8), which was subsequently brominated using N-bromosuccinimide (NBS) to afford the monomer 5,8-bis(5-bromo-4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (9). The other quinoxaline-based monomer, 5,8-bis(5-bromothiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline (19) was synthesized in a multi-step synthesis as outlined in Scheme 1. In brief, 1,2-bis(3-(octyloxy)phenyl)ethane-1,2-dione (13) was synthesized starting from 3-bromophenol (10) in two steps, similar to the method described for 2-bis-[4-(2-ethyl-hexyloxy)-phenyl]-ethane-1,2-dione by Andersson et al.,57 while 3,6-bis(5-bromothiophen-2-yl)cyclohexa-3,5-diene-1,2-diamine (18) was generated from 4-dibromo-2,1,3-benzothiadiazole (14).58 The intermediates 18 and 13 were subjected to a condensation reaction in the presence of Zn/acetic acid to yield the target monomer 5,8-bis(5-bromothiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline (19). The purity of all intermediates and the final monomers was confirmed by 1H NMR, 13C NMR and elemental analysis. The synthesis of polymer PTNDTT-QX-I was accomplished by Stille coupling copolymerization between 2,7-bis(3-hexadecylthiophene-2-yl)naphtho[1,2-b:5,6-b′]dithiophene (5) and 5,8-bis(5-bromo-4-hexadecylthiophen-2-yl)-2,3-bis(4-(octyloxy)phenyl)quinoxaline (9) using Pd2(dba)3/P(o-tolyl)3 as a catalyst. A similar copolymerization between monomers 5 and 5,8-bis(5-bromothiophen-2-yl)-2,3-bis(3-(octyloxy)phenyl)quinoxaline (19) afforded the other quinoxaline-based polymer, PTNDTT-QX-II. Differential scanning calorimetry was performed at a heating rate of 10 °C min−1 under an N2 atmosphere to measure the phase transition temperatures of the copolymers. In differential scanning calorimetry analyses, none of the samples exhibited distinct thermal transition peaks in the scanning range of 20 °C to 300 °C, clearly suggesting that both the polymers were mostly amorphous in nature (Fig. 1). Thermogravimetric analysis (TGA) was used to investigate the thermal stabilities of the polymers (Fig. 1, inset). Table 1 summarizes the corresponding decomposition temperatures (Td, 5% weight loss). The decomposition temperatures of PTNDT-QX-I and PTNDT-QX-II were 414 °C and 418 °C, respectively, indicating suitable thermal stability for photovoltaic applications.
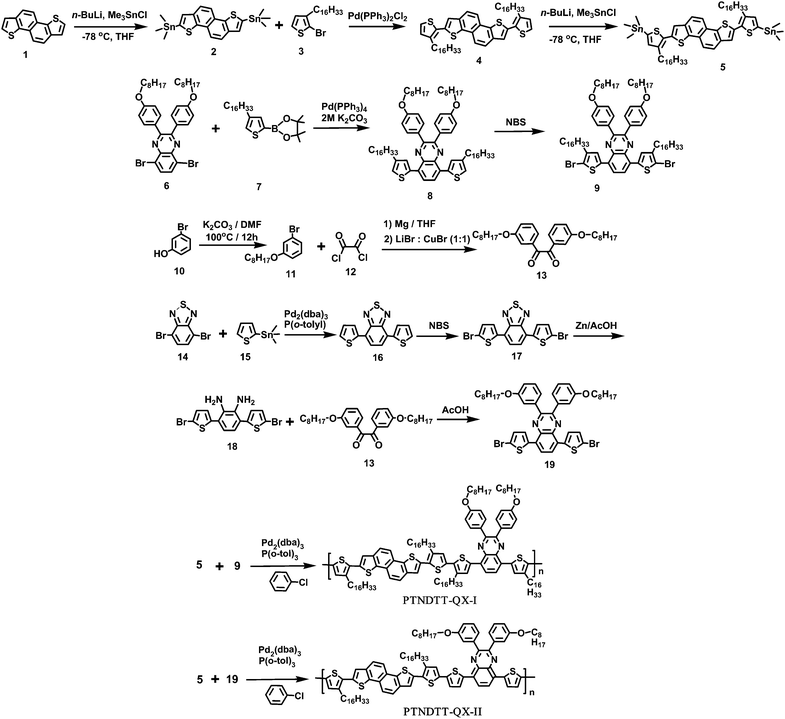 |
| | Scheme 1 Synthetic scheme for monomers and copolymers. | |
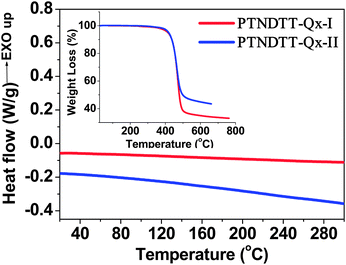 |
| | Fig. 1 DSC traces of the polymers at a scanning rate of 10 °C min−1 in the second heating cycle (inset: TGA curves of the polymers with a heating rate of 10 °C min−1). | |
Table 1 Molecular weight and thermal properties of copolymers
| Polymers |
Yield (%) |
M
n
(kD) |
M
w
(kD) |
PDI |
T
d (°C) |
|
Data from ref. 51.
