DOI:
10.1039/C3PY00727H
(Paper)
Polym. Chem., 2014,
5, 105-115
Facile synthesis of chain end functionalized polyethylenes via epoxide ring-opening and thiol–ene addition click chemistry†
Received
4th June 2013
, Accepted 19th July 2013
First published on 19th July 2013
Abstract
A wide range of low-molecular-weight, narrow-molecular-weight-distribution chain end functionalized polyethylenes (Cef-PEs), including hydroxyl-, amino-, carboxyl-, sulfo-, chloro-, azide- and trimethoxysilane-terminated polyethylenes, were synthesized under mild conditions via epoxide ring-opening and thiol–ene addition reactions with epoxy- and vinyl-terminated PEs as starting materials, respectively. The selectivities of the functionalizations were excellent. Similarly, amphiphilic polyethylene-block-poly(ethylene glycol) copolymer (PE-b-PEG) was prepared for the first time by simply treating epoxy terminated PE with hydroxyl terminated PEG and potassium hydroxide. A unique combination of primary (hindered-phenol) and secondary (thioester) antioxidants was introduced into the chain end of PE via successive thiol–ene addition and transesterification reactions. All Cef-PEs were characterized unambiguously by NMR, GPC, DSC and FTIR.
A Introduction
As one of the most widely used plastics, polyethylene (PE) possesses outstanding properties as well as some inherent drawbacks. Many efforts, taking advantage of the development of coordination catalysts, were devoted to PE functionalizations, aiming at introducing polar/reactive groups into PE chains. Chain end functionalized PEs (Cef-PEs) are of great importance in constructing complex macromolecular architectures (block and graft copolymers, star polymers, etc.) and PE property modifications (preparation of nanocomposites, e.g.). Cef-PEs can be obtained by three general pathways: (1) selectively end-capping living chains1,2 and/or applying functionalized catalysts3,4 in living coordination polymerisation of ethylene; (2) utilizing chain transfer reactions during coordination polymerisation (including coordination chain transfer polymerization);5–24 and (3) modifying preformed unsaturated chain ends.25–29 In addition, living polymerization of ylides,30–36 living anionic polymerisation of butadiene37,38 and ring-opening metathesis polymerisation of cycloolefins39,40 are alternative approaches to synthesize (analogues of) Cef-PEs. Numerous important reviews covered synthesis of Cef-PEs.41–46 Despite many recent advances, synthesis of Cef-PEs in a highly controlled manner remains challenging and has limited practical values with respect to efficiency, cost control and operability.
Click chemistry, characterized by efficiency, cleanliness, selectivity and simplicity, shows powerful potential in polymer and materials science.47,48 As K. B. Sharpless stated,49 “the most reliable and most useful organic reactions all involve olefins,… or their uniquely accessible heteroatom oxidative addition products.” “Both the oxidations (of olefins) to the (highly reactive, yet stable) intermediates (epoxides, e.g.) and their subsequent fusion with nucleophiles are stereospecific, and thus very predictable.” While azide–alkyneclick reaction is an ideal route to functionalization of atom transfer radical polymerisation produced polymers, whose terminal halogens could be readily transformed to azide groups, thiol–ene and epoxide ring-opening click reactions may be the best choices for functionalization of polyolefins bearing unsaturated double bonds. Neither of the reactions (i.e., thiol–ene and epoxide ring-opening click reactions) is new to polymer scientists considering vulcanization via radical addition of thiols across the double bonds along the main rubber chains48 and industrial productions of poly(ethylene glycol) (PEG) and epoxy resins that involve epoxide ring-opening reactions. Yet rare chain end functionalizations of PEs have been performed through these robust reactions with readily available starting materials at hand. Only recently D'Agosto et al.29 prepared a series of Cef-PEs with high functionalization degrees viathiol–ene click reactions between vinyl terminated PEs (v-PEs) and thiol–containing molecules (–SH). Alternatively, with FI catalyst produced v-PEs as starting materials, Fujita et al.27 illustrated a convenient way to produce Cef-PEs via epoxy terminated PEs.
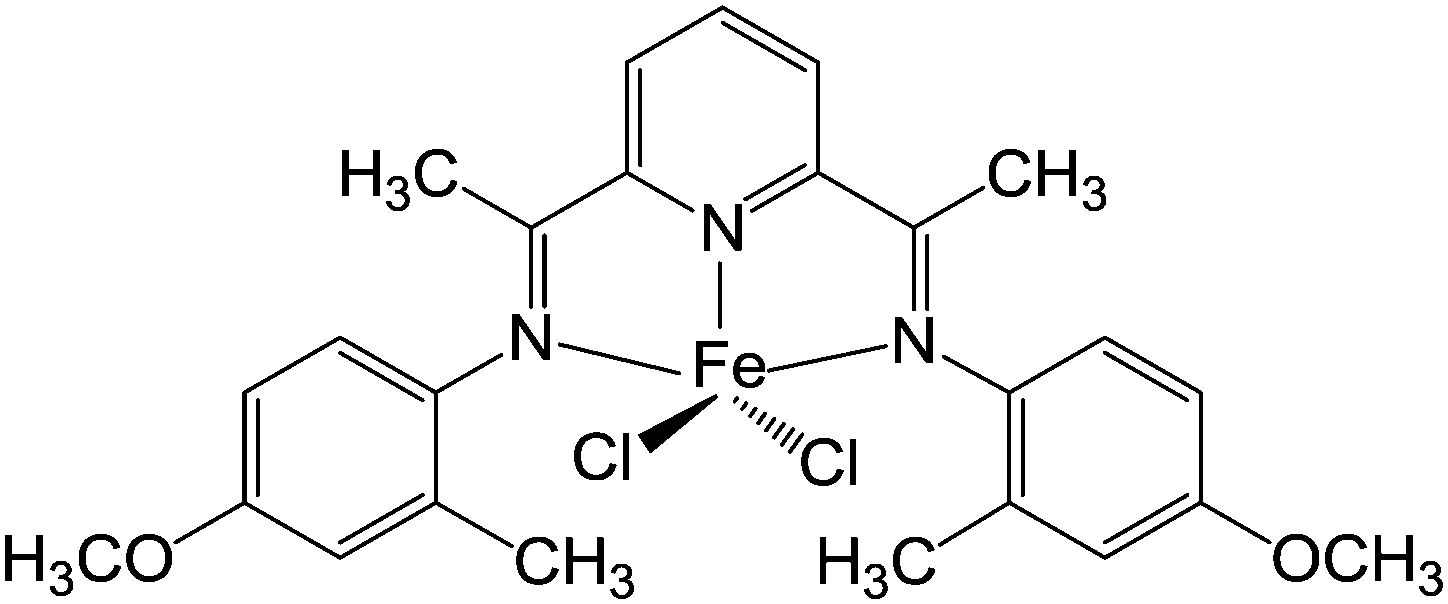
In our previous work, a novel iron(II) complex (1) (Fig.1) with methylaluminoxane (MAO) as the cocatalyst was applied in oligomerizations of ethylene to obtain linear α-olefins with remarkably high activity and selectivity.50 Tremendously useful click reactions, either thiol–ene addition or epoxide ring-opening type, catch our eyes for reasons mentioned above. Exploring the realm of click reactions leads to not only useful Cef-PEs or versatile functionalization methodology, but also better understanding of the philosophy and significance of click reactions. Given a special emphasis on convenience and economy, functionalization pathways reported herein are promising for large-scale production of Cef-PEs and thus are of great value in many practical areas.
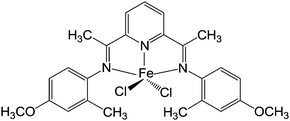 |
| | Fig. 1 The catalyst used to prepare v-PEs. | |
B Experimental section
B.1 Materials
Toluene and anhydrous THF were deoxygenated by nitrogen purge before refluxing for 48 h, and then distilled over sodium. Hexamethylphosphoramine (HMPA) and 1,2-ethylenediamine (EDA) were dried over CaH2 before distillation. High-purity nitrogen was used as received.
meta-Chloroperbenzoic acid (m-CPBA) (85%), sodium azide (NaN3), potassium hydroxide (KOH, AR, >85%), HCl acid (35%), 1,3-propane sultone (1,3-PS), methyl 3-(3,5-di-tert-butyl-4-hydroxyphenyl) propionate (3,5-methyl ester), hexane and cyclohexane from Sinopharm Chemical Reagent Beijing, methoxy poly(ethylene glycol) (m-PEG, Mn = 750 g mol−1), sodium bis(2-methoxyethoxy)aluminumhydride (Red-Al, 70 wt% in toluene), cerium(III) chloride heptahydrate (CeCl3, 99.99% metals basis), ammonium phosphomolybdate hydrate (AMP, AR), 1,1′-azobis(cyclohexanecarbonitrile) (ABCN, 98%), salicylic acid (AR), (3-mercaptopropyl)trimethoxysilane (MPTMS, 95%), chromium acetate (CrAc3, AR) and aluminiumisopropoxide (Al(iPrO)3, 99%) from Allddin, tetrabutylammonium chloride ([Bu4N]Cl, AR) from Beijing Datianfengtuo Chem. Ltd., thioglycol (98%) from Chengdu Aikeda Chem. Ltd., were used as received.
B.2 Analytical techniques
Room- and high-temperature NMR spectra were recorded on Bruker AVANCE 400 and DMX 300 instruments, respectively. The melting temperature of the polymers was measured by Differential Scanning Calorimetry (DSC) using a Perkin-Elmer DSC-7 instrument controller at a heating rate of 10 °C min−1. The molecular weight (MW) and Polydispersity Index (PDI) of polymers were determined by Gel Permeation Chromatography (GPC) using an Alliance GPC PL-220 instrument equipped with a refractive index detector (UV, 254 nm) and three PLgel 10 μm MIXED-B columns. The measurement was performed at 150 °C with 1,2,4-trichlorobenzene as the eluent at a flow rate of 1.0 ml min−1. Narrow-molecular-weight-distribution polystyrene samples were used as standards for calibration (Mp ranging from 580 to 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 870000 g mol−1).
870000 g mol−1).
B.3 General procedure for synthesis of v-PE
All procedures in Segment B.3–B.8 with air-sensitive compounds were performed under inert gas atmosphere using glovebox, syringe, or standard Schlenk techniques.
The catalyst (1) was synthesized as described before.50 In a typical ethylene polymerization, 100 ml of toluenesolvent was injected into a 250 ml flask, which, equipped with gas inlet, outlet tubes and a stir bar, had been vacuumed and filled with ethylene gas. Ethylene was continuously fed to keep a constant pressure at 1 atm throughout the entire polymerization process. MAO and the catalyst solution were charged into the flask in order. Temperature was kept constant at 50 °C. After the polymerization was done, the mixture was opened to air and cooled down to 0 °C. The precipitated polymer was filtered and washed with acidic ethanol and excess ethanol and dried under a vacuum to give crude v-PE.
