Binding and supramolecular organization of homo- and heterotelechelic oligomers in solutions†
Jessalyn
Cortese
,
Corinne
Soulié-Ziakovic
* and
Ludwik
Leibler
*
Matière Molle et Chimie (UMR 7167 ESPCI-CNRS), Ecole Supérieure de Physique et Chimie Industrielles de la Ville de Paris ESPCI-ParisTech, 10 rue Vauquelin, 75005 Paris, France. E-mail: Ludwik.Leibler@espci.fr; Corinne.Soulie@espci.fr
First published on 19th July 2013
Abstract
Solvent can subtly influence the organization of supramolecular polymers. We synthesized homotelechelic and heterotelechelic oligomers of poly(propylene oxide) (PPO) equipped with complementary hydrogen bonding functional ends, thymine (Thy) and diaminotriazine (DAT). In a solvent that dissociates Thy–DAT hydrogen bonds, such as DMSO, the viscosity was low for all functional telechelic oligomers. In non-dissociative solvents, the addition of functional oligomers increased the viscosity. For both the homotelechelic blends and the heterotelechelics, the viscosity in toluene was about two times higher than that in chloroform. Additionally, the Thy–DAT association constant was 22 times higher. Carbon relaxation times measured by NMR and viscosity variation for solutions of different concentrations suggest a distinct supramolecular organization in chloroform and toluene: linear and cyclic supramolecular chains in chloroform and small π-stacked objects with a PPO shell and a Thy, DAT core in toluene.
Introduction
Supramolecular chemistry, defined as the chemistry of noncovalent bonds (such as hydrogen bonding, π–π stacking, hydrophobic interactions, etc.),1 is a promising tool to create functional materials.2 Indeed, noncovalent bonds introduce reversibility and stimuli-responsiveness to materials. Therefore, compared to high-molecular-weight covalent polymers, processing and recycling of supramolecular polymers could be easier. Furthermore, incorporating noncovalent bonds into materials can impart original properties, such as self-healing.3 The simplest situation occurs when oligomers are equipped with complementary or self-complementary functional end-groups. They can in principle associate to form linear chain supramolecular polymers. The strength of the association constant controls the length of the polymer.4 Furthermore, supramolecular organizations at mesoscopic scales often appear and they lead to a richer and interesting behavior in comparison with classical covalent polymers. Complex mesoscopic organizations were observed both in the bulk5 and in solution.6 In solution, for telechelic oligomers, the aggregation is often induced by aromatic ring stacking. Thus, Hirschberg et al.6a observed helical columns, Kolomiets et al.6c a fibrillar nanostructure, and Iwaura et al.6d,e helical nanofibers. Moreover, the nanostructure can be solvent-dependent as in the self-complementary ureidotriazine-based supramolecular polymers that form random coils via hydrogen bonding in chloroform, but helical columns via cooperative and solvophobically induced π-stacking of the hydrogen-bonded pairs in dodecane and water.6a,b For small molecules, such a solvent-dependence has been shown, in particular by Bouteiller's group.7Introducing multiple parallel hydrogen bonding groups, either self-complementary (C with C), as ureidopyrimidone8 or imidazolidone,9 or hetero-complementary (A with B), as nucleobases,10 on both ends of an oligomeric spacer allows the formation of main-chain supramolecular polymers4 or supramolecular block copolymers.11 Three constructions are possible: (i) self-assembly of a homotelechelic oligomer (C-spacer-C)n, (ii) complementary assembly of two homotelechelic oligomers (A-spacer-A/B-spacer-B)n, or (iii) complementary assembly of a heterotelechelic oligomer (A-spacer-B)n. In each case, a thermodynamic equilibrium takes place between monomers, oligomers and polymers. In solution, the degree of polymerization (DP = n) and its distribution depend on the thermodynamic association constant of the associating groups (KCC or KAB), on the associative group concentrations and on the supramolecular polymerization mechanism.4 Higher association constants and/or associative group concentrations yield higher DP, and thus higher solution viscosity. However, to obtain a high DP with homotelechelic blends (A-spacer-A/B-spacer-B)n, the A and B associative group stoichiometry needs to be carefully controlled to 1 to 1, because the excess of one associative group has a chain stopper effect that limits DP.12
To avoid this issue of stoichiometry in hometelechelic blends, Zimmerman's13 and Meijer's14 groups synthesized supramolecular polymers with an associative group A not only capable of hetero-complementary association, but also of self-complementary dimerization, i.e. both KAB and KAA association constants were high. As a result, if A's concentration is higher than B's, DP is still large. The limitation of this strategy is that if it is B that is in excess, a chain stopper effect takes place. With the heterotelechelic unit (A-spacer-B), stoichiometry is always 1 to 1, so high dimerization constants are no longer needed. However, synthetic pathways are more tedious. Three main routes have been explored: (i) oligomerization of the spacer from the stickers, (ii) convergent coupling, and (iii) asymmetric end-functionalization post-oligomerization.
The first path has been developed by the Weck group.15 They used ring-opening metathesis polymerization (ROMP) with an associative group A-functionalized ruthenium initiator (A = Hamilton receptor) and an associative group B-based chain-terminator (B = 2,7-diamido-1,8-naphthyridine (DAN)), to synthesize a heterotelechelic A-poly(norbornene imide)-B. With a similar idea, Mansfeld et al. used a heterodifunctionalized alkoxyamine initiator to prepare, via nitroxide-mediated radical polymerization, heterotelechelics bearing the 2-ureido-4[1H]-pyrimidinone (UPy) sticker on one side and the terpyridine (tpy) group on the other.16 Reversible addition-fragmentation chain transfer (RAFT)17 or atom transfer radical polymerization (ATRP)18 processes have also been developed to prepare heterotelechelic polymers. Compared to ROP or ROMP techniques, which are limited to strained cyclic monomers, the radical based processes are more versatile with respect to the spacer monomer choice. These elegant routes offer a high-yielding preparation of asymmetrically functionalized supramolecular units. However, these approaches require a multistep synthesis of the initiators, transfer agents and/or chain terminators.