|
|
PTNDTT-QX
|
65 |
12.3 |
78.04 |
6.32 |
411 |
|
PTNDTT-QX-I
|
74 |
13.57 |
32.25 |
2.4 |
414 |
|
PTNDTT-QX-II
|
68 |
8.3 |
68.10 |
8.2 |
418 |
Optical properties
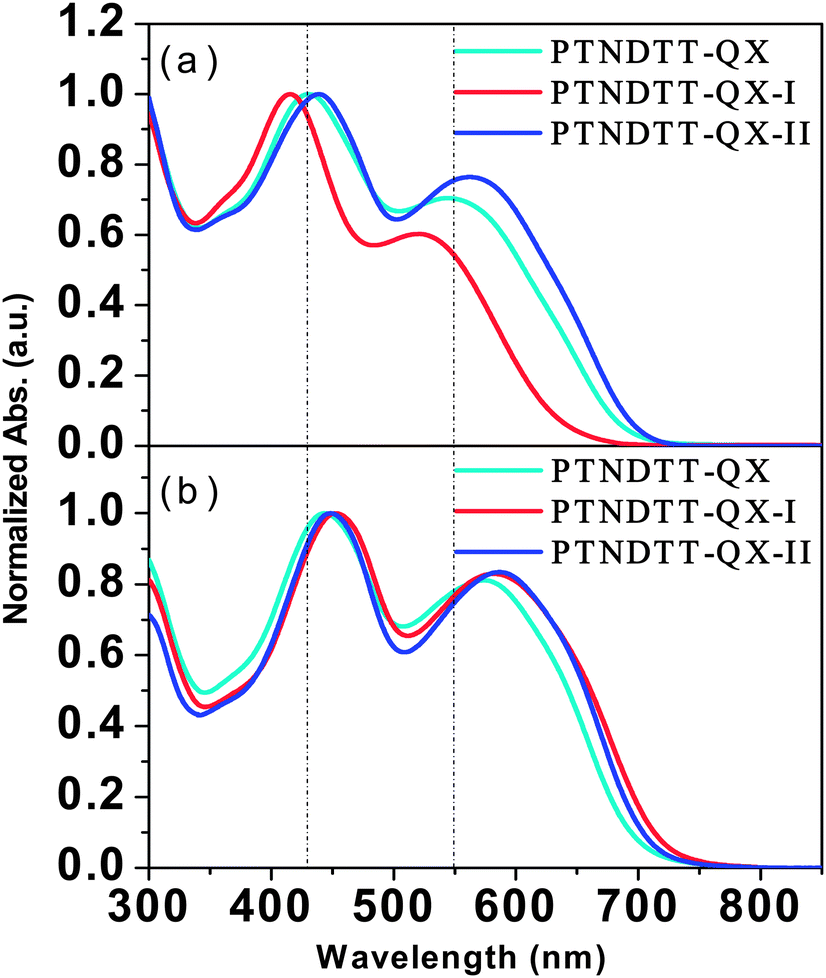
The absorption spectra of the new copolymers PTNDTT-QX-I and PTNDTT-QX-II were recorded both in ortho-dichlorobenzene (ODCB) solvent and as solid thin films (Fig. 2). We compared these absorption spectra to that of our previously reported copolymer PTNDTT-QX,53 to fully understand the effect of the positioning of the alkyl/alkyloxy groups in the copolymers on the corresponding optical properties both in solution and as thin films. Table 2 summarizes the related optical properties of the copolymers. Like PTNDTT-QX, the copolymers PTNDTT-QX-I and PTNDTT-QX-II both absorb broadly in the 350–700 nm region, with two distinct absorption bands corresponding to the π–π* transition of the NDT–QX-based conjugated backbone and their intramolecular charge transfer transition (ICT). PTNDTT-QX and PTNDTT-QX-II have quite similar absorption spectra in ODCB solution; the main absorption peaks of PTNDTT-QX-II are at 439 and 563 nm (Fig. 2a). In contrast, both absorption peaks of PTNDTT-QX-I were found to be blue-shifted relative to those of PTNDTT-QX, originating at 415 and 522 nm; the band maxima at longer wavelength (∼522 nm) were also relatively weak. A seemingly reasonable explanation for this blue shift and relatively weaker absorption is that the incorporation of the hexadecanyl group at the 4-position of the thiophene units attached to the quinoxaline acceptor moiety distorted the conjugated backbone, sterically hindering the effective intermolecular stacking, and reducing the effective conjugation length. However, simply changing the alkoxy group from the para (PTNDTT-QX) to the meta (PTNDTT-QX-II) position of the phenyl rings of the quinoxaline units did not produce any such adverse effect, which is quite similar to the results observed for the copolymers containing thieno[3,2 b]thiophene–quinoxaline units.59
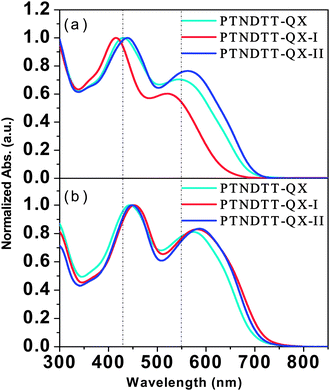 |
| | Fig. 2 Normalized UV-vis absorption spectra of the copolymers at room temperature: (a) in dilute o-dichlorobenzene solution and (b) as thin films. | |
Table 2 Optical, electrochemical and field effect transistor properties of the copolymers
| Polymers |
λ
max
(nm) |
λ
max
(nm) |
λ
onset
(nm) |
E
optg
(eV) |
E
ox
(V)/eHOMO (eV) |
LUMOf (eV) |
μ
(cm2 V−1 s−1) |
I
on/Ioffg |
|
Measured in ortho-dichlorobenzene solution.
Film spin-coated from ortho-dichlorobenzene solution.
Optical band gap, Eoptg = 1240/(λonset)film.
Potential determined by cyclic voltammetry in 0.10 M Bu4NPF6–CH3CN.
HOMO = −e(4.4 + Eoxonset) (eV).
LUMO = Eoptg + HOMO.
Hole mobilities (μ) and on–off ratios (Ion/Ioff) were extracted from transfer curves at the saturation regime.
Data from ref. 51.
|
|
PTNDTT-QX
|
430, 544 |
444, 572 |
698 |
1.77 |
0.84/−5.24 |
−3.47 |
5 × 10−5 |
5 × 102 |
|
PTNDTT-QX-I
|
415, 522 |
453, 578 |
726 |
1.70 |
0.74/−5.14 |
−3.14 |
7.6 × 10−4 |
102 |
|
PTNDTT-QX-II
|
439, 563 |
448, 585 |
714 |
1.73 |
0.89/−5.29 |
−3.55 |
7.6 × 10−5 |
103 |
The absorption spectra for both PTNDTT-QX-I and PTNDTT-QX-II in solid films were red shifted as compared with their solution spectra (Fig. 2b). A greater red shift (approximately ∼40 nm at the longer wavelength) was observed in the case of PTNDTT-QX-I. The larger spectral shift for PTNDTT-QX-I in its solid state as compared with its solution was probably related to the increased extent of π–π stacking by alignment of the backbone through efficient chain planarization. The absorption onsets of PTNDTT-QX-I and PTNDTT-QX-II were located at about ∼726 nm and ∼714 nm, respectively, which correspond to optical band gaps of 1.70 eV and 1.73 eV. The thin-film optical band gaps thus obtained for PTNDTT-QX-I and PTNDTT-QX-II were 0.07 and 0.04 eV lower, respectively, than that of PTNDTT-QX. The slightly lower thin film band gap (0.03 eV) of PTNDTT-QX-I relative to that of PTNDTT-QX-II could have been due to the higher molecular weight of PTNDTT-QX-I, as a larger molecular weight can translate into a greater conjugation length.