B.4 General procedure for synthesis of e-PE
A solution of 4.0 g of low MW v-PE1 (see Results and discussions) in 60 ml of toluene was first stirred for 30 min at 75 °C to form a visually clear suspension and then cooled to 65 °C. 3.0 g m-CPBA was then added under nitrogen. After 2.5 h, the reaction mixture was precipitated in methanol, and filtered, and the product was washed with methanol and dried under vacuum to give e-PE.
B.5 General procedure for e-PE ring-opening reactions
(a) Solution.
A solution of e-PE in toluene (THF when R = OH in Scheme 1; HMPA when R = OCH2CH2CH2SO3H in Scheme 1) was first stirred for 30 min at elevated temperature to form a visually clear suspension. Corresponding reactants and HMPA, if needed, were added under nitrogen. The resulting polymer was washed and collected as described in B.4 or otherwise. Detailed reaction conditions are listed in Table 2.
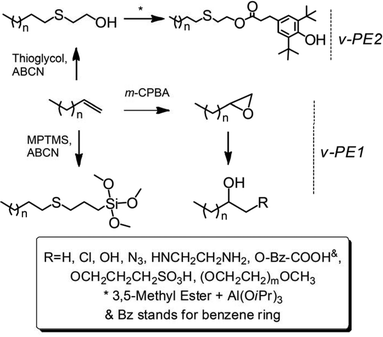 |
| | Scheme 1 Synthesis of chain end functionalized polyethylenes and polyethylene-block-poly(ethylene glyol) copolymer. | |
(b) Molten state.
(1) Ring-opening of e-PE with EDA.
A 25 ml round-bottom flask equipped with a magnetic stirrer was charged with e-PE and EDA. The mixture was stirred at 100 °C for 5 h and the product was washed with methanol and dried under a vacuum.
(2) Ring-opening of e-PE with m-PEG.
A 25 ml round-bottom flask equipped with a magnetic stirrer was charged with e-PE, m-PEG and KOH. The mixture was stirred at 130 °C for 5 h. After cooling down to ambient temperature, the solid mixture was collected and extracted by boiling water for 24 h to remove KOH and unreacted m-PEG. Consequently, the solid was extracted by boiling hexane. The solvent was removed under pressure to gain pure PE-b-PEG.
B.6 General procedure for synthesis of potassium 3-hydroxypropanesulfonate
20 g of KOH was slowly added into a mixture of 25 g 1,3-PS and 120 ml deionized water. The exothermic reaction caused the temperature to rise to around 50 °C. A clear light-yellow solution was formed. Then the reaction mixture was immersed into a 70 °C oil bath and stirred for 2 h to complete the reaction. Finally, the aqueous solution was poured into a 1000 ml beaker and heated at 80 °C to evaporate water away. 45 g of a white powder was collected and used without further purification.
B.7 General procedure for thiol–ene reactions
To a solution of v-PE (1 eq.) in toluene, –SH (10 eq.) and ABCN (1.2 eq.) were added. The mixture was then heated to 90 °C. Resulting polymers were washed and collected as described in Segment B.4.
B.8 General procedure for transesterification reaction
A 50 ml round-bottom flask equipped with a Dean–Stark trap and a magnetic stirrer was charged with 0.6 g of sulfur-containing, hydroxyl-terminated PE (PE-S-OH) and 10 ml of toluene which formed a visually clear suspension after 0.5 h mixing at 100 °C. 33 mg Al(iPrO)3 and 1.0 g 3,5-methyl ester were then introduced into the flask which was kept under stirring for 19 h at 120 °C. The resulting polymer was washed and collected as described in Segment B.4.
C Results and discussion
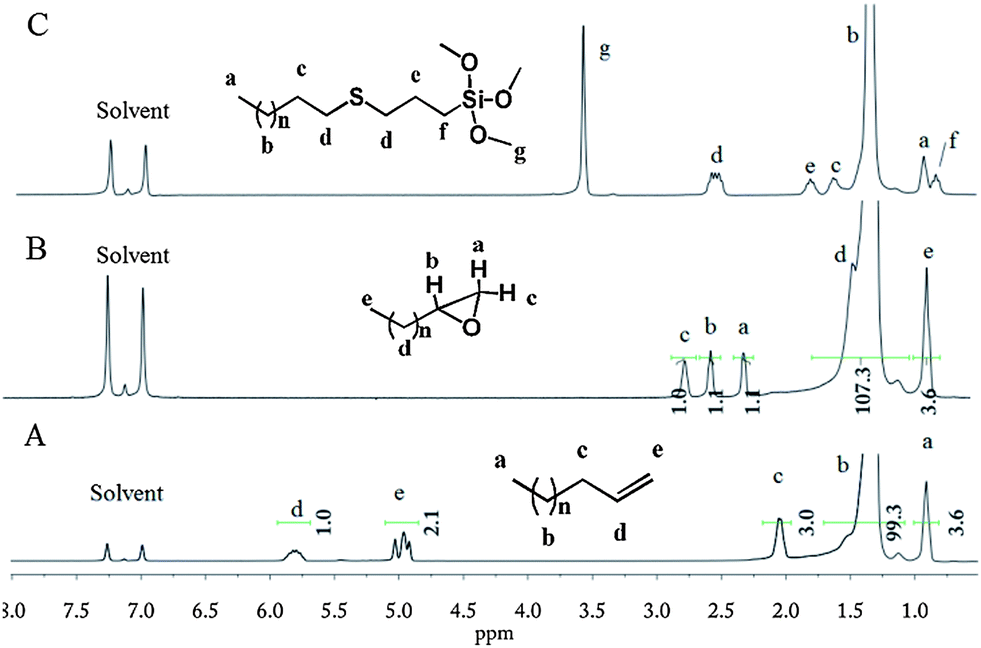
Crude v-PE was pretreated by successive Soxhlet extractions using boiling hexane and cyclohexane to gain low MW v-PE1 and v-PE2 with narrow molecular weight distribution, respectively. GPC and DSC results of crude v-PE and its different segments are listed in Table 1. Terminal vinyl groups are clearly evidenced by 1H NMR analysis (see Fig. 2A for v-PE1 and Fig. S2† for v-PE2). Taking Fig. 2A for example, the major peak (δ = 1.35 ppm) corresponds to CH2proton absorptions of the PE backbone. Around it, two minor peaks (δ = 0.92 ppm and δ = 2.05 ppm) correspond to proton absorptions of terminal CH3 and CH2 next to the vinyl group, respectively. In the downfield region, there are two iconic peaks at 4.96 and 5.80 ppm corresponding to –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and –CHCH2proton absorptions of the vinyl group. Peak integration of the vinyl groups relative to terminal methyl groups gives the selectivity of vinyl groups (for the calculation procedure, see Fig. S2;† for calculation results, see Table 1). Generally, 13C NMR provides the richest information for chain-end structural identifications. 13C NMR of v-PE1 and v-PE2 are given in Fig. S4† (with assignments) and Fig. S5,† respectively. As depicted in Fig. S4,† the main peak δ = 29.9 ppm definitely corresponds to CH2 carbon absorptions of the PE main chain while carbons in unsaturated bonds, –CHCH2 and –CHCH2, are responsible for downfield weak absorptions at δ = 139.0 ppm and δ = 114.1 ppm, respectively. Besides, various weak peaks between 14 and 34 ppm correspond to absorptions of carbons at the end of chains (not including carbons in vinyl groups).
CH2 and –CHCH2proton absorptions of the vinyl group. Peak integration of the vinyl groups relative to terminal methyl groups gives the selectivity of vinyl groups (for the calculation procedure, see Fig. S2;† for calculation results, see Table 1). Generally, 13C NMR provides the richest information for chain-end structural identifications. 13C NMR of v-PE1 and v-PE2 are given in Fig. S4† (with assignments) and Fig. S5,† respectively. As depicted in Fig. S4,† the main peak δ = 29.9 ppm definitely corresponds to CH2 carbon absorptions of the PE main chain while carbons in unsaturated bonds, –CHCH2 and –CHCH2, are responsible for downfield weak absorptions at δ = 139.0 ppm and δ = 114.1 ppm, respectively. Besides, various weak peaks between 14 and 34 ppm correspond to absorptions of carbons at the end of chains (not including carbons in vinyl groups).
Table 1
GPC and DSC results of v-PEs (v-PE1, hexane-soluble part; v-PE2, hexane-insoluble and cyclohexane-soluble part; v-PE3, cyclohexane-insoluble part; v-PE0, crude v-PE.)
| Sample |
Weight fraction |
x
a/% |
M
n (g mol−1) |
PDI |
T
m/(°C) |
ΔHm/(J g−1) |
T
c/(°C) |
ΔHc/(J g−1) |
|
x Stands for vinyl group selectivity.