The second route, convergent coupling, has been followed by Xu et al.19 for oligonucleotides and by Scherman et al.20 Scherman and his coworkers used selective olefincross-metathesis coupling of an A-functionalized acrylate with a B-functionalized olefin (both also obtained by coupling21) to get heterotelechelic A-spacer-B supramolecular oligomers (A = UPy, B = NaPy).
The third strategy, employed here, is based on the asymmetric end-functionalization of an oligomeric spacer, which on the contrary, is versatile but not trivial. Because of the oligomeric nature of spacers, generally larger than 5 carbons, both ends present the same reactivity, making difficult the synthesis of differently functionalized ends by grafting. To circumvent this difficulty, Shimizu et al.22 reacted a large excess (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of a difunctional spacer with the associative group A (A = thymine). Under these conditions, A-bifunctionalized spacer is scarcely formed. Then, they separated the A-monofunctionalized spacer from the excess spacer by column chromatography, and finally reacted it with sticker B (B = adenine), leading to an A-spacer-B heterotelechelic unit. However, they used very well-defined spacers (diaminodecane, diaminododecane, etc.), convenient for column chromatography purification. Such a purification process is hardly applicable to oligomeric or polymeric spacers because the polydispersity generally precludes efficient product separation.
:1) of a difunctional spacer with the associative group A (A = thymine). Under these conditions, A-bifunctionalized spacer is scarcely formed. Then, they separated the A-monofunctionalized spacer from the excess spacer by column chromatography, and finally reacted it with sticker B (B = adenine), leading to an A-spacer-B heterotelechelic unit. However, they used very well-defined spacers (diaminodecane, diaminododecane, etc.), convenient for column chromatography purification. Such a purification process is hardly applicable to oligomeric or polymeric spacers because the polydispersity generally precludes efficient product separation.
Here we report on the four step synthesis (Scheme 1) of heterotelechelic oligomers consisting of poly(propylene oxide) oligomeric spacers (1a-b, denoted as NH2-PPO-X-NH2, with X the molecular weight in g mol−1, X = 460, 2200), capped with a thymine group (Thy) on one end and a 2,6-diamino-1,3,5-triazine (DAT) on the other (denoted as Thy-PPO-X-DAT, 5a-b). The first step is the protection of one of the two amines by a BOC group of the PPO spacer. Purification was achieved by successive liquid–liquid extractions and aqueous wash processes. Indeed, since BOC is hydrophobic and amines are hydrophilic especially at low pH, BOC-PPO-NH2 preferred the organic phase, whereas NH2-PPO-NH2 preferred the acidic aqueous phase. After this key step, Thy was grafted by amidation on the unprotected amine,5 then the protected amine is deprotected, and finally DAT was grafted by aromatic nucleophilic substitution.23
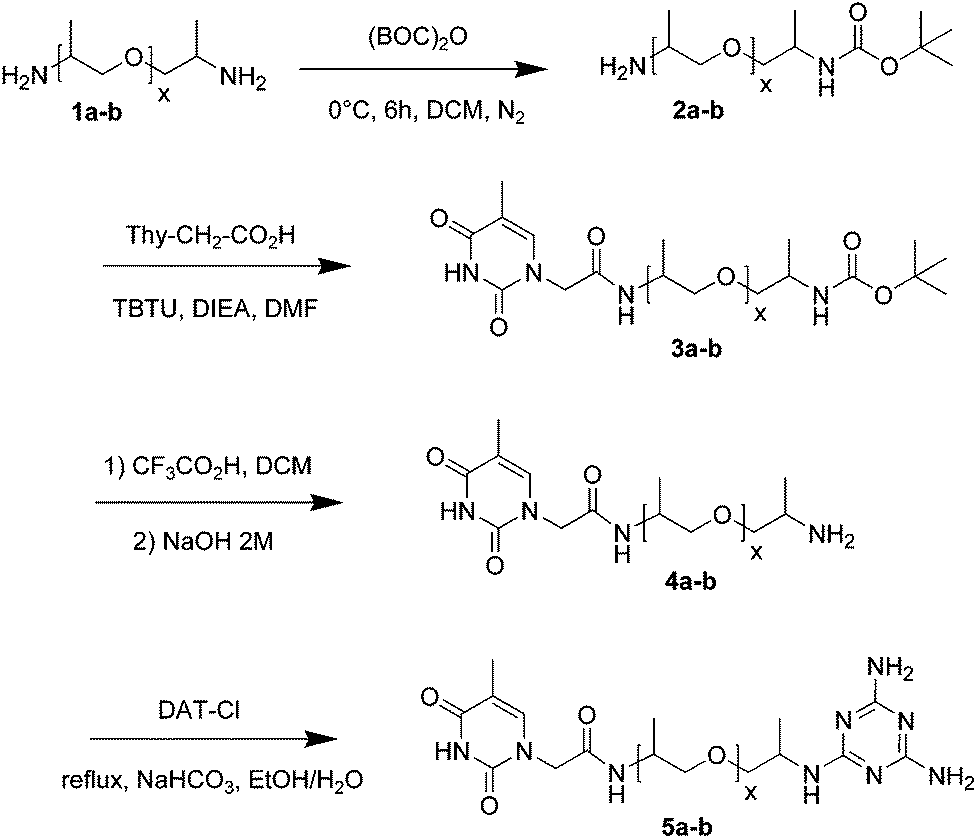 | ||
| Scheme 1 Synthesis of heterotelechelic oligomers, Thy-PPO-X-DAT (a: X = 460, b: X = 2200). | ||
We also report on the solution behavior of these heterotelechelic oligomers (Thy-PPO-X-DAT 5a-b), as well as homotelechelic oligomers (Thy-PPO-X-Thy 6a-b and DAT-PPO-X-DAT 7a-b), and their 50/50 mixture (50/50-M-X8a-b, X = 460, 2200), in aprotic solvents of different polarities (dimethylsulfoxide (DMSO), chloroform and toluene). Since they are amphiphilic molecules, with polar H-bonding associative groups and a less polar PPO spacer, solvent nature has a strong impact on their association constant, their supramolecular polymerization mechanism, and thus their mesoscopic organization, as evidenced by spectroscopic and rheological characterizations performed at different temperatures and concentrations.