Electrochemical properties
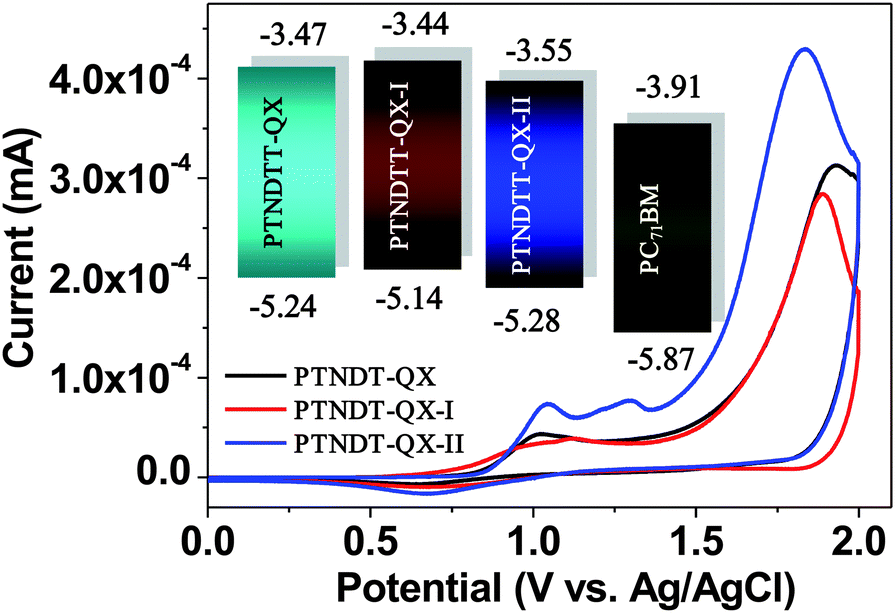
To examine the electrochemical properties of the copolymers, cyclic voltammetry (CV) was carried out using Ag/AgCl as a reference electrode in a 0.1 M solution of Bu4NPF6 in acetonitrile at room temperature under argon, with a scan rate of 50 mV s−1. Prior to CV measurements, the reference electrode was calibrated using the ferrocene/ferrocenium (Fc/Fc+) redox couple as an external standard. The onset potential of the Fc/Fc+ redox couple was detected at 0.4 V relative to the Ag/AgCl reference electrode. Thus, the HOMO energy levels of the polymers were calculated according to the following equation: EHOMO = −e(Eoxonset + 4.40) (eV). Since we are unable to obtain reproducible reduction peaks, the LUMO energy levels of the polymers were calculated based on the correlation between the optical band gap and the HOMO (ELUMO = EHOMO + Eg). The CV curves for the polymers, including a reference curve of copolymer PTNDTT-QX from a previous report, are shown in Fig. 3. Table 2 summarizes the corresponding electrochemical characteristics, including onset oxidation (Eoxonset) and the estimated energy levels (both HOMO and LUMO) for all three copolymers. Similar to PTNDTT-QX, both PTNDTT-QX-I and PTNDTT-QX-II demonstrate irreversible oxidation peaks. Eoxonset was observed in PTNDTT-QX-I and PTNDTT-QX-II at 0.74 V and 0.89 V, respectively; while the respective HOMO levels estimated from Eoxonset were −5.14 and −5.29 eV. The energy levels of PTNDTT-QX-I and PTNDTT-QX-II were further compared with those of PTNDTT-QX using the energy level diagram (Fig. 3 inset). The HOMO energy level of PTNDTT-QX-I shifted upwards relative to that of PTNDTT-QX. Therefore, the incorporation of electron-donating alkyl substituents at the thiophene rings adjacent to the quinoxaline moiety renders the resulting conjugated backbone of PTNDTT-QX-I more electron-rich. Conversely, the HOMO level of PTNDTT-QX-II was lowered with respect to that of PTNDTT-QX, which could be attributed to the less electron-rich character of PTNDTT-QX-II, since the alkoxy groups present at the meta-position of the phenyl rings, which were attached as side chain substituents to the quinoxaline base, exert a weaker electron-donating effect relative to the para-position, as a result of lower resonance stabilization energy.
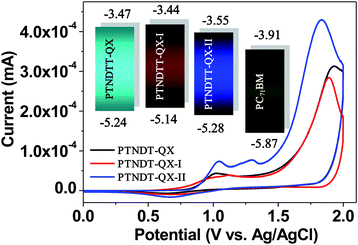 |
| | Fig. 3 Cyclic voltammograms of copolymer films deposited on ITO glass in an acetonitrile solution of 0.1 M n-Bu4NF6 at a scan rate of 50 mV s−1; inset: energy level diagram showing the HOMO and LUMO energy levels of the copolymers and PC71BM. | |
Field-effect transistor characteristics
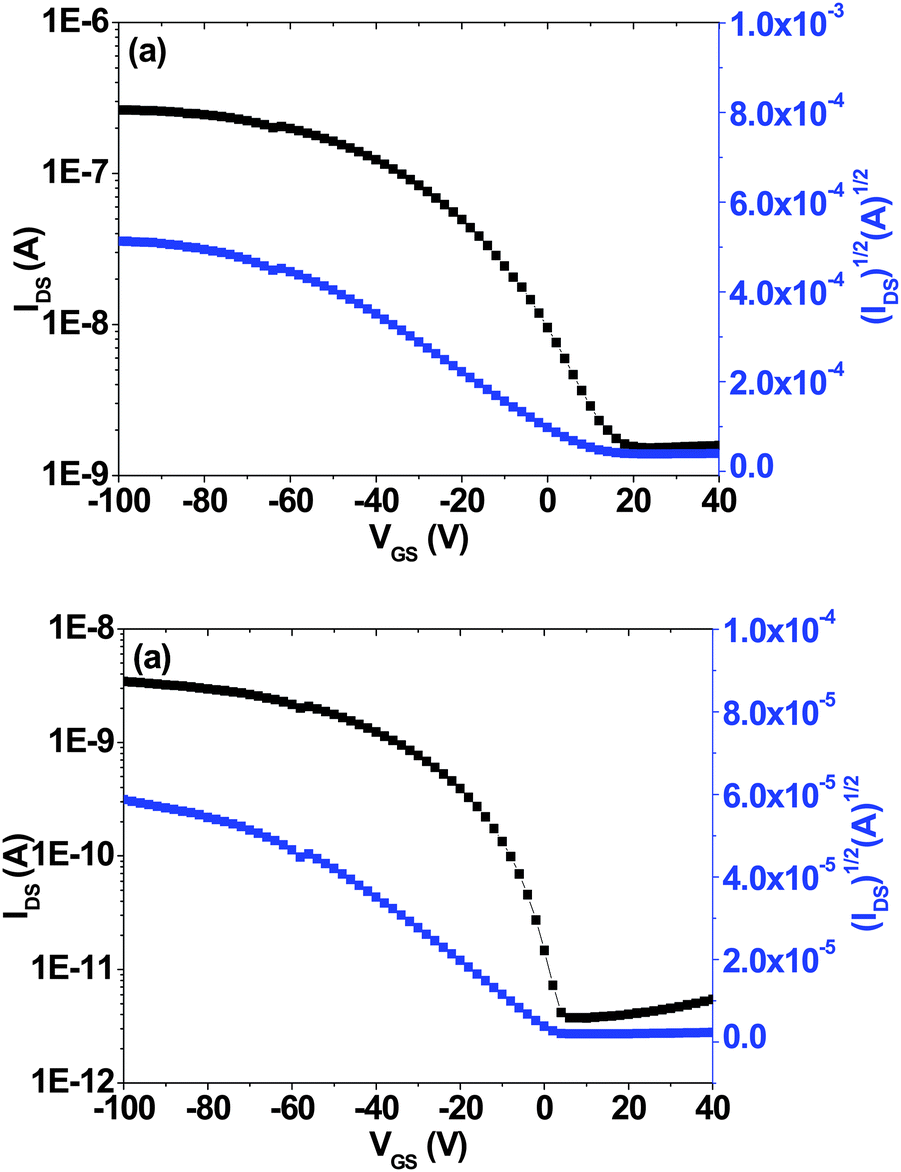
The field-effect hole mobilities of the copolymers were investigated by fabricating thin-film organic field-effect transistors (OFETs). These devices were fabricated in a bottom-contact geometry (channel length = 12 μm, width = 120 μm). Both PTNDTT-QX-I and PTNDTT-QX-II showed typical p-channel output characteristics. In the saturated region, the source–drain current (ID) and gate voltage (VG) are related to the device and hole mobility of the materials by the equation: ID = (W/2L)μCi(VG − VT),2 where ID is the drain current in the saturated regime, W and L are the channel width and length, respectively, μ is the field-effect mobility, Ci is the capacitance per unit area of the gate dielectric layer, and VG and VT are the gate and threshold voltages, respectively. The saturation region field-effect mobility of the copolymers was calculated from the transfer characteristics of the OFETs by plotting IDS1/2vs. VGS (Fig. 4). Table 2 summarizes the resulting hole mobilities from measurements of the corresponding devices. The hole-mobility of PTNDTT-QX-II was found to be 7.6 × 10−5 cm2 V−1 s−1 with an on–off current ratio of 103 in the PTNDTT-QX-II OFET, which is quite