|
| v-PE1 |
∼0.30 |
91 |
390 |
1.17 |
74.6 |
196.3 |
67.3 |
197.6 |
| v-PE2 |
∼0.25 |
88 |
1293 |
1.31 |
107.5/116.8 |
193.2 |
107.4 |
191.8 |
| v-PE3 |
∼0.45 |
N.A. |
3280 |
4.26 |
129.8 |
241.0 |
116.3 |
243.7 |
| v-PE0 |
1.0 |
N.A. |
823 |
6.75 |
124.0 |
218.1 |
111.4 |
209.4 |
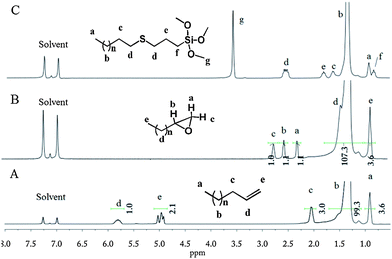 |
| | Fig. 2
1H NMR (1,2-dichlorobenzene-d4) of v-PE1 (A); e-PE (B); and TMS-t-PE (C). | |
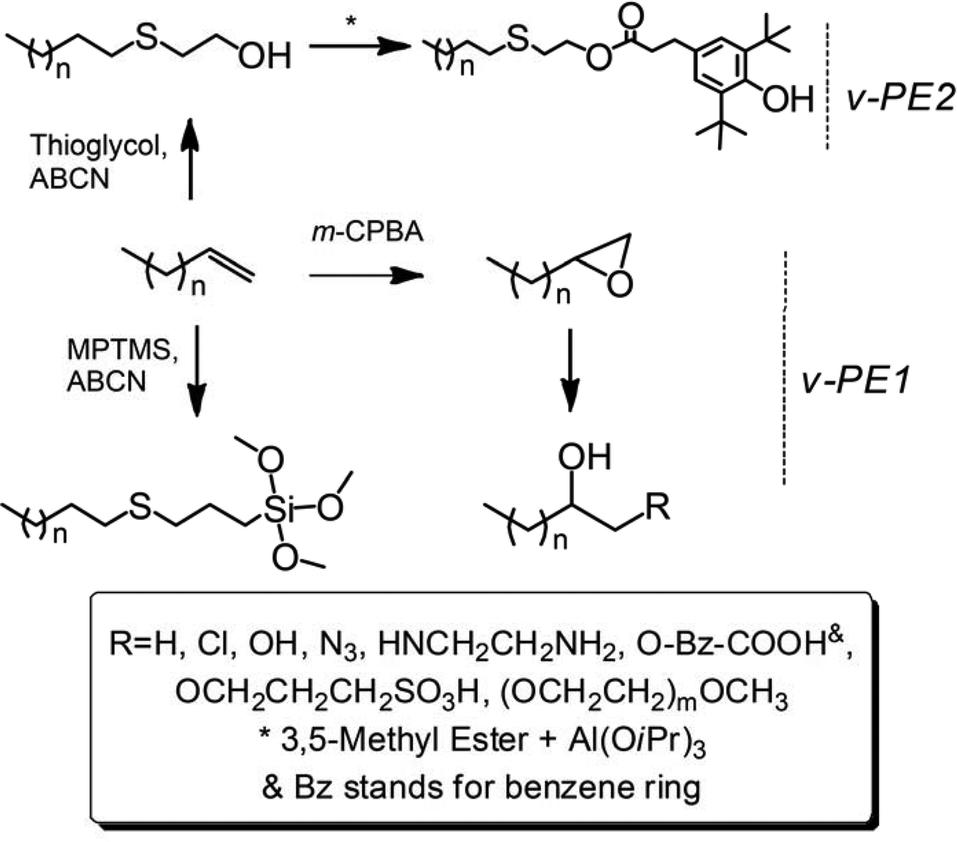
The synthetic strategies to prepare Cef-PEs are presented in Scheme 1. Detailed conditions of e-PE ring-opening reactions and thiol–ene addition reactions are given in Tables 2 and 3, respectively. Results of GPC and DSC analyses of Cef-PEs are summarized in Table 4 (for GPC curves, see Fig. S30–33;† for DSC curves, see Fig. 7, S34 and 35†).
Table 2 Detailed conditions of e-PE ring-opening reactions
| Sample |
Reagent |
Solvent(s) |
Conditions |
e-PE g−1 |
| Chloro-t-PE |
CeCl3 |
Toluene/HMPA (5 ml/5 ml) |
1.5 eq., 85 °C, 5 h |
0.5 |
| Azide-PE |
AMP/NaN3 |
|
0.5/1.5 eq., 90 °C, 6 h |
0.4 |
| Carboxy-t-PE |
Salicylic acid/CrAc3 |
|
10/0.5 eq., 85 °C, 7 h |
0.4 |
| 2-Hydroxy PE |
Red-Al |
5 ml Toluene |
3 eq., 90 °C, 5 h |
0.4 |
| Diol-PE |
H2O/TfOH |
4 ml THF |
22/0.09 eq., 70 °C, 3 h |
0.5 |
| NH2-t-PE |
EDA |
\ |
20 eq., 100 °C, 6 h |
0.9 |
| Sulfo-t-PE |
KPS/KOH |
15 ml HMPA |
25/10 eq., 120 °C, 7 h |
0.67 |
| PE-b-PEG |
m-PEG/KOH |
\ |
5/4 eq., 130 °C, 5 h |
1.0 |
Table 3 Conditions and results of thiol–ene click reactions
| v-PE |
[v-PE]/(mol L−1) |
Reaction time/(h) |
Conversion |
| v-PE1 |
0.1 |
5 |
85% |
| 8 |
∼100% |
| v-PE2 |
0.03 |
4 |
96% |
Table 4
GPC and DSC results of Cef-PEs (1. e-PE; 2. chloro-t-PE; 3. azide-t-PE; 4. 2-hydroxy PE; 5. diol-PE; 6. NH2-t-PE; 7. carboxy-t-PE; 8. sulfo-t-PE; 9. PE-b-PEG; 10. TMS-t-PE; 11. PE-S-OH; 12. AO-t-PE)
| Sample |
M
n (g mol−1) |
PDI |
T
m/(°C) |
ΔHm/(J g−1) |
T
c/(°C) |
ΔHc/(J g−1) |
|
PE segment, ΔH normalized to PE.
PEG segment, ΔH normalized to PEG.
|
| 1 |
406 |
1.32 |
77.7 |
229.2 |
69.5/88.1 |
230.3 |
| 2 |
477 |
1.24 |
80.6 |
229.0 |
77.9/90.4 |
224.2 |
| 3 |
464 |
1.25 |
84.0 |
185.9 |
78.8/91.3 |
184.4 |
| 4 |
466 |
1.25 |
83.3 |
194.5 |
78.6/90.3 |
192.2 |
| 5 |
444 |
1.92 |
102.3 |
197.1 |
94.1 |
193.3 |
| 6 |
493 |
1.49 |
93.2. |
179.6 |
88.3 |
180.1 |
| 7 |
481 |
1.17 |
81.1 |
168.2 |
77.8 |
167.2 |
| 8 |
435 |
1.28 |
83.0/93.3 |
180.6 |
77.5 |
180.4 |
| 9 |
755 |
1.36 |
82.7a |
222.6a |
82.4a |
225.2a |
| 27.9b |
104.4b |
12.3b |
95.2b |
| 10 |
649 |
1.13 |
77.9 |
168.5 |
79.4 |
164.9 |
| 11 |
1359 |
1.33 |
118.0 |
194.9 |
108.6 |
191.6 |
| 12 |
1576 |
1.27 |
114.6 |
210.3 |
106.7 |
215.7 |
C.1 Synthesis of e-PE
e-PE was synthesized according to the literature.51,52m-CPBA, one of the most widely used oxidation agents for research-scale applications, was chosen to convert v-PE1 into e-PE. Reaction time must be controlled carefully to avoid undesired epoxide ring-opening side reaction caused by the slight acidity of the reaction milieu. The 1H NMR spectrum (Fig. 2B) clearly shows that e-PE was quantitatively formed at 2.5 h, evidenced by the disappearance of peaks at 4.9–5.8 ppm corresponding to proton absorptions of the vinyl groups and the emergence of three new peaks at 2.30, 2.55 and 2.75 ppm corresponding to proton absorptions of the epoxy groups. The successful synthesis of e-PE is also proved by the 13C NMR of e-PE (Fig. S6†) since former downfield absorptions (δ = 139.0 and 114.1 ppm) of vinyl groups in parent v-PE1 disappear completely and two new peaks at 47.2 and 52.7 ppm, corresponding to carbons in three-membered epoxy rings, emerge. Meanwhile, absorption peaks at 914 cm−1 and 848 cm−1 in FTIR (Fig. S18†) due to C–O–C (in the epoxy group) stretching are observed. Analyses of GPC and DSC results reveal that the main chain of PE was not affected since MW and Tm of e-PE (Mn = 406 g mol−1, PDI = 1.32, 77.7 °C; Table 4) show no obvious changes compared to those of parent v-PE1 (Mn = 390 g mol−1, PDI = 1.17, 74.6 °C; Table 1).
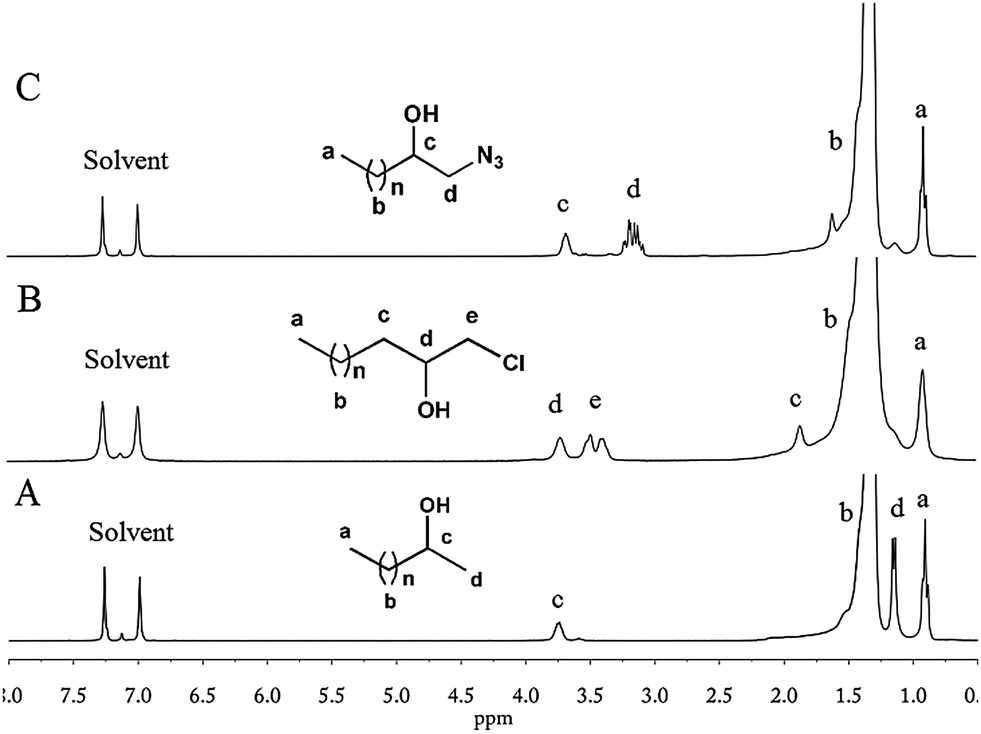
C.2 Ring-opening of e-PE with Red-Al (R = H)
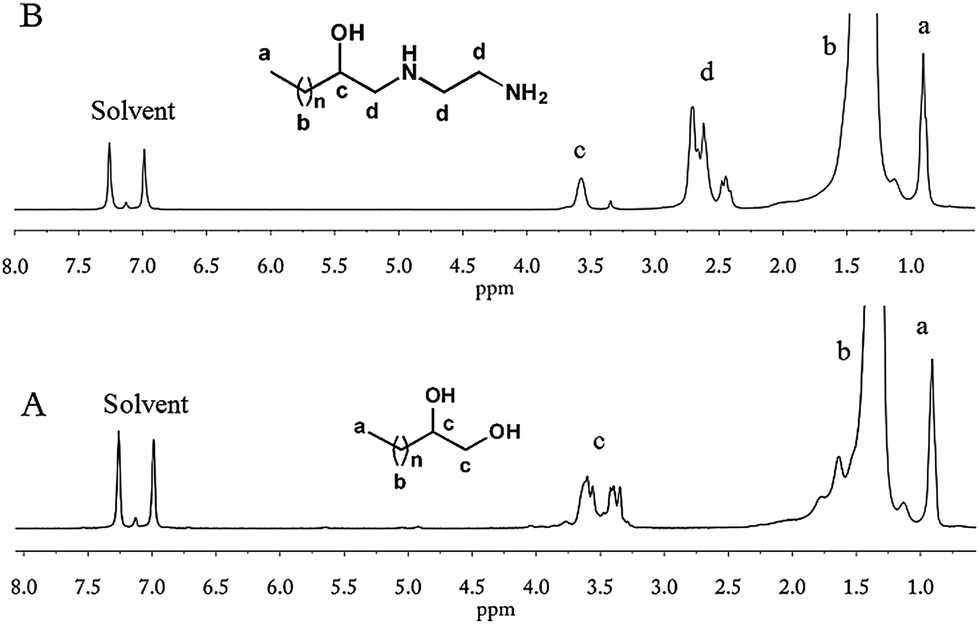
Red-Al, a versatile reduction reagent in organic chemistry with an excellent balance between reactivity and stability, can be used to reduce epoxide into alcohol.53Reduction of e-PE by Red-Al was conducted to yield hydroxyl terminated PE. NMR spectra clearly show that e-PE was transformed into desired 2-hydroxy PE (R = H in Scheme 1) highly regioselectively and efficiently. The 1H NMR of 2-hydroxy PE is given in Fig. 3A. No absorptions are observed at 2.3–2.8 ppm, indicating the complete reduction of epoxy groups. Furthermore, two new peaks at δ = 3.70 and δ = 1.10 ppm, with the latter resolved into two minor ones by neighboring –CH–, are clearly noticed due to the formation of hydroxyl groups. The presence of hydroxyl is affirmed both by carbon absorption of CHOH at 67.9 ppm in 13C NMR (Fig. S7†) and FTIR absorption at 3363 cm−1 due to O–H stretching (Fig. S19†). GPC analysis of 2-hydroxy PE recorded a slight increase in MW (Mn = 466 g mol−1) without altering the value of PDI (for GPC data, see Table 4; for GPC curve, see Fig. S31†). Enthalpy values of melting and crystallization of 2-hydroxy PE are found to be lower than those of e-PE (Table 4).