Experimental section
Reagents and solvents
NH2-PPO-X-NH2 (1a-b, Jeffamine® D Series) was kindly provided by Huntsman. The other reagents were purchased from Sigma-Aldrich, Acros or Alfa-Aesar. All chemicals were used as received unless otherwise stated. Thy-PPO-X-Thy (6a-b), DAT-PPO-X-DAT (7a-b), and their 50:50 mixture, 50/50-M-X (8a-b), were synthesized as previously reported.5,23
NMR experiments
1H and 13C NMR spectra were recorded on a Bruker AC300 400 MHz spectrometer. The solvents used were: CDCl3, DMSO-d6, or toluene-d8. The chemical shifts (δ) are expressed in ppm relative to TMS, with the solvent signal being used as an intern reference (1H: δCDCl3 = 7.26 ppm, δDMSO-d6 = 2.50 ppm, and δtoluene-d8 = 2.09 ppm; 13C: δCDCl3 = 77.16 ppm, δDMSO-d6 = 39.52 ppm, and δtoluene-d8 = 20.4 ppm). The signal multiplicity is indicated as follows: s: singlet; d: doublet; t: triplet; q: quadruplet; m: multiplet. The numbering of atoms, used to attribute the NMR signals, does not correspond to the nomenclature.Rheology experiments
The rheological properties of the solutions were investigated on an Anton Paar Physica MCR 501 rheometer, equipped with a double-gap Couette cell (external diameter: 26.7 mm; gap: 0.45 mm, sample volume: 3.8 mL).Synthesis of Thy-PPO-X-DAT (X = 460, 2200) 5a-b
A general procedure for the synthesis of heterotelechelic oligomers is shown with the spacer X = 2200 (5b) (for 5a (X = 400) see ESI†).Results and discussion
Synthesis of heterotelechelic supramolecular polymers
The key step of our heterotelechelic supramolecular polymer synthesis is the protection of only one of the two symmetrical amino groups, into an N-tertio-butoxycarbonyl (BOC) group (Scheme 1). Tertio-butyl carbonylation is a very popular protection reaction of amines,24 including the monoprotection of symmetrical diamines.25 To increase the yield of mono-BOC protected diamines, as opposed to unreacted and di-BOC protected diamines, several procedures have been developed: low concentrations, prior acid addition, slow addition of di-tertio-butyl dicarbonate ((BOC)2O) solution and low temperatures.25,26 Still, a mixture of unreacted, mono-BOC protected and di-BOC protected α,ω-diamines was obtained. Unreacted diamines could be separated by taking advantage of their affinity and basic property differences. Indeed, the BOC group is strongly lipophilic, therefore it enhances the hydrophobic nature of N-BOC products. On the other hand, the hydrophilic nature of unreacted diamines was reinforced in acidic aqueous phases, which turned them into ammonium salts. Thus, by successive organic extractions and acidic aqueous wash processes, unreacted diamines mainly went into aqueous phases, while N-BOC products mostly went into organic ones. Di-BOC protected α,ω-diamines were still there, but could be separated after deprotection, also by liquid–liquid extraction. Moreover, the quantity of the di-BOC protected compounds could be reduced by proceeding in dilute media, at a low temperature (0 °C), without any catalyst, in a short reaction time (6 hours), and by taking advantage of the sterical hindrance of the end-chain amine due to the α-methyl group of the PPO chain.The reactions were followed by 1H NMR analysis (see Fig. 1 for the X = 2200 series, see ESI† for X = 460 series).
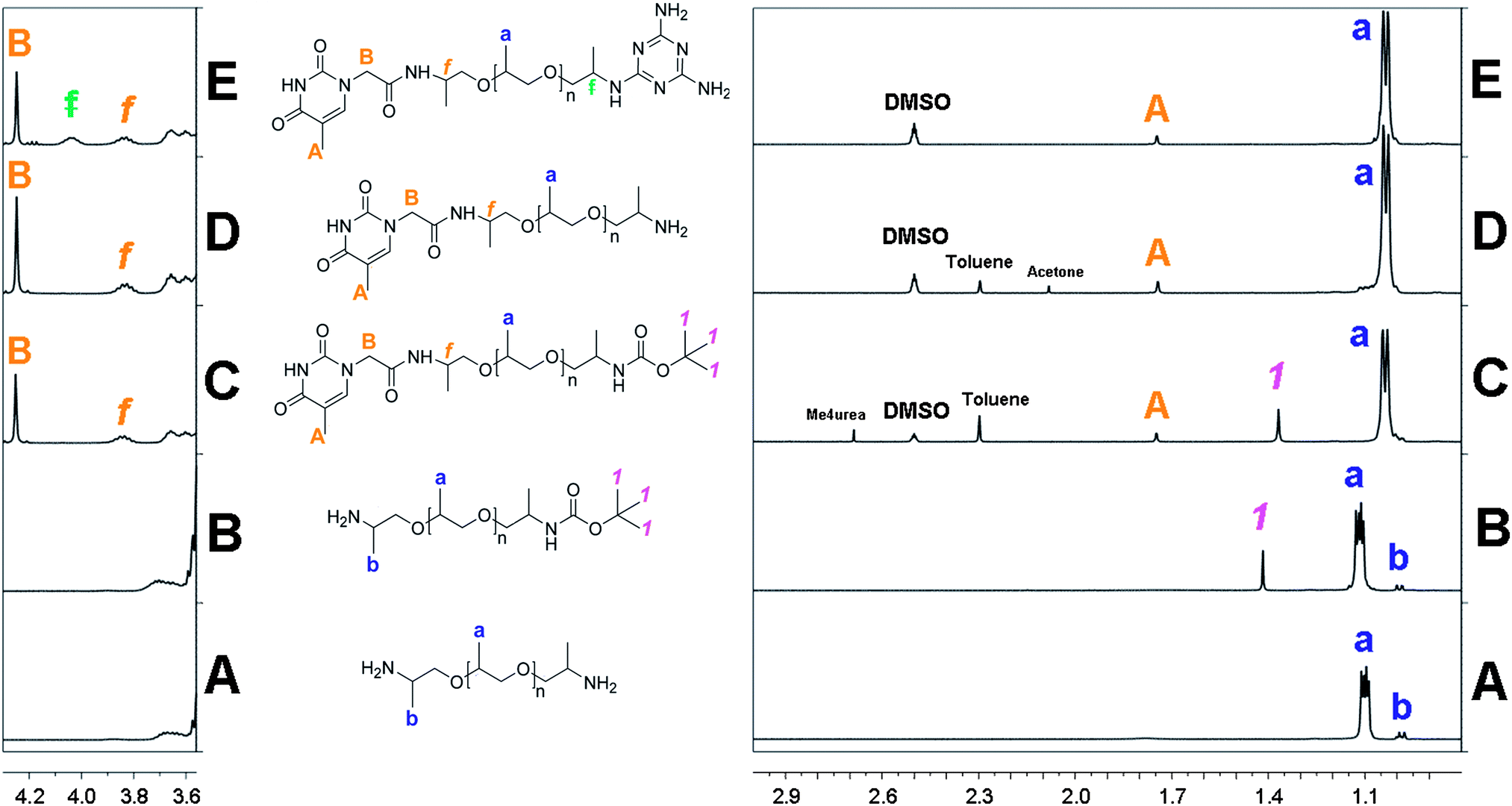 | ||
| Fig. 1 1H NMR spectra at 25 °C: (A) NH2-PPO-2200-NH21b (CDCl3); (B) NH2-PPO-2200-BOC 2b (CDCl3); (C) Thy-PPO-2200-NH23b (DMSO-d6); (D) Thy-PPO-2200-NH24b (DMSO-d6); (E) DAT-PPO-2200-Thy 5b (DMSO-d6). | ||
For the protection reaction, integrating the methyl BOC signal (1) over the α-methyl of the end-functionalized NH2 PPO signal (b) (2b, Fig. 1B) showed that the final organic phase 2b contained 1.5 mol% of di-BOC protected amine and no unreacted diamine1b. This confirmed that the di-BOC protection reaction has been limited.