similar to that of our reference PTNDTT-QX polymer (5 × 10−5 cm2 V−1 s−1). On the other hand, the saturation region mobility of PTNDTT-QX-I was found to be 7.6 × 10−4 cm2 V−1 s−1, which is one order of magnitude higher than that of our previously reported copolymer PTNDTT-QX. It is interesting to note that the incorporation of an alkyl substituent on the thiophene (attached to the quinoxaline unit), which conceivably could impede close chain packing by twisting the conjugated backbone, did not have any negative impact on hole mobility for PTNDTT-QX-I. Rather, the hole mobility of PTNDTT-QX-I was found to be quite improved as compared to copolymer PTNDTT-QX. The higher hole mobility of PTNDTT-QX-I was likely related to its higher molecular weight with well-controlled polydispersity, since carrier mobility depends not only on the planarity of polymers, which facilitates intra- and interchain charge transfers through enhanced π⋯π overlap, but also depends strongly on the molecular weight of the polymer.60,61
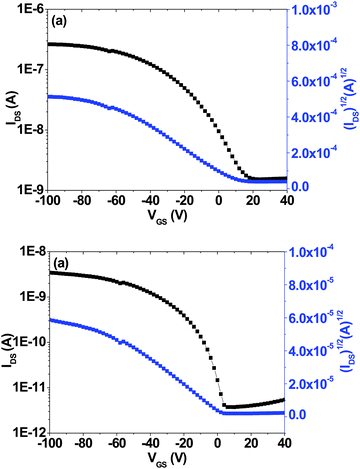 |
| | Fig. 4 Transfer curves of bottom-gate top-contact thin film transistor (TFT) devices based on (a) PTNDTT-QX-I and (b) PTNDTT-QX-II. | |
Photovoltaic properties
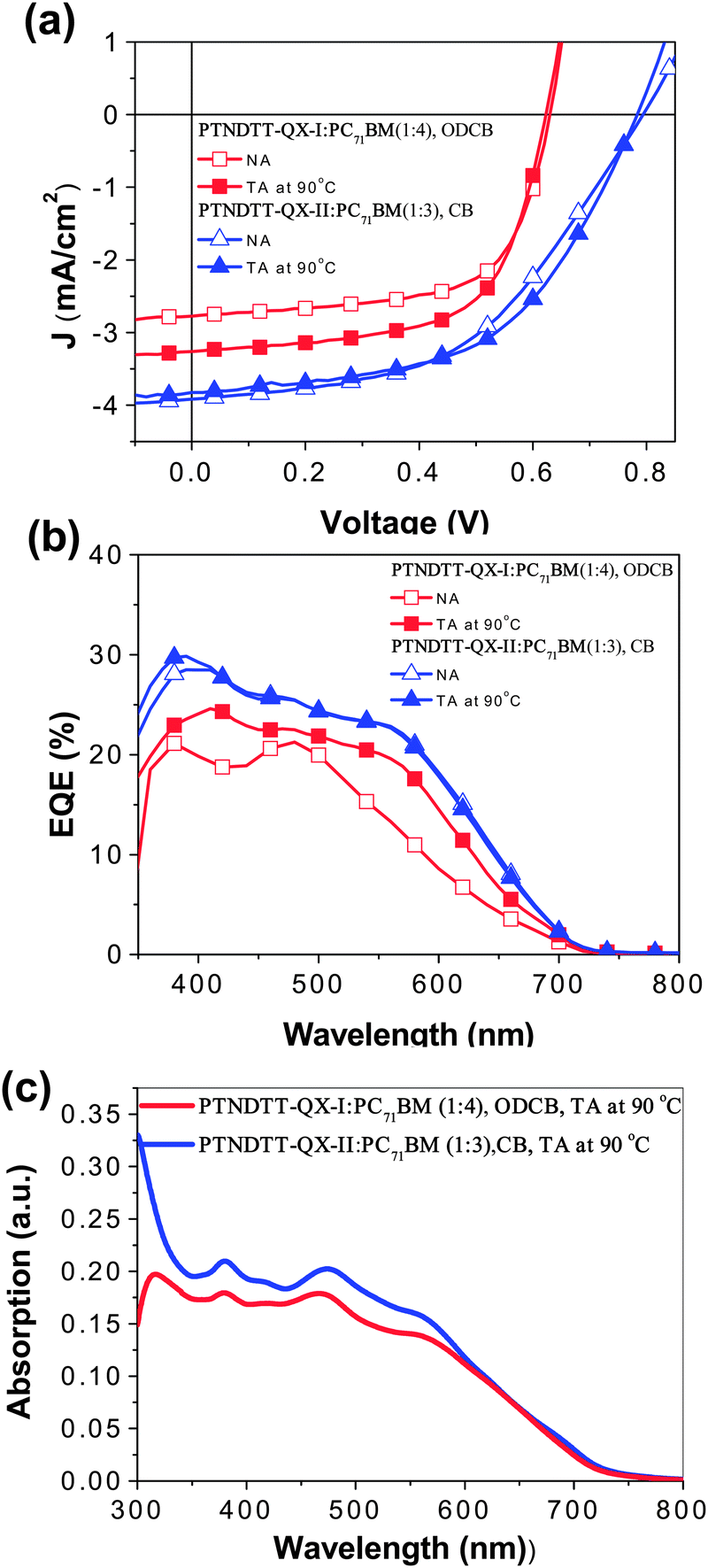
To investigate the potential use of the copolymers for photovoltaic applications, bulk-heterojunction photovoltaic cells based on blends of PTNDTT-QX-I and PTNDTT-QX-II with PC71BM at different weight ratios (1:1 to 1:4 w/w) were constructed initially using the conventional device structure of ITO/PEDOT:PSS/copolymer:PC71BM/LiF/Al. Current density–voltage (J–V) curves of these devices measured under simulated AM 1.5 G illumination (100 mW cm−2) are presented in Fig. S4 in the ESI,† and the devices' photovoltaic parameters, short circuit current (Jsc), open circuit voltage (Voc), fill factor (FF), and PCE, are summarized in Table 3. Table 3 also includes previously reported data of our reference copolymer PTNDTT-QX in its best PSC device conditions. Among the solar cell devices based on blends of PTNDTT-QX-I and PC71BM from o-DCB solvent, the best device performance was observed at a ratio of 1:4 (w/w). Without any special treatment, the maximum PCE obtained was 1.13% for PTNDTT-QX-I, with Jsc of 2.77 mA cm−2, Voc of 0.63 V, and FF of 0.64. In contrast, blends of PTNDTT-QX-II and PC71BM at a ratio of 1:3 (w/w) exhibited the highest PCE of up to 1.23% with Jsc of 2.99 mA cm−2, Voc of 0.78 V, and FF of 0.52. In the case of the copolymer PTNDTT-QX under similar conditions, the best PCE was 1.44%, which was obtained by blending with PC71BM at a 1:4 (w/w) ratio, with Jsc of 2.99 mA cm−2, Voc of 0.78 V, and FF of 0.52. Comparing the photovoltaic properties of the solar cell devices of PTNDTT-QX, PTNDTT-QX-I, and PTNDTT-QX-II, it can be observed that the Voc of the devices containing PTNDTT-QX-I (0.63 V) and PTNDTT-QX-II (0.78 V) are 0.06 V lower and 0.09 V higher, respectively, than that of the device based on PTNDTT-QX (0.69 V). It is well known that Voc is directly related to the difference between the HOMO energy level of the donor (copolymer) and the LUMO energy level of the acceptor (PCBM). Due to the presence of additional electron donating alkyl groups at the 4-positions of the thienyl units attached to the quinoxaline acceptor, the copolymer PTNDTT-QX-I acted as a stronger electron donor than the copolymer PTNDTT-QX; consequently, the HOMO of PTNDTT-QX-I was higher, at −5.14 eV, resulting in a lower Voc. In contrast, changing the alkyl groups from the para to the meta positions in the outward phenyl rings of PTNDTT-QX-II made the conjugated backbone less electron-rich, lowering the HOMO of PTNDTT-QX-II to −5.29 eV and increasing the Voc. More interestingly, the FF of the copolymer PTNDTT-QX-I was significantly higher (0.64) than that of the copolymer PTNDTT-QX, which could be a result of the higher molecular