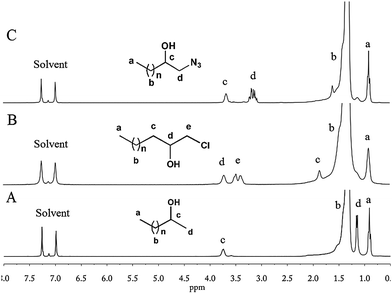 |
| | Fig. 3
1H NMR (1,2-dichlorobenzene-d4) of 2-hydroxy PE (A); chloro-t-PE (B); and azide-t-PE (C). | |
C.3 Ring-opening of e-PE with CeCl3 (R = Cl)
1-Chloro-2-hydroxy-PE (chloro-t-PE, R = Cl in Scheme 1) was synthesized according to ref. 54. A cheap, nontoxic and water-tolerant reagent CeCl3 was found to be useful in converting epoxides to the corresponding β-halohydrins highly regioselectively and efficiently with excellent yields under mild reaction conditions.54Polar solvent CH3CN was used in this research since only small molecules were involved. To adapt such a reaction to PE functionalization, the solvent is always the first challenge encountered. A HMPA–toluene mixture, which is believed to meet the demand for both dissolving capacity for PE and polarity needed in nucleophile-attacking ring-opening of epoxides, turned out to be a universal solvent system. The reaction was conducted under rather mild conditions and lasted for only a few hours. The 1H NMR spectrum of the resulting polymer clearly shows that former absorptions at 2.3–2.8 ppm have disappeared and absorptions of the same protons move downfield due to the introduction of chloride (peaks d and e in Fig. 3B). The chain end structure of chloro-t-PE is identified by the absorptions at 71.8 ppm and 50.4 ppm in the 13C NMR spectrum (Fig. S8†). FTIR (Fig. S20†) analysis provides evidence of the terminal hydroxyl group (O–H and C–O stretching) but misses the absorption peak of C–Cl stretching, which is expected to range from 785 to 540 cm−1. The minor increase in MW of chloro-t-PE could be attributed to the introduction of functional groups (Table 4; for the GPC curve, see Fig. S31†). Given that Tm and ΔHm of chloro-t-PE (Table 4) are nearly the same as those of parent e-PE and that PDI of chloro-t-PE is well maintained, a conclusion can be drawn that the ring-opening reaction of e-PE had little effect on the main chain of PE.
C.4 Ring-opening of e-PE with AMP/NaN3 (R = N3)
The past decade has witnessed an explosive growth of post-polymerization modifications via Cu-catalysed azide–alkynecycloaddition (CuAAC) reaction, one of the most representative click reactions, in polymer science.55,56 Even in the field of polyolefins, Dong et al.57,58 employed azide terminated isotatic polypropylene (iPP) in synthesis of well-defined star-shaped iPP and iPP-based diblock copolymers. Das et al.59 recently reported that AMP could be applied for cleavage of epoxides with NaN3 to form the corresponding 1,2-azidoalcohols in high yields with good regio- and stereoselectivity under mild reaction conditions. In their work, CH3CN was chosen as a polar solvent similar to ref. 54, which prompted us to conduct the reaction under conditions similar to Segment C.3. However, it turned out that AMP did not dissolve well in the HMPA–toluene mixture even at elevated temperatures. Therefore, [N(Bu4)]Cl, a phase transfer catalyst, was added. Hydrogen peroxide was used to wash AMP away since it is insoluble either in water or methanol. NMR spectra (for 1H and 13C NMR, see Fig. 3C and S9,† respectively) of the resultant polymer, which are very similar to those of chloro-t-PE, provided direct evidence of successful synthesis of 1-azide-2-hydroxy-PE (azide-t-PE). To be succinct, detailed explication of NMR spectra is omitted intentionally (see assignments in Fig. 3C and S9†). FTIR of azide-t-PE (Fig. S21†) serves as a valid proof of the existence of functional terminal hydroxyl (3388 cm−1) and azide (2104 cm−1) groups. Similarly, a slight increase in MW of azide-t-PE was detected when PDI of azide-t-PE maintained a low value (Table 4). DSC analysis shows that the introduction of the azide group might have a negative effect on melting and crystallization behaviors of azide-t-PE (Table 4).
C.5 Ring-opening of e-PE with salicylic acid (R = O–Bz–COOH, Bz for benzene)
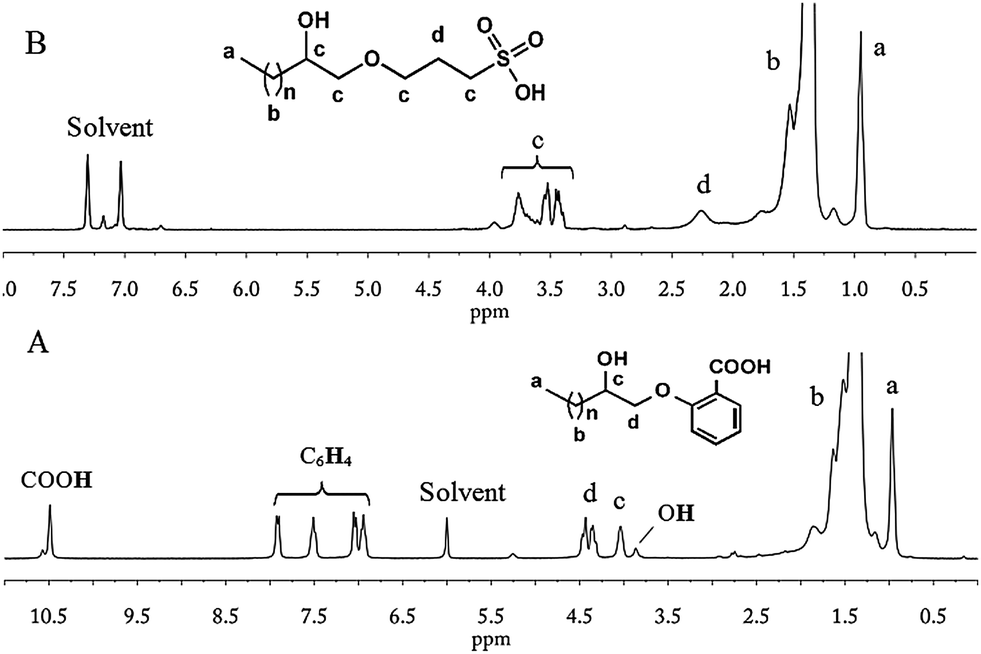
Bukowski and Bukowska60 thoroughly studied the reactions between acetic acid and epoxides in the presence of CrAc3, which promoted the addition of carboxylic acid to the epoxy group. Similarly, e-PE was treated with salicylic acid and CrAc3. Nevertheless, the 1H NMR shows that the terminal epoxy group was selectively opened by the phenol hydroxyl group rather than the carboxyl group, which, therefore, was successfully introduced into the chain end of PE. Thus, carboxyl terminated PE (carboxy-t-PE) was obtained. The 1H NMR of carboxy-t-PE (Fig. 4A) can be separated into four parts: (I) (0.9–2.0 ppm), absorptions of CH2 in PE main chains and terminal CH3 groups; (II) (3.6–4.6 ppm), absorptions of –CHOH– and –CH2OBzCOOH; (III) (6.8–8.0 ppm), absorptions of protons on benzene rings; and (IV) (10.5 ppm), absorptions of COOH. The FTIR spectrum (Fig. S22†) also confirms the existence of carboxyl groups. Firstly, the absorption peak of carboxyl O–H stretching is observed at a relatively shorter wavelength (3207 cm−1). Secondly, strong absorption at 1678 cm−1 due to carboxyl CO stretching affirms the absence of an ester structure that would be generated by addition of carboxyl to the epoxy group.60 The 13C NMR clearly confirms the chain-end structures of carboxy-t-PE (Fig. S10†). Assignments of peaks at 14–72 ppm are similar to those in the 13C NMR of azide- or chloro-t-PE. Absorption peaks of six carbons in benzene rings spread in a wide range from 112 to 162 ppm. The first peak from the left (171.6 ppm) corresponds to COOH carbon absorptions in carboxyl groups. To identify the role of CrAc3, a blank experiment was carried out in which e-PE was treated with salicylic acid without CrAc3. The 1H NMR of the resulting polymer (carboxy-t-PE1) shows that e-PE was partially opened by (phenolhydroxyl of) salicylic acid even without CrAc3, which, however, has limited efficiency and selectivity (see Fig. S3†). The GPC result of carboxy-t-PE (Mn = 481 g mol−1, PDI = 1.17, Table 4) further confirms the successful introduction of the functional group into the chain end of PE. Moreover, relatively lower ΔHm and ΔHc of carboxy-t-PE compared to those of starting e-PE reveal that terminal carboxyl groups might disturb the melting and crystallization behaviors of the main chain of PE (Table 4).