As mono-protected amine2b is an amphiphilic compound, it was also found in aqueous phases, and the reaction yield was estimated to be 78 mol% from NH2-PPO-NH21b.
In the second step, amidation of the unreacted amine of 2b by thymine-1-acetic acid was conducted in DMF, in the presence of TBTU as a coupling agent.5
Purification was facilitated by the overall hydrophobicity of the thymine moiety. Indeed, even though polar and able to form H-bonds with polar and/or protic solvents (DMSO, methanol, etc.), thymine is only slightly water-soluble, due to the hydrophobic nature of its methyl substituent.27 On the 1H NMR spectrum (3b, Fig. 1C), ratio of BOC (1) and α-methyl of end-functionalized NH2 PPO (b) signal integrations with the thymine CH3 (A) and CH2 (B) showed that 3b was obtained in 90 mol% and that the 1.5 mol% of di-protected diamine were still present. Protected products 3b were deprotected with trifluoroacetic acid/dichloromethane (DCM) blend, neutralized with a 2 M soda solution,24 and the desired product 4b was extracted with toluene. The 1H NMR spectrum (4b, Fig. 1D) confirmed that the deprotection reaction was complete since the BOC signal (1) had disappeared. Yet, the ratio of PPO methyl (a) signal integration with the thymine CH3 (A) and CH2 (B) revealed an excess of starting diamine1b, produced from the deprotection of diprotected diamine (1.5 mol% from step 1) and unreacted mono-protected diamine2b (10 mol% from step 2).
The heterotelechelic unit Thy-PPO-2200-DAT 5b was synthesized by the nucleophilic substitution of an amine end-function of 4b with 4-chloro-DAT in a water–ethanol 1/1 blend.23 The reaction could easily be followed since, even at reflux, 4-chloro-DAT was insoluble: as the reaction was taking place, the mixture was getting less and less cloudy until it was perfectly clear. Like thymine, even polar and H-bonding able, DAT is roughly hydrophobic due to its aromatic nature. The heterotelechelic oligomer was thus hydrophobic and was isolated with DCM. The 1H NMR spectrum (5b, Fig. 1E) revealed 5.7 mol% excess of DAT moiety. Excess of DAT could be attributed to homotelechelic DAT-PPO-2200-DAT (i.e. 2.8 mol%) produced from reaction of NH2-PPO-2200-NH21b recovered by deprotection of N-BOC products in step 3. MALDI-TOF experiments were performed on 5b and led to the same conclusion (see ESI†).
Association constant in DMSO, chloroform and toluene
Hydrogen bonds could take place between NH (D) and NH2 (Φ, Ψ). However, as the other donor sites (N, O) are more nucleophilic, interactions between NH (D) and N (DAT) and NH2 (Φ, Ψ) and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C are favored and stronger. Hydrogen bond orientations are better.28,29 Association constants can be determined by 1H NMR titration.1b,30 Indeed, the chemical shifts of Thy and DAT protons implicated in the hydrogen bonds (D, Φ, Ψ, Scheme 2) varied greatly between the free, self-associated, and Thy–DAT associated states. Since the equilibria were fast on the NMR spectroscopic time scale, the observed chemical shift for Thy NH (D) and DAT NH2 (Φ, Ψ) was a weighted average between the chemical shifts of the associated, self-associated, and free states (Fig. 2). Thus, Thy–Thy and DAT–DAT self-association constants (KThy–Thy and KDAT–DAT), and the Thy–DAT association constant (KThy–DAT) can be measured by monitoring the Thy NH (D) or DAT NH2 (Φ, Ψ) chemical shift as a function of species concentration. The titration curves were then analyzed by computer fitting with the least-squares method (EQNMR program).31 More details on this procedure (as well as equations, titration curves and fitting) are available in the ESI.† The results are gathered in Table 1.