weight of the former, since it has been established that molecular weight plays a significant role in determining the effective FF of a PSC device.61 However, quite surprisingly, the Jsc of copolymer PTNDTT-QX-I was not satisfactory, and was indeed lower than that of the structurally related copolymer PTNDTT-QX. This poor Jsc limited the PCE of devices using copolymer PTNDTT-QX-I, despite its other advantages of a smaller band gap as well as the broader absorption that resulted from the increase in molecular weight from the added alkyl chains at the thienyl units attached to quinoxaline acceptor. The low Jsc of PTNDTT-QX-I might be attributable to poor charge transport, insufficient morphology, or both at the same time. Comparing devices using PTNDTT-QX-II and PTNDTT-QX as the donor, the Voc of the former was about ∼0.10 V higher than that of the latter, and the FF of the former was also relatively higher. However, as before, the Jsc of the PTNDTT-QX-II device was lower than that of the PTNDTT-QX device, also resulting in a lower PCE in the former. The atomic force microscopy (AFM) images of the corresponding devices (Fig. 6a and c) indicated that the low PCE of PTNDTT-QX-I and PTNDTT-QX-II was mainly caused by the poor film morphology formed in the active layer. Considering the poor film morphology, we anticipated that the device performance of these new copolymers can be further enhanced if a suitable morphology could be achieved. Hence, the optimization of the polymer solar cells was further carried out by applying processing solvent, additive and thermal annealing conditions to the solar cell devices of PTNDTT-QX-I and PTNDTT-QX-II, since both processing additives and thermal annealing can significantly influence the morphology of the active layers and consequently improve the performances of the photovoltaic devices. To optimize the device performance of the PSCs based on PTNDTT-QX-I:PC71BM and PTNDTT-QX-II:PC71BM with the weight ratios of 1:4 and 1:3, respectively, we used different concentrations of 1,8-diiodooctane (DIO) as a processing additive, and the cells were annealed at three different temperatures (90, 120, and 150 °C). In addition, the optimization of the device performance of the copolymers was also accomplished by changing the processing solvent from ODCB to chlorobenzene (CB). J–V curves of all the photovoltaic cells based on all three copolymers are displayed in Fig. S5 and S6,† and the detailed photovoltaic parameters have been provided in Table S1.† Representative J–V curves of the optimized photovoltaic cells for copolymers are displayed in Fig. 5a and the summarized photovoltaic parameters of the optimized devices are included in Table 4 for fair comparison. It was observed that the overall performance of the PTNDTT-QX-I:PC71BM (1:4) solar cell remained unaffected by the addition of DIO, although its Jsc was improved slightly. Contrary to the DIO effect, the thermal annealing of the PTNDTT-QX-I:PC71BM (1:4) film did improve the performance of the PTNDTT-QX-I copolymer. The PCE of the PTNDTT-QX-I:PC71BM (1:4) cell could be increased up to 1.27–1.28% when the active layer was annealed at 90 °C from either ODCB or CB solvent. Unlike the PTNDTT-QX-I:PC71BM solar cell, the device containing PTNDTT-QX-II:PC71BM (1:3) was found to be quite strongly influenced by the processing additive. Following an increasing trend with the increase in DIO concentration, the PCE of PTNDTT-QX-II:PC71BM (1:3) cell levelled off at 1.45% with 1.0% v/v DIO concentration. A similar PCE value (1.47%) was also found for the PTNDTT-QX-II:PC71BM (1:3) composite when the active layer was thermally annealed at 120 °C from ODCB solvent. More impressive performance with a maximum PCE of 1.61% and Jsc of 3.83 mA cm−2, Voc of 0.78 V, and FF of 0.54 resulted when the active layer of PTNDTT-QX-II:PC71BM (1:3) was processed from CB solvent with annealing at 90 °C.