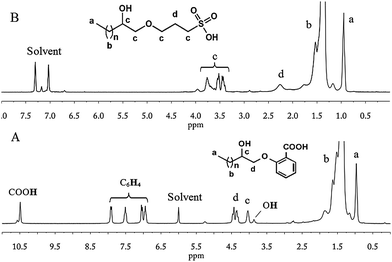 |
| | Fig. 4
1H NMR of carboxy-t-PE (A, 1,1,2,2-tetrachloroethane-D2) and sulfo-t-PE (B, 1,2-dichlorobenzene-d4). | |
C.6 Ring-opening of e-PE with potassium 3-hydroxypropanesulfonate (R = OCH2CH2CH2SO3H)
Potassium 3-hydroxypropanesulfonate (KPS) was prepared by heating 1,3 PS with KOH in aqueous solution. A clear light-yellow solution was formed finally, indicating that 1,3 PS was completely converted since 1,3-PS is only slightly water-soluble. After evaporation of water, a mixture of KPS and KOH residue was collected and used without further purification. The 1H NMR of 1,3-PS (for comparison) and KPS–KOH mixture are given in Fig. S1.† Three sets of proton absorptions are observed both before and after ring-opening of 1,3-PS, which, however, differ dramatically due to the altered chemical environment of corresponding protons.
The hydroxyl group of KPS, as a nucleophilic agent, attacked terminal epoxy groups of e-PE resulting in sulfopropylation of PE; KOH might serve as a catalyst. After the reaction, HCl was added to adjust the PH value to 2–3 generating sulfonic group terminated PE (sulfo-t-PE). The 1H NMR of sulfo-t-PE is given in Fig. 4B. In the upfield region (0.9–2.0 ppm), similarly, two sets of peaks corresponded to CH3(CH2)n–proton absorptions of PE main chains. In the downfield region (3.3–4.2 ppm), a group of peaks should be assigned as –CHOH, –CH2O–, and –CH2SO3H absorption of protons in terminal functional groups. In the middle part, one single peak is found at δ = 2.19 ppm in contrast to three peaks at 2.30, 2,55 and 2.75 ppm in the 1H NMR of parent e-PE, indicating that terminal epoxy groups were converted completely. However, a questionable 13C NMR spectrum of sulfo-t-PE was recorded since absorption peaks of carbons labelled with h and i are not found (Fig. S11†) and a duplicated experiment gave the same result. On the other hand, other peaks can be assigned with great confidence. Especially, at 25.5 ppm two peaks of different heights (thus intensities) are actually absorptions of carbons (e and j) which belong to different groups. To put it another way, carbon labelled with j is always accompanied by carbons labelled with g and k but not necessarily e or f. Therefore, the higher peak is assigned to carbon absorption of carbon labelled with e and the lower j. In the FTIR spectrum (Fig. S23†), weak but distinguishable absorption peaks at 1377 cm−1 and 1158 cm−1 correspond to OSO asymmetrical and symmetrical stretching, respectively. GPC and DSC results of sulfo-t-PE (Table 4) are similar to those of carboxy-t-PE, which have been thoroughly discussed in Segment C.5.
The significance of this functionalization method is believed to be not limited to facile synthesis of highly hydrophilic sulfo group terminated PE. Many organic acids tend to exist in their salt forms and often bear OH or NH2 groups, dyes like alizarin red for example, thus sharing a similar chemical structure (in the sense of reactivities toward e-PE) with KPS. Being aware of this, one can imagine that many other amazing Cef-PEs can be obtained through the right functionalization route demonstrated herein. D'Agosto et al.22 recently prepared α-[Cu(II)-porphyrin]-PE viaCuAAC and pointed out the potential utility of chromophore-labelled PE (PE chemically linked with a dye). We are now working on dye terminated PE.
C.7 Ring-opening of e-PE with H3+O (R = OH)
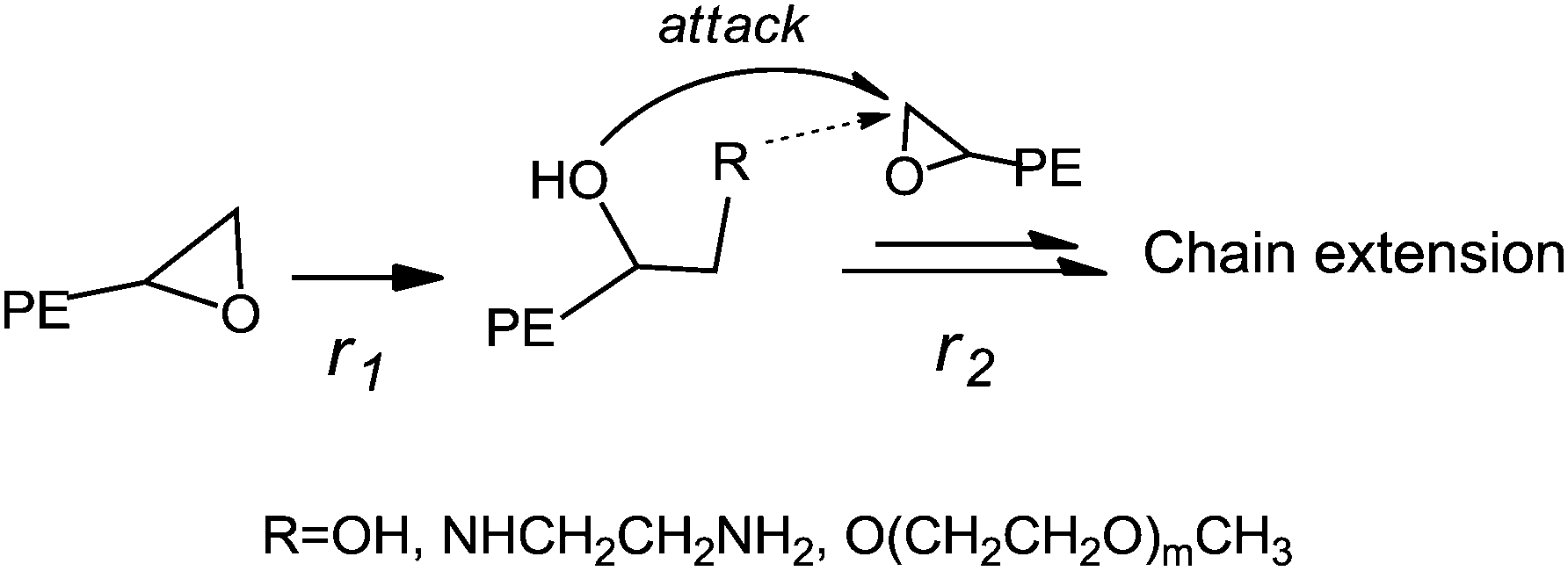
Riffle et al.38 reported that successive reactions, including termination with chlorodimethylsilane of poly(butadiene) anionic living chains, hydrogenation and hydrosilation, could be applied to synthesize an e-PE oligomer, which was subsequently hydrolyzed by TfOH (H3+O) yielding 1,2-dihydroxyl PE (diol-PE). Accordingly, ring-opening of terminal e-PE with H3+O was conducted to obtain diol-PE in the presence of TfOH. The 1H NMR spectrum shows that the reaction was completed within 3 h (Fig. 5A). New peaks emerge at 3.2–3.7 ppm while former peaks at 2.3–2.8 ppm disappear. The 13C NMR spectrum of diol-PE confirms the chain end structure of diol-PE and shows no significant differences with that of azide- or chloro-t-PE (Fig. S12†). In the FTIR (Fig. S24†), strong absorption peaks are observed at 3383 cm−1 and 1072 cm−1, corresponding to O–H and C–O stretching respectively. Though relatively low concentration of TfOH acid (9% mol/mol relative to e-PE) was used, chain extension seemed to be unavoidable, which caused obvious increases in MW, PDI and Tm (Table 4). Similar phenomena were reported by Riffle et al.38 and also observed when e-PEs reacted with EDA and m-PEG under molten conditions (vide infra). The proposed mechanism of chain extension is illustrated in Scheme 2.
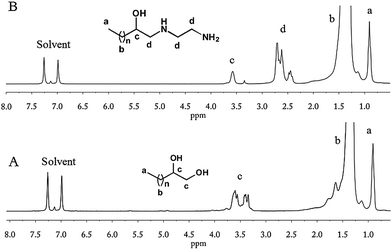 |
| | Fig. 5
1H NMR (1,2-dichlorobenzene-d4) of diol-PE (A) and NH2-t-PE (B). | |
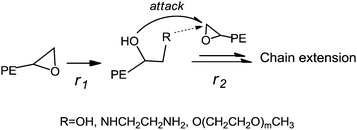 |
| | Scheme 2 Proposed mechanism of chain extension. | |
C.8 Ring-opening of e-PE with EDA (R = NHCH2CH2NH2)
Fujita et al.27 reported that e-PE reacted with diethanolamine and 2-aminomethylaniline under molten conditions to give corresponding triol-terminated PE and diamino-terminated PE (NH2-t-PE), respectively. The reaction between e-PE and EDA was examined for reasons to be discussed below. NMR spectra show that the terminal epoxy group was opened completely by EDA and NH2-t-PE was formed. The 1H NMR of NH2-t-PE is given in Fig. 5B. A group of (new) peaks are found at 2.3–2.8 ppm, which might overlap with peaks due to CH and CH2 absorptions of protons of epoxy groups, if they existed. Thus, no direct information about the conversion of epoxy groups can be collected here. The relative integration of peak c (CHOH absorptions of protons in terminal CHOH groups) to peak a (CH3 absorptions of protons in terminal methyl groups) is 3.9:1 (87% selectivity) while relative integration of peak b (CH absorptions of protons in terminal epoxy groups) to peak e (CH3 absorptions of protons in terminal methyl groups) in Fig. 2B is 3.6:1 (91% selectivity). Thereby, a conclusion can be drawn that terminal epoxy groups were quantitatively opened by EDA. The chain end structure of NH2-t-PE is well identified by the 13C NMR spectrum (Fig. S13†), in which two ‘new’ peaks (53.2 and 43.6 ppm) emerge due to NHCH2CH2NH2 carbon absorptions (other peaks are similar to peaks discussed elsewhere and thus discussions are omitted intentionally). FTIR also provides evidence of the existence of terminal amine groups (Fig. S25†). Strong peaks at 1576 cm−1 and 1319 cm−1 correspond to N–H bending and C–N stretching respectively. The broad peak at 3600–3000 cm−1 is a bit complicated since N–H stretching overlaps with O–H stretching (even C–H stretching when hydrogen bonds take effect, which makes N–H and O–H absorption peaks shift to a longer wavelength region).