C are favored and stronger. Hydrogen bond orientations are better.28,29 Association constants can be determined by 1H NMR titration.1b,30 Indeed, the chemical shifts of Thy and DAT protons implicated in the hydrogen bonds (D, Φ, Ψ, Scheme 2) varied greatly between the free, self-associated, and Thy–DAT associated states. Since the equilibria were fast on the NMR spectroscopic time scale, the observed chemical shift for Thy NH (D) and DAT NH2 (Φ, Ψ) was a weighted average between the chemical shifts of the associated, self-associated, and free states (Fig. 2). Thus, Thy–Thy and DAT–DAT self-association constants (KThy–Thy and KDAT–DAT), and the Thy–DAT association constant (KThy–DAT) can be measured by monitoring the Thy NH (D) or DAT NH2 (Φ, Ψ) chemical shift as a function of species concentration. The titration curves were then analyzed by computer fitting with the least-squares method (EQNMR program).31 More details on this procedure (as well as equations, titration curves and fitting) are available in the ESI.† The results are gathered in Table 1.
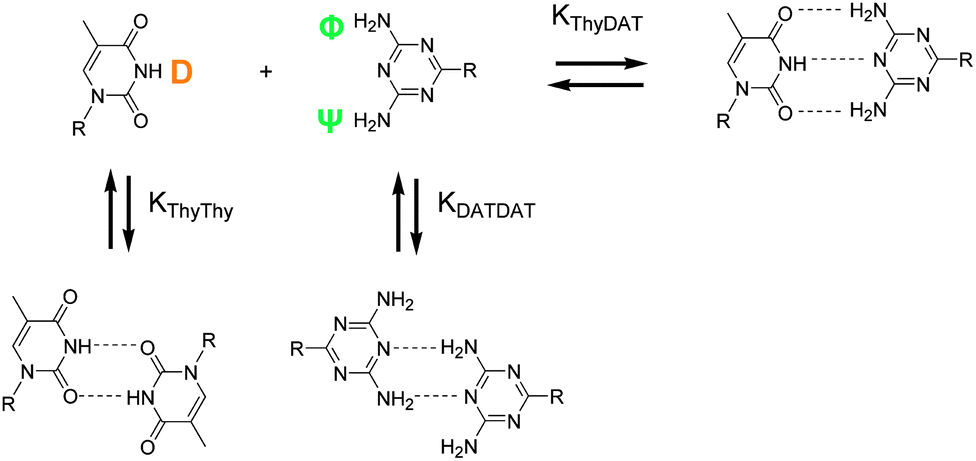 | ||
| Scheme 2 Thy–DAT complementary association and Thy–Thy, DAT–DAT self-associations. | ||
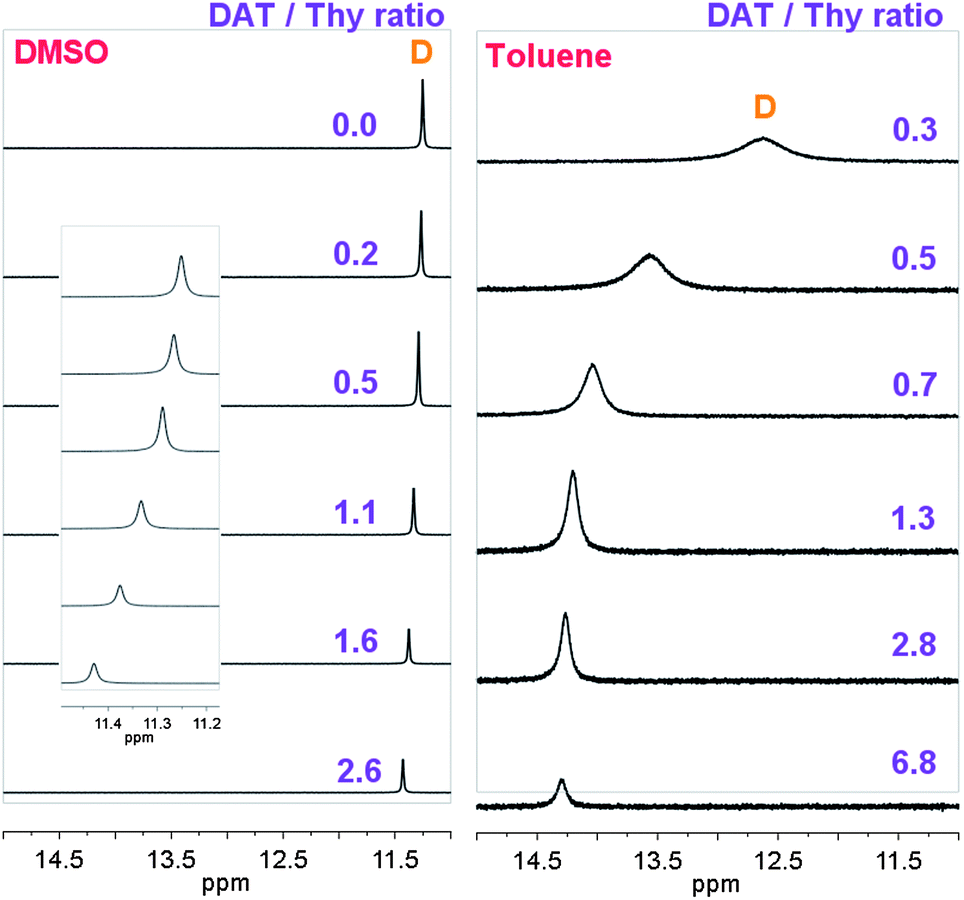 | ||
| Fig. 2 1H NMR spectra at 25 °C (A) of BuThy (at 9.0 × 10−2 mol L−1), MeDAT (from 0 to 2.3 × 10−1 mol L−1) mixtures in DMSO-d6, and (B) of Thy-PPO-2200-Thy 6b (from 3.0 × 10−3 to 6.2 × 10−2 mol L−1), DAT-PPO-2200-DAT 7b (at 2.1 × 10−2 mol L−1) mixtures in toluene-d8. | ||
The values of association constants (KThy–Thy, KDAT–DAT and KThy–DAT) obtained in CDCl3 were close to what can be found in the literature.32,33
To correctly fit the experimental data, and thus determine the Thy–DAT association constant KThy–DAT, it was necessary to take the DAT–DAT self-association constant KDAT–DAT into account, even though it was 300 times lower (Thy–Thy self-association could be neglected because DAT was in excess in this set of experiments).