Table 3 Photovoltaic characteristics of the polymer solar cells at different polymer:PC71BM weight ratios from o-dichlorobenzene (ODCB) solution
| Active layer |
Blend ratio |
J
sc (mA cm−2) |
V
oc (V) |
FF |
PCE (%) |
|
Data from ref. 51.
|
|
PTNDTT-QX-I:PC71BM |
1:1 |
2.17 |
0.67 |
0.58 |
0.85 |
| 1:2 |
2.20 |
0.66 |
0.60 |
0.89 |
| 1:3 |
2.48 |
0.65 |
0.63 |
1.03 |
| 1:4 |
2.77 |
0.63 |
0.64 |
1.13 |
|
PTNDTT-QX-II:PC71BM |
1:1 |
2.72 |
0.8 |
0.42 |
0.92 |
| 1:2 |
2.90 |
0.79 |
0.48 |
1.12 |
| 1:3 |
2.99 |
0.78 |
0.52 |
1.23 |
| 1:4 |
2.75 |
0.77 |
0.52 |
1.10 |
|
PTNDTT-QX:PC71BMa |
1:2 |
4.52 |
0.69 |
0.46 |
1.44 |
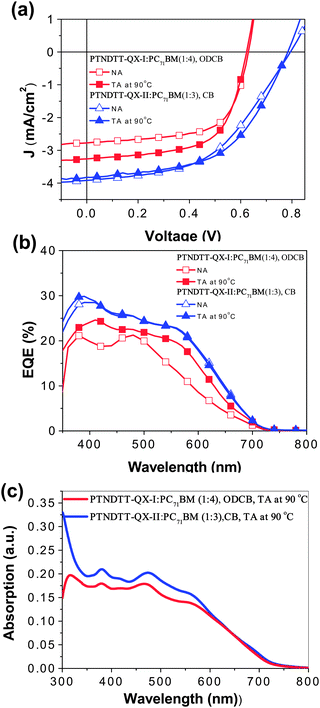 |
| | Fig. 5 (a) J–V curves and (b) EQE curves of the photovoltaic cells incorporating PTNDTT-QX-I:PC71BM and PTNDTT-QX-II:PC71BM blends without (N/A) and with thermal annealing (T/A). (c) UV-vis absorption spectra of PTNDTT-QX-I:PC71BM and PTNDTT-QX-II:PC71BM blended thin films. | |
Table 4 Optimized device characteristics of PTNDTT-QX-I:PC71BM (1:4)- and PTNDTT-QX-II:PC71BM (1:3)-based solar cells
| Active layer |
Solvent |
DIOa (v/v%) |
T/Ab temp. (°C) |
J
sc (mA cm−2) |
V
oc (V) |
FF |
PCE (%) |
|
Processing additive: 1,8-diiodooctane.
Thermal annealing.
|
|
PTNDTT-QX-I:PC71BM (1:4) |
ODCB |
0.2 |
w/o |
3.22 |
0.59 |
0.60 |
1.14 |
| ODCB |
w/o |
90 |
3.26 |
0.62 |
0.63 |
1.28 |
| ODCB |
0.2 |
90 |
3.27 |
0.62 |
0.63 |
1.28 |
| CB |
w/o |
w/o |
2.98 |
0.62 |
0.62 |
1.14 |
| CB |
0.2 |
w/o |
3.07 |
0.58 |
0.60 |
1.06 |
| CB |
w/o |
90 |
3.15 |
0.63 |
0.64 |
1.27 |
| CB |
0.2 |
90 |
3.25 |
0.61 |
0.63 |
1.24 |
|
PTNDTT-QX-II:PC71BM (1:3) |
ODCB |
1.0 |
w/o |
3.83 |
0.76 |
0.50 |
1.45 |
| ODCB |
w/o |
120 |
3.83 |
0.77 |
0.50 |
1.47 |
| ODCB |
1.0 |
120 |
3.83 |
0.76 |
0.48 |
1.42 |
| CB |
w/o |
w/o |
3.92 |
0.79 |
0.49 |
1.52 |
| CB |
1.0 |
w/o |
3.98 |
0.77 |
0.48 |
1.48 |
| CB |
w/o |
90 |
3.83 |
0.78 |
0.54 |
1.61 |
| CB |
1.0 |
90 |
3.86 |
0.77 |
0.51 |
1.53 |
To investigate the devices' photoresponses, external quantum efficiency (EQE) spectra of the optimized devices were collected under monochromatic light. Both the devices based on PTNDTT-QX-I:PC71BM (1:4, ODCB, annealed at 90 °C) and PTNDTT-QX-II:PC71BM (1:3, CB, annealed at 90 °C) exhibited broad spectral responses ranging from 300 to 700 nm (Fig. 5b), strongly correlating with their absorption spectra (Fig. 5c). The maximum EQE of the device consisting of the PTNDTT-QX-I:PC71BM composite was 24% at 408 nm. In contrast, the device based on PTNDTT-QX-II:PC71BM (1:3) had a much better spectral response, with a maximum EQE value of 29% at 388 nm. The higher EQE of the device based on PTNDTT-QX-II:PC71BM than that of the device based on PTNDTT-QX-I:PC71BM was likely attributable to the higher Jsc of the former.
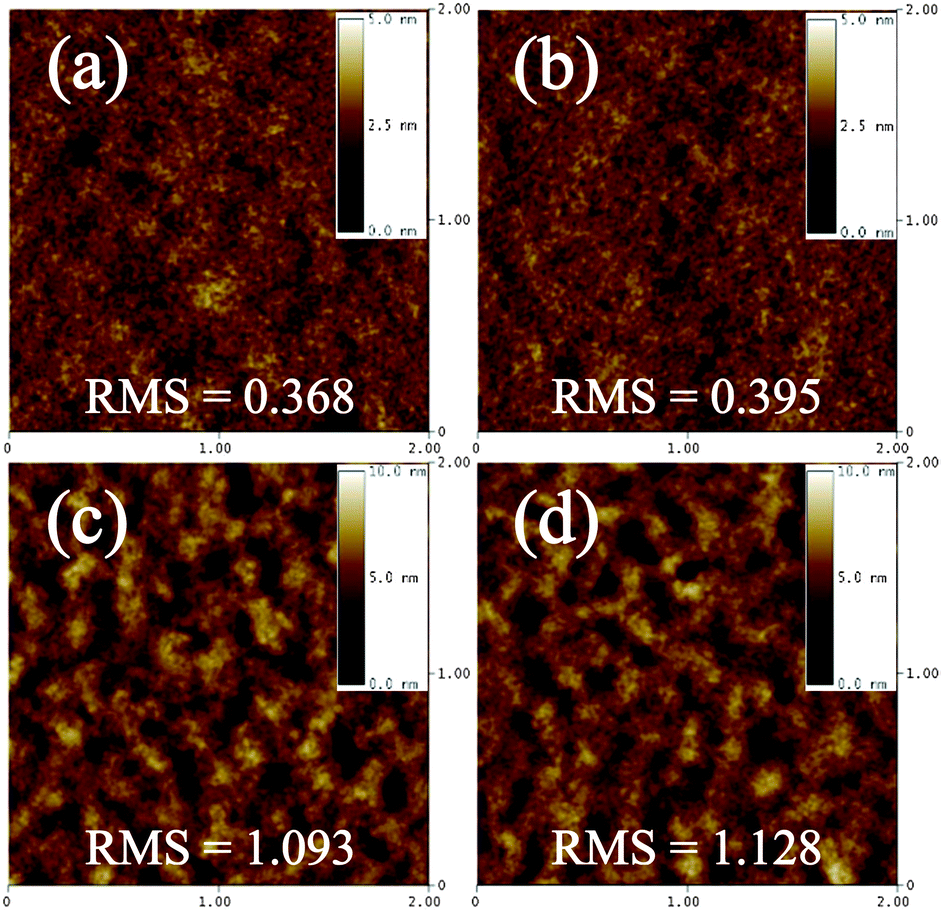
We characterized the PTNDTT-QX-I:PC71BM (1:4, ODCB) and PTNDTT-QX-II:PC71BM (1:3, CB) blended films using atomic force microscopy (AFM) to investigate the relationship between the morphology and the photovoltaic characteristics of the optimized devices. Fig. 6 shows the surface topography of PTNDTT-QX-I:PC71BM (1:4, ODCB) and PTNDTT-QX-II:PC71BM (1:3, CB) blended films with and without thermal annealing at 90 °C. The PTNDTT-QX-I:PC71BM film has a smooth and homogeneous surface with a root mean square (RMS) roughness of 0.368 nm. In contrast, the surface of the PTNDTT-QX-II:PC71BM blended film (Fig. 6c) is not very uniform, and aggregated domains with an average size of more than 100 nm are very common on the film surface. The RMS roughness of the PTNDTT-QX-II:PC71BM blended film (RMS = 1.093 nm) is also relatively high compared to the film based on the PTNDTT-QX-I:PC71BM blend, which is not favorable for charge separation and the formation of percolation paths, allowing excitons generated in the film to be trapped and preventing their easy transport and collection at the respective electrodes. Thus, although the PTNDTT-QX-II:PC71BM blended film shows relatively higher Jsc as a result of the stronger absorption feature of the PTNDTT-QX-II:PC71BM composite, which is supported by the UV-vis absorption spectrum of the PTNDTT-QX-II:PC71BM blended thin film (Fig. 5c), the unfavorable film morphology of the PTNDTT-QX-II:PC71BM blend resulted in a slightly lower FF (∼0.10) of the corresponding devices relative to the devices based on the PTNDTT-QX-I:PC71BM blend. On annealing, although the surface morphology of both polymer:PC71BM blended films was not dramatically changed, the RMS surface roughness values of the polymer:PC71BM composites were slightly increased with respect to the unannealed films (i.e. from 0.368 to 0.395 nm for PTNDTT-QX-I:PC71BM and 1.093 to 1.128 nm for PTNDTT-QX-II:PC71BM) (Fig. 6b and d), thus it may be possible that the thermal annealing process enhanced the preferred interpenetrating bicontinuous morphology that is essential for efficient charge transport, which consequently lead to slightly higher PCEs for the PTNDTT-QX-I:PC71BM (1:4, ODCB) and PTNDTT-QX-II:PC71BM (1:3, CB) blends under the employed annealing conditions.