Yet further GPC and DSC analyses reveal that remarkable chain extension occurred under the given reaction conditions for increases in MW; PDI and Tm of resulting polymers were detected. As illustrated in Scheme 2, both the newly formed terminal OH and NH2 groups, especially the latter, can be relatively active toward terminal epoxy groups in the molten state and be apt to react with remaining e-PE, which finally caused chain extension. Either higher ratio of EDA to e-PE or relatively lower reaction temperature is believed to be conducive to enhancing r1/r2 ratio (see Scheme 2) and, if aiming at amine group terminated PE, suppressing undesired side reactions. Reversely, hyper-branched or star-like PEs are expectable when e-PEs react with multi-amine functional groups bearing poly(ethylene imine)61 or poly(propylene imine)62 dendrimers, which is now under research and will be reported elsewhere.
C.9 Ring-opening of e-PE with m-PEG
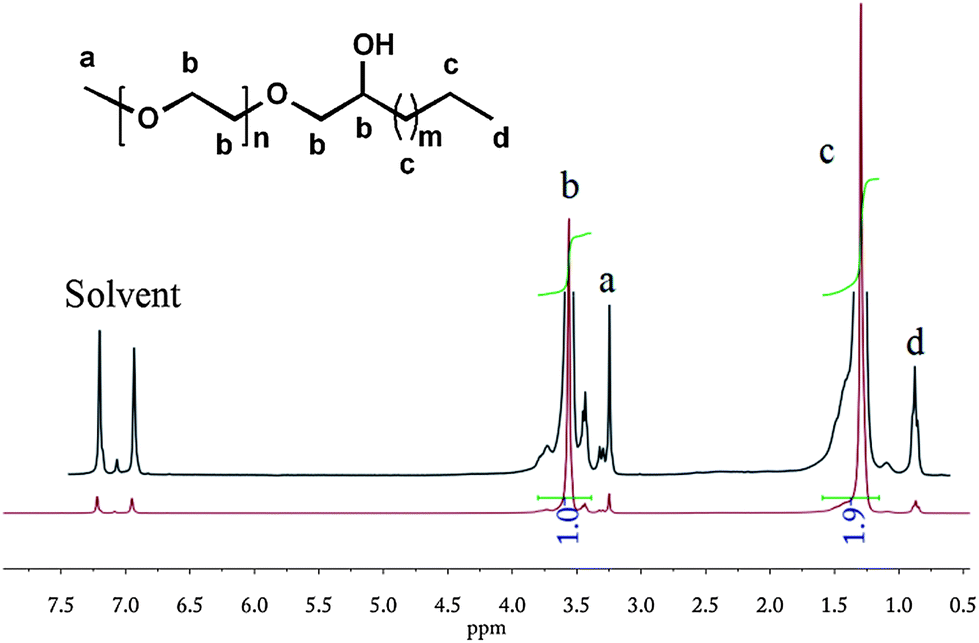
Aforementioned triol-terminated PE on reaction with ethylene oxide using KOH as a catalyst resulted in the formation of PE/PEG AB3-type hybrid materials that display attractive features.63 D'Agosto et al.64 recently synthesized PE-b-PEG using thia-Michael addition chemistry between thiol terminated PE and acrylate terminated PEG. Although they claimed its efficiency, the starting materials were not so readily available, toluene as a solvent was needed and the reaction lasted for 24 h. All these ‘parameters’ push such a process away from click chemistry. In our research, PE-b-PEG was synthesized by simply treating e-PE with m-PEG and KOH in the molten state. A visually clear suspension was formed at evaluated temperature indicating that KOH, serving as a catalyst, is partially ‘soluble’ in a polar m-PEG melt. Terminal epoxy groups in e-PE were opened by terminal hydroxyl groups in m-PEG so that PE-b-PEG, with a hydroxyl group pendant near the conjunction of PE and PEG segments, was obtained via a rather simple and efficient method. The hydroxyl group is of great significance and may find usage in synthesis of star polymers. Such PE-b-PEG may act as a macroinitiator for the ring-opening polymerization of ε-caprolactone resulting in a miktoarm star terpolymer. In another way, by initiating monomers like 4,4′-bioxepanyl-7,7′-dione,65 star polymers with a degradable cross-linked “core” and PE/PEG “arms” can be obtained viaring-opening polymerization using the “arm first” method. 1H NMR (Fig. 6) and FTIR (Fig. S26†) of resulting PE-b-PEG roughly can be seen as superpositions of corresponding spectra of PEG and PE (no proton characteristics of the epoxy function of e-PE). The 13C NMR contains more significant and detailed information about the microstructure of PE-b-PEG. Especially, the presence of peak d (δ = 76.4 ppm, see Fig. S14†) confirms that terminal epoxy groups of PE were opened by terminal hydroxyl groups of m-PEG.
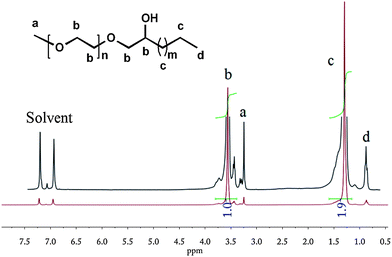 |
| | Fig. 6
1H NMR of PE-b-PEG (1,2-dichlorobenzene-d4). | |
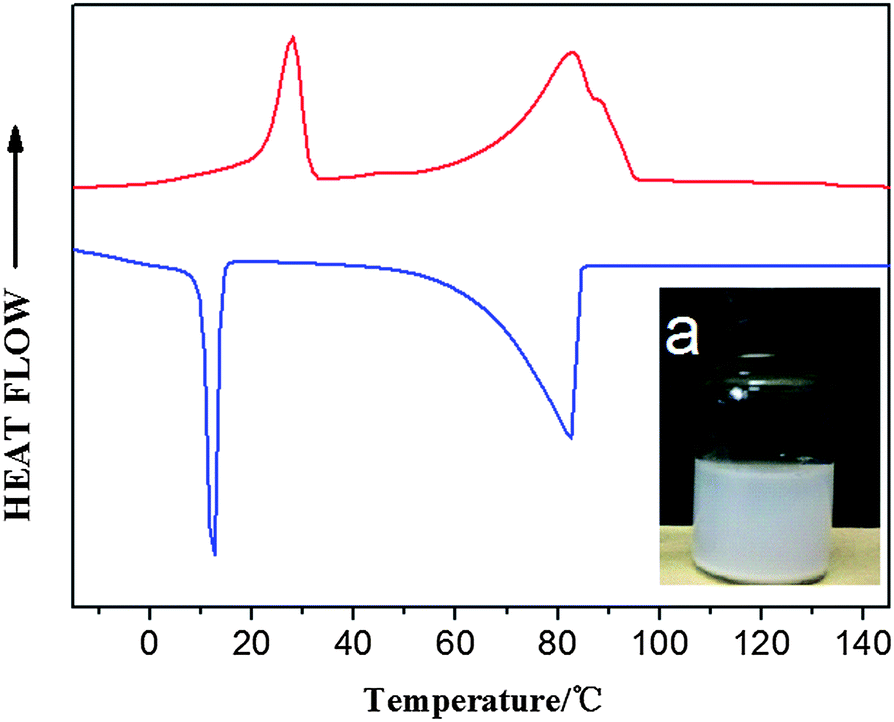
A remarkable increase in (apparent) MW shows the successful synthesis of a block copolymer. Meanwhile, the small shoulder peak of the GPC curve demonstrates the existence of a branched structure (Y-shaped) due to chain extension (see Fig. S32†). As one of the distinguished features of block copolymers, two isolated peaks corresponding to PE and PEG segments in both heating and cooling DSC curves are observed (Fig. 7). Melting and crystallization temperatures of the PE block of PE-b-PEG are close to those of parent e-PE. Enthalpy values of the PE segment in PE-b-PEG, normalized to PE according to relative intensities of resonance peaks of the 1H NMR, show little difference from those of parent e-PE (Table 4).
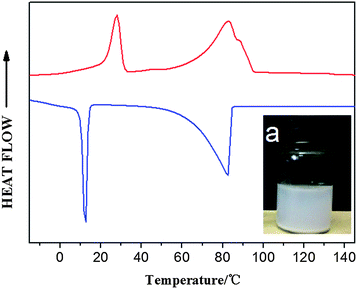 |
| | Fig. 7
DSC heating (upper) and cooling (lower) curves of PE-b-PEG – (a) inner picture: water dispersion (1 wt%) of PE-b-PEG; the mixture was ultrasonicated for 5 min and set still over 24 hours. | |
A unique molecular structure means excellent properties and thus, huge potential usages. Such hydrophilic–hydrophobic block copolymer PE-b-PEG is found to be completely ‘soluble’ in polar solvents such as CH2Cl2 and CH3Cl at ambient temperature. Moreover, the aqueous mixture of PEG-b-PE would stay in the emulsion state for a rather long time (over 1 day; see Fig. 7 inset) after ultrasonication for 5 min for quick dispersion, indicating that PEG-b-PE possesses unique amphiphilic properties. Telechelic PEG (t-PEG), bearing hydroxyl groups at both ends of the hydrophilic main chain, is more readily available and thus more suitable for large-scale synthesis of amphiphilic block copolymers (ABCs), which is the major concern in the case of polymeric surfactants.66 Systematic research on ABCs synthesized by treating e-PE with t-PEGs of different MW (600 to 6000 g mol−1) has been carried out and has given positive results. In general, reactions between e-PE and high MW t-PEGs still show high reactivities and e-PE can be converted efficiently into PE-b-PEGs. ABCs generated from higher MW t-PEGs are actually more likely to disperse in water, which is an important factor for potential usage in many fields.67 Detailed results will be covered elsewhere.