K Thy–DAT could not be determined in toluene-d8 by this method. Thy–Thy and DAT–DAT self-association constants (KThy–Thy and KDAT–DAT) measured in toluene-d8 were more than 10 times higher than in CDCl3. Therefore, KThy–DAT was expected to also be more than 10 times higher in toluene-d8 than in CDCl3 (i.e. over 8 × 103 L mol−1). However, the NMR titration method does not allow the determination of association constants greater than 104 L mol−11b and 105 L mol−1.30 Moreover, in toluene-d8, the Thy NH (D) chemical shift was very high and did not vary with DAT concentration above the equimolar DAT–Thy ratio, but only started to shift when Thy was in excess (Fig. 2). Furthermore, the Thy NH (D) chemical shift of Thy-PPO-2200-DAT 5b was independent of concentration in toluene but not in chloroform. These results confirmed that KThy–DAT was much higher in toluene-d8 than in CDCl3.
In contrast, KThy–DAT was very low in DMSO-d6 (Table 1). Indeed, the Thy and DAT hydrogen bond donors and acceptors are highly solvated in this polar solvent (relative polarity of DMSO = 0.444),34 and scarcely assemble.1b Similarly, KThy–DAT may be much higher in toluene than in chloroform because the hydrogen bond donors and acceptors are more poorly solvated in the non-polar toluene (relative polarity = 0.099)34 than in the relatively polar chloroform (relative polarity = 0.259).28
Although the KThy–DAT could not be measured in toluene-d8 by NMR titration, temperature-dependent NMR measurements of solutions of Thy-PPO-2200-Thy 6b, DAT-PPO-2200-DAT 7b, and Thy-PPO-2200-DAT 5b allowed the estimation of KThy–DAT, as well as KThy–Thy and KDAT–DAT, as a function of temperature (Table 2) (see ESI† for detailed procedure and equations).35
| T (°C) | CDCl3 | Toluene-d8 | ||||
|---|---|---|---|---|---|---|
| K Thy–Thy | K DAT–DAT | K Thy–DAT | K Thy–Thy | K DAT–DAT | K Thy–DAT | |
| 0 | 21 | 10 | 7.4 × 103 | 6.7 × 103 | 493 | 3.3 × 105 |
| 10 | 12 | 6 | 2.7 × 103 | 374 | 134 | 8.3 × 104 |
| 24 | 7 | 4 | 835 | 95 | 17 | 2.2 × 104 |
| 30 | 7 | 3 | 552 | 36 | 10 | — |
| 40 | 4 | 2 | 174 | 11 | 3 | 2.9 × 103 |
| 50 | 2 | 2 | 82 | 5 | 1 | 960 |
| 60 | — | — | — | 4 | 0,6 | 322 |
| 70 | — | — | — | 1 | 0,3 | 152 |
If values of dimerisation constants KThy–Thy and KDAT–DAT are close to those recently reported by Binder36 (70 ± 14 L mol−1 and 22 ± 7 L mol−1 resp. at 27 °C), the value of the association constant KThy–DAT was 10 times higher (2.6 ± 1.3 × 103 L mol−1). This could be explained by the fact that we did not use the same fit as Binder did, i.e. we took into account possible dimerisations in the association constant fit. Indeed, as we have mentioned above, even though weak, these self-complementary associations cannot be ignored, especially in toluene in which constants are enhanced.
Mesoscopic organizations in DMSO, toluene and chloroform
These solvent-dependent association and self-association constants seemed to have impact on the solution properties. Indeed, in chloroform and toluene, the relative viscosities of 0.096 g cm−3 solutions containing both Thy and DAT groups (Thy-PPO-2200-DAT 5b and 50/50-M-2200 8b) were higher than those containing only Thy or only DAT groups (Thy-PPO-2200-Thy 6b and DAT-PPO-2200-DAT 7b) (Fig. 3B and C). These viscosity measurements were thus in agreement with the NMR results: weak Thy–Thy and DAT–DAT self-associations and strong Thy–DAT complementary associations, in chloroform and toluene.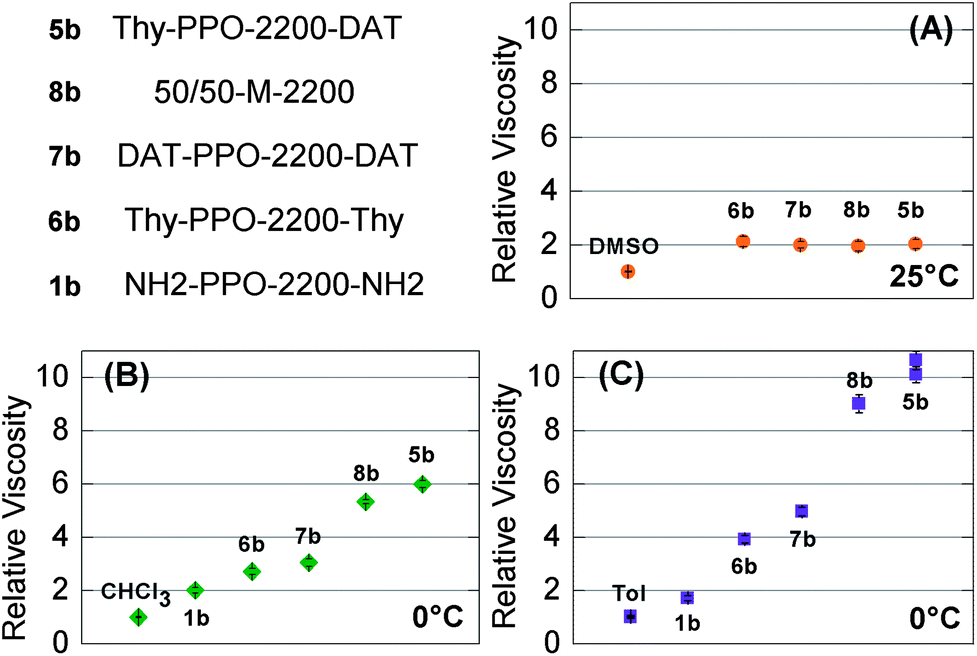 | ||
| Fig. 3 Relative viscosity of 9.6 × 10−2 g mL−1 solutions of the heterotelechelic unit Thy-PPO-2200-DAT (5b), homotelechelic units Thy-PPO-2200-Thy (6b) and DAT-PPO-2200-DAT (7b), 50:50 mixture of Thy-PPO-2200-Thy and DAT-PPO-2200-DAT (8b), and precursor NH2-PPO-2200-NH2 (1b) in: (A) DMSO at 25 °C, (B) chloroform at 0 °C, and (C) toluene at 0 °C. | ||