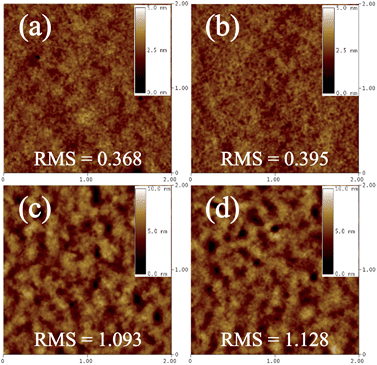 |
| | Fig. 6 Atomic force microscopy images of blended films of (a and b) PTNDTT-QX-I:PC71BM (1:4, ODCB) and (c and d) PTNDTT-QX-II:PC71BM (1:3, CB); (a and c): N/A and (b and d): T/A at 90 °C. Scan size: 2 × 2 μm. | |
Conclusions
In conclusion, we synthesized and characterized two new D–A conjugated copolymers, PTNDTT-QX-I and PTNDTT-QX-II, which incorporate the same naphtho[1,2-b:5,6-b′]dithiophene donor unit but different alkyl chain substituted quinoxaline acceptor units. We investigated the effect of the positioning of the alkyl chains on the thermal, optical, electrochemical, field-effect transistor, and photovoltaic properties of the resulting polymers. Varying their positions on the quinoxaline monomer could significantly influence the electrochemical properties of these new naphtho[1,2-b:5,6-b′]dithiophene-containing copolymers. The HOMO and LUMO energy levels of the copolymers could be tuned in the ranges of −5.14 to −5.29 eV and −3.44 to −3.55 eV, respectively. In contrast with these electrochemical differences, alkyl chain substitution had a negligible effect on the optical properties of the copolymers. Both PTNDTT-QX-I and PTNDTT-QX-II displayed a similar absorption profile, with absorption bands in the range of 300–700 nm and optical band gaps of about 1.70–1.73 eV. Solution-processed organic field-effect transistors fabricated with these polymers exhibited the characteristics of p-type organic thin-film transistors. The field-effect mobilities of PTNDTT-QX-I and PTNDTT-QX-II were measured to be 7.6 × 10−4 and 7.6 × 10−5 cm2 V−1 s−1, respectively; this difference was due primarily to the difference in molecular weight rather than the difference in alkyl chain position. The polymers were investigated as electron donors in photovoltaic cells using PC71BM as an electron acceptor. The optimized PCEs of these cells were 1.28% and 1.61% for the devices containing PTNDTT-QX-I and PTNDTT-QX-II, respectively. The superior photovoltaic performance of PTNDTT-QX-II is attributable to its higher Jsc and higher Voc; while its higher Jsc can be ascribed to its higher charge transport mobility, the higher open circuit potential of PTNDTT-QX-II is attributable to its deep-lying HOMO energy level. These results indicate once again that in addition to the main constituent structural units of D–A conjugated polymers, the positioning of alkyl side chains on the D–A conjugated main chain can also significantly influence the optoelectronic and photovoltaic properties of the resulting copolymers.
Acknowledgements
This research was supported by the Pioneer Research Center Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (contract no. 2008-05103).
Notes and references
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70 RSC.
- T.-A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087 CrossRef CAS.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- B. S. Ong, L. Y. Wu, P. Liu and S. Gardner, J. Am. Chem. Soc., 2004, 126, 3378 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. Macronald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. Mcgehee and M. F. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS.
-
(a) C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC;
(b) Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS;
(c) D. Gendron and M. Leclerc, Energy Environ. Sci., 2011, 4, 1225 RSC.
-
(a) H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS;
(b) R. L. Uy, S. C. Price and W. You, Macromol. Rapid Commun., 2012, 33, 1162 CrossRef CAS.
- F. C. Krebs, J. Fyenbo, D. M. Tanenbaum, S. A. Gevorgyan, R. Andriessen, B. Remoortere, Y. Galagan and M. Jørgensen, Energy Environ. Sci., 2011, 4, 4116 CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817 CrossRef CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617 CrossRef CAS.