C.10
Thiol–ene addition of v-PE1 with MPTMS
Trialkoxysilanes are valuable reagents for surface property modifications of materials and devices while thiol–ene click reaction provides a simple and robust way to functionalize either trialkoxysilanes68 or PEs.29 Parent and Sengupta examined the radical-mediated addition of MPTMS and mercaptopropyltriethoxysilane to the terminal unsaturation of polypropylene69 and the unsaturation within PE resins,70 respectively, generating moisture-curing polyolefin derivatives. A silane coupling agent bearing a long tail of a PE chain, trimethoxysilane terminated PE (TMS-t-PE), was prepared for the first time viathiol–ene click reaction. ABCN, with a 10 h half-life temperature of 88 °C, was chosen as a radical initiator to ensure a high level radical concentration throughout the reaction time. The conversion of v-PE was over 80% at 5 h reaction time (not shown) and nearly 100% at 8 h (1H NMR, see Fig. 2C). Both 1H and 13C NMR (Fig. S15†) spectra provide definite proof of the terminal structure of TMS-t-PE and assignments of absorption peaks were rather clear-cut. The FTIR of TMS-t-PE exhibits no peak at 3500–3100 cm−1 due to the absence of O–H or N–H bonds. A relatively strong absorbing peak is observed at 816 cm−1 (Fig. S27†), which, after comparison with a couple of FTIR spectra of different functional silanes, is identified to be possibly related to the Si–O–C bond.71 Due to the addition of MPTMS (Mn = 196 g mol−1), a reasonable increase in (apparent) MW of resulting TMS-t-PE (Mn = 649 g mol−1, PDI = 1.13, Table 4) was detected compared to that of parent v-PE1 (Mn = 390 g mol−1, PDI = 1.17, Table 1). The maintenance of low PDI reveals that the main chain of PE was unaffected during the thiol–ene click reaction. Analysis of DSC depicts that the introduction of MPTMS into the chain end of PE seems to have a negative influence on the crystallization and melting behaviors of PE since lower ΔHm (168.5 J g−1) and ΔHc (164.9 J g−1) were recorded (Table 4).
C.11
Thiol–ene addition of v-PE2 with thioglycol and subsequent transesterification
Polypropylene and some PE resins in their natural state (without additives) are inherently unstable. Free radicals initiated by heat, mechanical stress, metal catalyst residues, and radiation, can cause severe degradation of polyolefins and thus remarkable losses of desired mechanical properties, making an antioxidant (AO) an indispensable additive added into polyolefin resins to influence the resulting compound's properties. Based on their mode of action, AOs can be classified as primary and secondary. Hindered-phenol AOs (e.g., 3,5 methyl ester) are primary AOs that scavenge free radicals by donating a hydrogen atom. Secondary AOs, typically sulfur- or phosphorus-based compounds, are peroxide decomposers and often used in combination with primary AOs to achieve synergistic performance.72 AOs with low MW are susceptible to physical loss through migration, evaporation, and extraction.73 Moreover, safety concern arises when polyolefins are used in food packaging materials. Polymer-bound AOs have gained much interest to enhance compatibility and thus achieve long term stabilizing efficiency and avoid potential toxicity.74–76
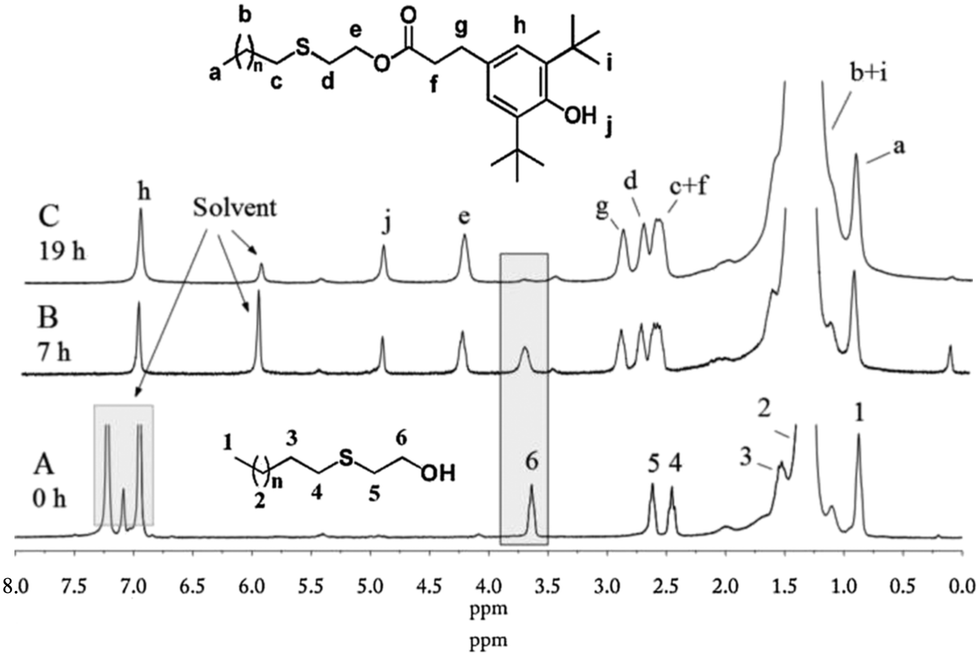
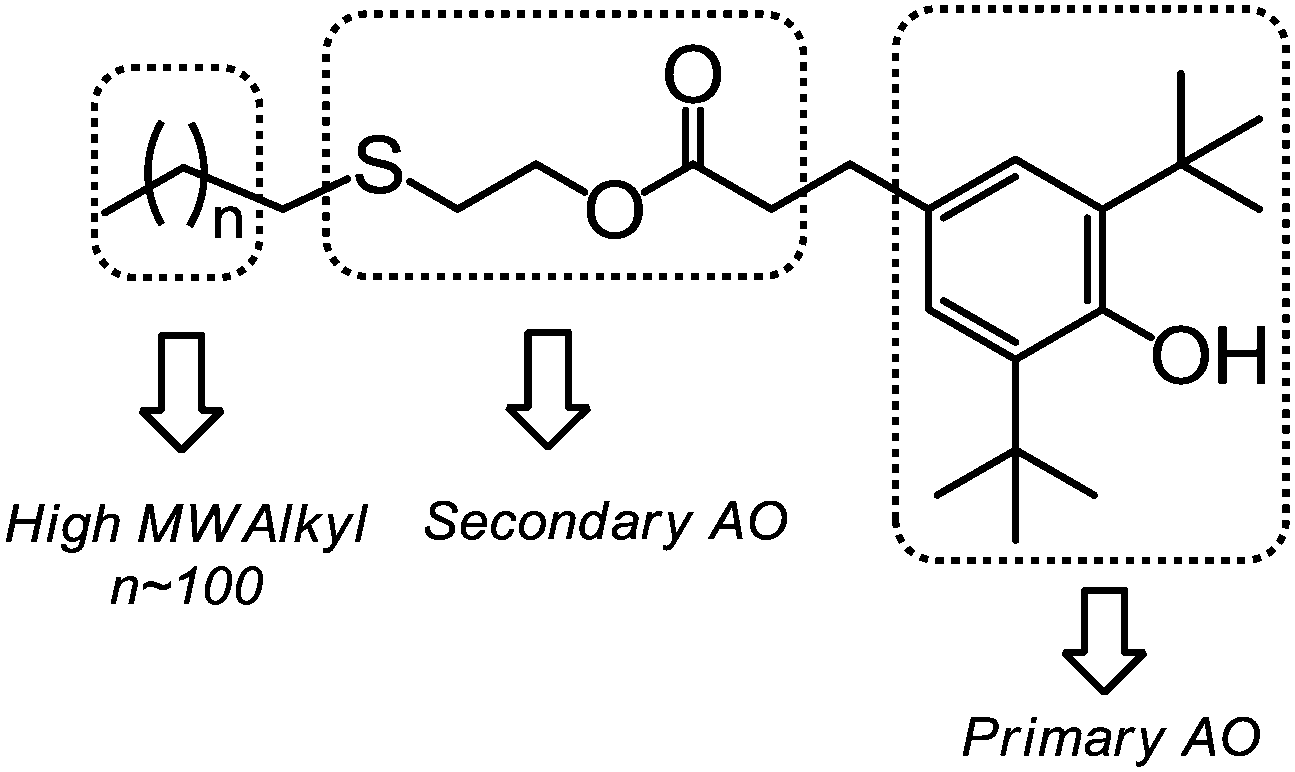
In this study, a new route was developed to prepare PE-bound AOs via successive thiol–ene addition and transesterification reactions. v-PE2 was first added by thioglycol viathiol–ene click chemistry to generate sulfur-containing, hydroxyl-terminated PE (PE-S-OH) according to ref. 29. In the 1H NMR (Fig. 8A), the appearance of three characteristic isolated peaks at 3.64, 2.62 and 2.45 ppm and the disappearance of peaks between 4.9 and 5.8 ppm corresponding to the vinyl groups illustrate the success of the thiol–ene addition reaction. 13C NMR (Fig. S16†) and FTIR (Fig. S28†) spectra provide further information for the chain-end structural identification of PE-S-OH. Consequently, PE-S-OH underwent Al(OiPr)3 catalysed transesterification reaction with 3,5-methyl ester in a toluene solution to obtain AO terminated PE (AO-t-PE). The reaction was monitored by 1H NMR and its conversion was 80% at 7 h (Fig. 8B) and 95% at 19 h (Fig. 8C). A steady decrease in intensities of resonances corresponding to CH2OH in PE-S-OH is observed. The detailed chain end structure of AO-t-PE is also confirmed by 13C NMR (Fig. S17†). Besides, the existence of a phenolic hydroxyl group is evidenced by a sharp peak at 3647 cm−1 in FTIR (Fig. S29†). A characteristic absorption at 1740 cm−1 corresponds to (ester) CO stretching. Constant increases in MW (see Tables 1 and 4, Fig. S33†) are observed from v-PE2 (Mn = 1293 g mol−1, PDI = 1.31) to PE-S-OH (Mn = 1359 g mol−1, PDI = 1.33) and to AO-t-PE (Mn = 1576 g mol−1, PDI = 1.27). The GPC result of PE-S-OH is consistent with the literature29 and the increase in MW of AO-t-PE compared to that of PE-S-OH, apparently, is due to the introduction of hindered phenol into the chain end of PE. Moreover, DSC analysis of PE-S-OH and AO-t-PE (Table 4) presents further proof that the procedure of transesterification did not affect the main chain of PE. Resultant AO-t-PE, possessing a unique structure that contained both primary and secondary AO (see Fig. 9), is readily soluble in polyolefins without migration and will find great utility in stabilizing polyolefin resins. Further research on its applications is now under way.
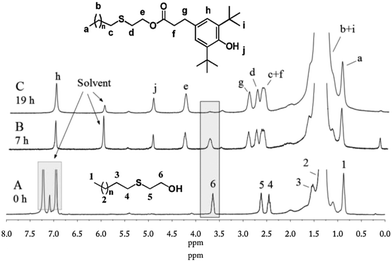 |
| | Fig. 8
1H NMR of PE-S-OH and AO-t-PE. | |
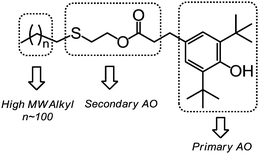 |
| | Fig. 9 Distinguished and useful segments combined together in AO-t-PE. | |
D Conclusions
In conclusion, we have demonstrated a range of synthetic opportunities through the development of robust, efficient, and orthogonal organic transformations, such as epoxide ring-opening and thiol–ene click reactions. A library of chain end functionalized PEs (Cef-PEs) were prepared with high selectivity and efficiency and characterized unambiguously. All reactants involved are readily available and all reaction conditions are mild. The significance of strategies applied in functionalization of PE, either epoxide-intermediated or ene-based, has been emphasized by demonstrating their distinct advances compared with traditional approaches. Cef-PEs reported herein possess huge potentials in macromolecular design, surface modifications, and polymer material science. Further study is ongoing intensively.