In contrast, in DMSO, 10 wt% (0.122 g cm−3) solutions of Thy-PPO-2200-DAT 5b, 50/50-M-2200 8b, Thy-PPO-2200-Thy 6b and DAT-PPO-2200-DAT 7b all had the same relative viscosity (ηrel = 2.0) (Fig. 3A). Furthermore, this relative viscosity of 2.0 in DMSO was quite lower than in chloroform and toluene. All of this confirmed that hardly any H-bonding association occurs in DMSO, and that no linear supramolecular polymer grew in this solvent. However, since DMSO is a polar solvent and the PPO chain is mostly non-polar, hydrophobic interactions induce aggregation of the PPO chain. This aggregation could be evidenced by comparing the carbon NMR relaxation times T1 of Thy-PPO-2200-DAT 5b and NH2-PPO-2200-NH21b PPO chain in DMSO-d6, CDCl3 and toluene-d8 (Table 3). Indeed, T1 reflects the local mobility of the studied group, and a high T1 indicates a high mobility. The T1 results showed that chloroform and, especially, toluene, were good solvents of the PPO chain, since the T1 values were rather high. In contrast, the T1 values in DMSO were much weaker, indicating that the PPO chain mobility was strongly diminished. In fact, these results suggested that Thy-PPO-2200-DAT 5b, 50/50-M-2200 8b, Thy-PPO-2200-Thy 6b and DAT-PPO-2200-DAT 7b may form micelles in DMSO, with the PPO chains collapsed in the core and the Thy and DAT groups at the surface, solvated by the DMSO (Fig. 7A). Indeed, DMSO is a good solvent of the polar associative groups, but a bad solvent for the much less polar PPO chain. This image is consistent with the fact that 5b, 8b, 6b and 7b all had the same relative viscosity in DMSO.
The relative viscosities of 0.096 g cm−3 solutions of 50/50-M-2200 8b in toluene or chloroform were slightly lower than that of Thy-PPO-2200-DAT 5b. This effect could be either due to a Thy–DAT ratio closer to 1 in the asymmetric Thy-PPO-2200-DAT 5b than in the mixture 50/50-M-2200 8b (chain-stopper effect); or due to different mesoscopic organizations. Besides, the relative viscosities of 0.096 g cm−3 solutions of all compounds were higher in toluene than in chloroform. This was particularly obvious for Thy-PPO-2200-DAT 5b for which ηrel was 10 in toluenevs. 6 in chloroform at 0 °C. This difference was less marked as the temperature increased (5 vs. 4.3 at 20 °C). This result could be explained by the higher KThy–DAT, KThy–Thy, and KDAT–DAT in toluene compared to chloroform, leading to different supramolecular mesoscopic organizations in toluene and in chloroform. Furthermore, the temperature dependence could also be explained by lower association constants as demonstrated by 1H NMR measurements.
However, concentration-dependent viscosity measurements (Fig. 4) did not seem to be consistent with this idea, at least on the first approach. Indeed, specific viscosities (relative viscosities minus one) of all compounds in toluene and in chloroform were equal at low concentrations. If the higher values of association constants in toluene were responsible for the higher measured viscosities, one would also naïvely expect higher viscosities in toluene at lower concentrations. To see if this idea was correct, we have compared our experimental results with viscosity models of supramolecular polymers.
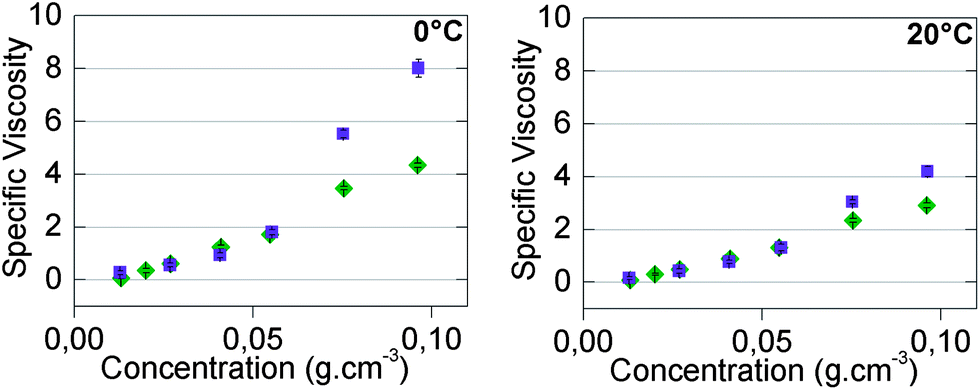 | ||
| Fig. 4 Concentration-dependent specific viscosity in chloroform (◆) and in toluene (■) at 0 °C (left) and 20 °C (right) of 50/50-M-2200 8b. | ||
In the diluted regime, assuming that the supramolecular polymerization is under thermodynamic control with an isodesmic mechanism, the specific viscosity of a monodisperse polymer solution is expressed as a function of the polymer radius of gyration Rg, its weight concentration Cg, and its molecular weight M, according to the Zimm model (eqn (1) from experiments on linear polymers under theta-conditions and more recent theories,37κ is a constant and Na, the Avogadro number):
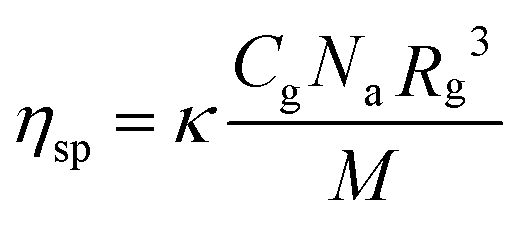 | (1) |
The specific viscosity of a supramolecular polymer solution can then be considered to be a weighted sum on all sizes (i.e. an integral) of eqn (1). Eqn (2) expresses ηsp as a function of the supramolecular weight concentration Cg and mean size N, N depending on association constants and concentration (see ESI† for detailed expressions). α is a constant, characteristic of the telechelic oligomer and ν is equal to 0.5 for short chains or in Θ-solvents.38
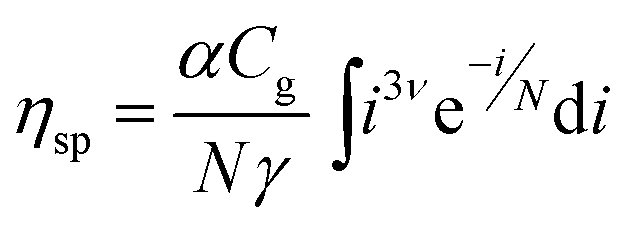 | (2) |
The results of this simple model which is applied mostly at low concentrations (below 0.06 g cm−3) were plotted in Fig. 5. The fit was good for chloroform (Fig. 5a) but not for toluene (Fig. 5b), since as naïvely expected, a higher association constant yields higher viscosities at any concentration. This very crude model does not consider intrachain associations, which are quite likely at low concentrations. We thus have confronted our experimental results with a viscosity model of supramolecular polymers with a ring-chain equilibrium.