- Y. J. He, H. Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377 CrossRef CAS.
- G. J. Zhao, Y. J. He and Y. F. Li, Adv. Mater., 2010, 22, 4355 CrossRef CAS.
- Y. J. Cheng, C. H. Hsieh, Y. J. He, C. S. Hsu and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 17381 CrossRef CAS.
- W. Y. Wong, X. Z. Wang, Z. He, A. B. Djurisic, C. T. Yip, K. Y. Cheung, H. Wang, C. S. K. Mak and W. K. Chan, Nat. Mater., 2007, 6, 521 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupre, S. Chao, N. Coates, J. S. Moon, D. Moses, M. Lecrelec, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297 CrossRef CAS.
- H.-Y. Chen, J. H. Hou, S. Q. Zhang, Y. Y. Liang, G. W. Yang, Y. Yang, L. P. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS.
- Y.-J. Chen, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. R. Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330 CrossRef CAS.
- L. Huo, X. Guo, Y. Li and J. Hou, Chem. Commun., 2011, 47, 8850 RSC.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638 CrossRef CAS.
- Z. He, C. Zhong, X. Huang, W. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- L. T. Dou, J. B. You, J. Yang, C.-C. Chen, Y. J. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180 CrossRef CAS.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115 CrossRef CAS.
- X. H. Li, W. C. H. Choy, L. J. Huo, F. X. Xie, W. E. I. Sha, B. F. Ding, X. Guo, Y. F. Li, J. H. Hou, J. B. You and Y. Yang, Adv. Mater., 2012, 24, 3046 CrossRef CAS.
- S. Chen, C. E. Small, C. M. Amb, J. Subbiah, T. H. Lai, S. W. Tsang, J. R. Manders, J. R. Reynolds and F. So, Adv. Energy Mater., 2012, 2, 1333 CrossRef CAS.
- J. L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691 CrossRef CAS.
- Y. Liu, Y. Liu and X. Zhan, Macromol. Chem. Phys., 2011, 212, 428 CrossRef CAS.
- J. S. Wu, C. T. Lin, C. L. Wang, Y. J. Cheng and C. S. Hsu, Chem. Mater., 2012, 24, 2391 CrossRef CAS.
- J. H. Kim, H. U. Kim, I. N. Kang, S. K. Lee, S. J. Moon, W. S. Shin and D. H. Hwang, Macromolecules, 2012, 45, 8628 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS.
- J. H. Hou, H. Y. Chen, S. Q. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144 CrossRef CAS.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616 CrossRef CAS.
- R. L. Qin, W. Li, C. Du, C. Veit, H. F. Schleiermacher, M. Andersson, Z. Bo, Z. Liu, O. Inganäs, U. Wuerfel and F. Zhang, J. Am. Chem. Soc., 2009, 131, 14612 CrossRef CAS.
- Y. Y. Liang and L. P. Yu, Acc. Chem. Res., 2010, 43, 1227 CrossRef CAS.
- Y. Huang, X. Guo, F. Liu, L. Huo, Y. Chen, T. P. Russell, C. C. Han, Y. Li and J. Hou, Adv. Mater., 2012, 24, 3383 CrossRef CAS.
- J. C. Bijleveld, M. Shahid, J. Gilot, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 3262 CrossRef CAS.
- M. Zhang, H. Fan, X. Guo, Y. He, Z. Zhang, J. Min, J. Zhang, X. Zhan and Y. Li, Macromolecules, 2010, 43, 5706 CrossRef CAS.
- Y. Chen, C. Yu, Y. Fan, L. Hung, C. Chen and C. Ting, Chem. Commun., 2010, 46, 6503 RSC.
- Y. Zhang, J. Zou, H. Yip, K. Chen, J. Davies, Y. Sun and A. Jen, Macromolecules, 2011, 44, 4752 CrossRef CAS.
-
(a) I. Osaka, T. Abe, S. Shinamura, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2010, 132, 5000 CrossRef CAS;
(b) I. Osaka, S. Shinamura, T. Abe and K. Takimiya, J. Mater. Chem. C, 2013, 1, 1297 RSC.
-
(a) S. Shi, P. Jiang, S. Yu, L. Wang, X. Wang, M. Wang, H. Wang, Y. Li and X. Li, J. Mater. Chem. A, 2013, 1, 1540 RSC;
(b) S. Shi, X. Xie, P. Jiang, S. Chen, L. Wang, M. Wang, H. Wang, X. Li, G. Yu and Y. Li, Macromolecules, 2013, 46, 3358 CrossRef CAS.
- S. R. Sanjaykumar, S. Badgujar, C. E. Song, W. S. Shin, S.-J. Moon, I.-N. Kang, J. Lee, S. Cho, S. K. Lee and J.-C. Lee, Macromolecules, 2012, 45, 6938 CrossRef.
- H. Zhou, Y. Liqiang, S. Liu and W. You, Macromolecules, 2010, 43, 10390 CrossRef CAS.
- L. Yang, H. Zhou and W. You, J. Phys. Chem. C, 2010, 114, 16793 CAS.
- N. Kleinhenz, L. Yang, H. Zhou, S. C. Price and W. You, Macromolecules, 2011, 44, 872 CrossRef CAS.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142 CrossRef CAS.
- P. Dutta, W. Yang, S. H. Eom, W.-H. Lee, I.-N. Kang and S.-H. Lee, Chem. Commun., 2012, 48, 573 RSC.
- P. Dutta, H. Park, W. H. Lee, K. Kim, I. N. Kang and S.-H. Lee, Polym. Chem., 2012, 3, 601 RSC.
- S. Shinamura, E. Miyazaki and K. Takimiya, J. Org. Chem., 2010, 75, 1228 CrossRef CAS.
- G. Zhang, Y. Fu, Q. Zhang and Z. Xie, Polymer, 2010, 51, 2313 CrossRef CAS.
- A. Gadisa, W. Mammo, L. M. Andersson, S. Admassie, F. Zhang, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2007, 17, 3836 CrossRef CAS.
- W. Mammo, S. Admassie, A. Gadisa, F. Zhang, O. Inganäs and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2007, 91, 1010 CrossRef CAS.
- L. J. Lindgren, F. Zhang, M. Andersson, S. Barrau, S. Hellström, W. Mammo, E. Perzon, O. Inganäs and M. R. Andersson, Chem. Mater., 2009, 21, 3491 CrossRef CAS.
- Y. Huang, M. Zhang, L. Ye, X. Guo, C. C. Han, Y. Li and J. Hou, J. Mater. Chem., 2012, 22, 5700 RSC.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Frechet and M. F. Toney, Macromolecules, 2005, 38, 3312 CrossRef CAS.
- P. T. Wu, T. Bull, F. S. Kim, C. K. Luscombe and S. A. Jenekhe, Macromolecules, 2009, 42, 671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures showing the NMR spectra of the monomers and polymers, and the J–V characteristics and device properties at different additive concentrations and temperatures. See DOI: 10.1039/c3py00911d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.