Acknowledgements
The financial support from PetroChina Co. Ltd. is greatly appreciated.
Notes and references
- W.-J. Wang, P. Liu, B.-G. Li and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3024 CrossRef CAS.
- H. Makio and T. Fujita, Macromol. Rapid Commun., 2007, 28, 698 CrossRef CAS.
- P. Liu, E. Landry, Z. Ye, H. Joly, W.-J. Wang and B.-G. Li, Macromolecules, 2011, 44, 4125 CrossRef CAS.
- K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2008, 41, 640 CrossRef CAS.
- H. Gao, Y. Tang, Z. Hu, Q. Guan, X. Shi, F. Zhu and Q. Wu, Polym. Chem., 2013, 4, 1107 RSC.
- X. Shi, Y. Zhao, H. Gao, L. Zhang, F. Zhu and Q. Wu, Macromol. Rapid Commun., 2012, 33, 374 CrossRef CAS PubMed.
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, J. Mater. Chem., 2012, 22, 5737 RSC.
- R. Liu, Z. Li, D. Yuan, C. Meng, Q. Wu and F. Zhu, Polymer, 2011, 52, 356 CrossRef CAS PubMed.
- W. Wang, R. Liu, Z. Li, C. Meng, Q. Wu and F. Zhu, Macromol. Chem. Phys., 2010, 211, 1452 CrossRef CAS.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311 CrossRef CAS PubMed.
- C. J. Han, M. S. Lee, D.-J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923 CrossRef CAS.
- A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 12764 CrossRef CAS PubMed.
- A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 6311 CrossRef CAS PubMed.
- S. B. Amin and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 10102 CrossRef CAS PubMed.
- Z. Li, R. Liu, B. Mai, W. Wang, Q. Wu, G. Liang, H. Gao and F. Zhu, Polymer, 2013, 54, 1663 CrossRef CAS PubMed.
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, J. Mater. Chem., 2012, 22, 5737 RSC.
- T. Li, W. J. Wang, R. Liu, W. H. Liang, G. F. Zhao, Z. Li, Q. Wu and F. M. Zhu, Macromolecules, 2009, 42, 3804 CrossRef CAS.
- A. Guimont, E. Beyou, P. Cassagnau, G. Martin, P. Sonntag, F. D'Agosto and C. Boisson, Polym. Chem., 2013, 4, 2828 RSC.
- I. German, W. Kelhifi, S. Norsic, C. Boisson and F. D'Agosto, Angew. Chem., Int. Ed., 2013, 52, 3438 CrossRef CAS PubMed.
- D. Damiron, J. Mazzolini, F. Cousin, C. Boisson, F. D'Agosto and E. Drockenmuller, Polym. Chem., 2012, 3, 1838 RSC.
- M. Bieligmeyer, S. M. Taheri, I. German, C. Boisson, C. Probst, W. Milius, V. Altstädt, J. Breu, H.-W. Schmidt, F. D'Agosto and S. Förster, J. Am. Chem. Soc., 2012, 134, 18157 CrossRef CAS PubMed.
- M. M. Unterlass, E. Espinosa, F. Boisson, F. D'Agosto, C. Boisson, K. Ariga, I. Khalakhan, R. Charvet and J. P. Hill, Chem. Commun., 2011, 47, 7057 RSC.
- H. Makio, T. Ochiai, J. Mohri, K. Takeda, T. Shimazaki, Y. Usui, S. Matsuura and T. Fujita, J. Am. Chem. Soc., 2013, 135, 8177 CrossRef CAS PubMed.
- W. Lin, H. Niu, T. C. M. Chung and J.-Y. Dong, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3534 CrossRef CAS.
- Y. Inoue and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 496 CrossRef CAS.
- A. V. Sesha Sainath, M. Isokawa, M. Suzuki, S. Ishii, S. Matsuura, N. Nagai and T. Fujita, Macromolecules, 2009, 42, 4356 CrossRef.
- K. Matoishi, K. Nakai, N. Nagai, H. Terao and T. Fujita, Catal. Today, 2011, 164, 2 CrossRef CAS PubMed.
- A. M. Anderson-Wile, G. W. Coates, F. Auriemma, C. De Rosa and A. Silvestre, Macromolecules, 2012, 45, 7863 CrossRef CAS.
- J. Mazzolini, O. Boyron, V. Monteil, D. Gigmes, D. Bertin, F. D'Agosto and C. Boisson, Macromolecules, 2011, 44, 3381 CrossRef CAS.
- B. B. Busch, M. M. Paz, K. J. Shea, C. L. Staiger, J. M. Stoddard, J. R. Walker, X.-Z. Zhou and H. Zhu, J. Am. Chem. Soc., 2002, 124, 3636 CrossRef CAS PubMed.
- K. J. Shea, B. B. Busch and M. M. Paz, Angew. Chem., Int. Ed., 1998, 37, 1391 CrossRef CAS.
- K. J. Shea, J. W. Walker, H. Zhu, M. Paz and J. Greaves, J. Am. Chem. Soc., 1997, 119, 9049 CrossRef CAS.
- C. Yuan, H.-C. Lu, Q.-Z. Li, S. Yang, Q.-L. Zhao, J. Huang, L.-H. Wei and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2398 CrossRef CAS.
- Q.-Z. Li, G.-Y. Zhang, J.-Z. Chen, Q.-L. Zhao, H.-C. Lu, J. Huang, L.-H. Wei, F. D'Agosto, C. Boisson and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 511 CrossRef CAS.
- J.-Z. Chen, Q.-L. Zhao, H.-C. Lu, J. Huang, S.-K. Cao and Z. Ma, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1894 CrossRef CAS.
- J.-Z. Chen, K. Cui, S.-Y. Zhang, P. Xie, Q.-L. Zhao, J. Huang, L.-P. Shi, G.-Y. Li and Z. Ma, Macromol. Rapid Commun., 2009, 30, 532 CrossRef CAS PubMed.
- L. Yin and M. A. Hillmyer, Macromolecules, 2011, 44, 3021 CrossRef CAS.
- R. B. Jayaraman, J. V. Facinelli, J. S. Riffle and S. E. George, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1543 CrossRef CAS.
- L. M. Pitet, J. Zhang and M. A. Hillmyer, Dalton Trans., 2013, 42, 9079 RSC.
- L. M. Pitet and M. A. Hillmyer, Macromolecules, 2011, 44, 2378 CrossRef CAS.
- C. T. Chung, Prog. Polym. Sci., 2002, 27, 39 CrossRef.
- J.-Y. Dong and Y. L. Hu, Coord. Chem. Rev., 2006, 250, 47 CrossRef CAS PubMed.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363 CrossRef CAS PubMed.
- S. B. Amin and T. J. Marks, Angew. Chem., Int. Ed., 2008, 47, 2006 CrossRef CAS PubMed.
- R. Godoy Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419 CrossRef CAS PubMed.
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2013, 42, 5809 RSC.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- H. Wang, W. D. Yan, T. Jiang, B. B. Liu, W. Q. Xu, J. J. Ma and Y. L. Hu, Chin. Sci. Bull., 2002, 47, 1616 CAS.
-
I. Kim, Reactive and Functional Polymers, 2001, vol. 49, p. 197 Search PubMed.
- N. N. Schwartz and J. H. Blumbergs, J. Org. Chem., 1964, 29, 1976 CrossRef CAS.
- D. Tanner and T. Groth, Tetrahedron, 1997, 53, 16139 CrossRef CAS.
- G. Sabitha, R. S. Babu, M. Rajkumar, C. S. Reddy and J. S. Yadav, Tetrahedron Lett., 2001, 42, 3955 CrossRef CAS.
- S. T. Milner and T. C. B. McLeish, Macromolecules, 1998, 31, 7479 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed.
- H. Huang, H. Niu and J.-Y. Dong, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2222 CrossRef CAS.
- H. Huang, H. Niu and J.-Y. Dong, Macromolecules, 2010, 43, 8331 CrossRef CAS.
- D. Biswanath, R. Vtukuri Saidi, K. Maddeboina and R. Yerra Koteswara, J. Mol. Catal. A: Chem., 2007, 270, 89 CrossRef PubMed.
- A. Bukowska and W. Bukowski, Org. Process Res. Dev., 2002, 6, 234 CrossRef CAS.
- O. Yemul and T. Imae, Colloid Polym. Sci., 2008, 286, 747 CAS.
- E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed., 1993, 32, 1308 CrossRef.
- K. Matoishi, S. Nakatsuka, K. Nakai, M. Isokawa, N. Nagai and T. Fujita, Chem. Lett., 2010, 39, 1028 CrossRef CAS.
- J. Mazzolini, O. Boyron, V. Monteil, F. D'Agosto, C. Boisson, G. C. Sanders, J. P. A. Heuts, R. Duchateau, D. Gigmes and D. Bertin, Polym. Chem., 2012, 3, 2383 RSC.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 9018 CrossRef CAS.
- S. Liu and S. P. Armes, Curr. Opin. Colloid Interface Sci., 2001, 6, 249 CrossRef CAS.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026 CrossRef CAS PubMed.
- J. S. Parent and S. S. Sengupta, Macromolecules, 2005, 38, 5538 CrossRef CAS.
- S. S. Sengupta and J. S. Parent, Polym. Eng. Sci., 2006, 46, 480 CAS.
- H. E.-R. Rami Al-Oweini, J. Mol. Struct., 2009, 919, 140 CrossRef PubMed.
-
M. Tolinski, in Additives for Polyolefins, William Andrew Publishing, Oxford, 2009, p. 23 Search PubMed.
- J. Pospíšil, W. D. Habicher and S. Nešpůrek, Macromol. Symp., 2001, 164, 389 CrossRef.
- P. Stagnaro, G. Mancini, A. Piccinini, S. Losio, M. C. Sacchi, C. Viglianisi, S. Menichetti, A. Adobati and S. Limbo, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1007 CrossRef CAS.
- L. Boragno, P. Stagnaro, S. Losio, M. C. Sacchi, S. Menichetti, C. Viglianisi, L. Piergiovanni and S. Limbo, J. Appl. Polym. Sci., 2012, 124, 3912 CrossRef CAS.
- M. S. Dopico-García, J. M. López-Vilariñó and M. V. González-Rodríguez, J. Agric. Food Chem., 2007, 55, 3225 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.