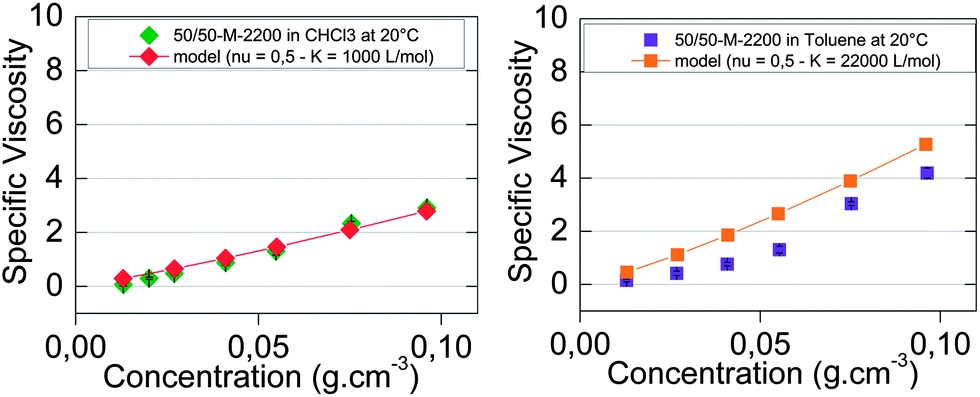 | ||
| Fig. 5 Concentration-dependent specific viscosity of 50/50-M-2200 8b at 20 °C (a) in chloroform (◆) and (b) in toluene (■). The viscosity model for an isodesmic supramolecular polymerization into linear chains with an association constant of (a) 1000 L mol−1 (◆) and (b) 22000 L mol−1 (■). | ||
Cyclic supramolecular polymers obey the same Zimm eqn (1) as linear ones except that, for the same molecular weight, the radius of gyration is lower. Taking into account the presence of rings, the specific viscosity becomes a weighted average of linear and cyclic supramolecular polymers, following eqn (3).39
| ηsp = ρwηringssp + (1 − ρw)ηringssp | (3) |
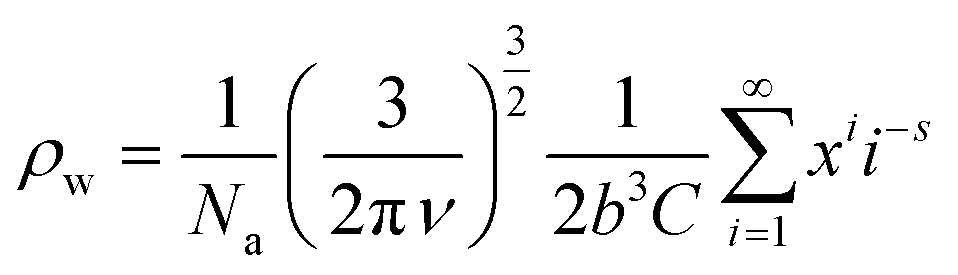 | (4) |
The results of this second model, also mostly applicable at low concentrations (below 0.06 g cm−3), were plotted in Fig. 6, assuming that all rings were made of the two supramolecular units (by far the predominant form in 50/50-M-2200 8b, see ESI† for detailed expressions and ring size distribution).
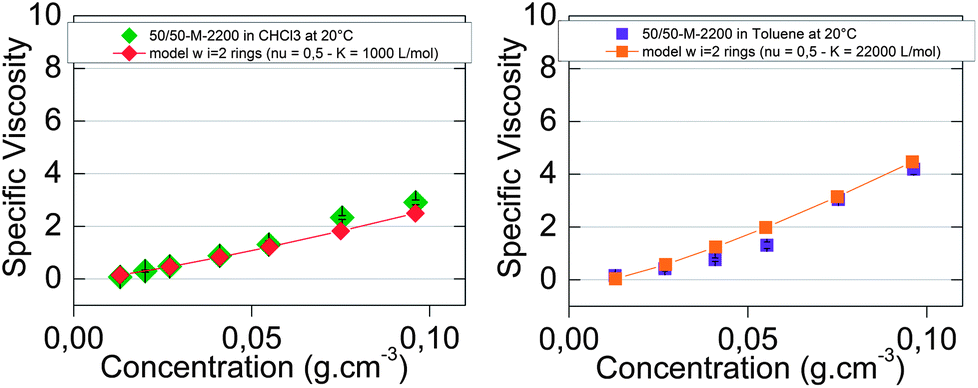 | ||
| Fig. 6 Concentration-dependent specific viscosity of 50/50-M-2200 8b in (a) chloroform (◆) and (b) toluene (■) at 20 °C. The viscosity model for a ring-chain equilibrium supramolecular polymerization with an association constant of (a) 1000 L mol−1 (◆) and (b) 22000 L mol−1 (■). | ||
Taking into account the presence of rings, the specific viscosity fit was still good for chloroform (Fig. 6a), especially at concentrations below 0.06 g cm−3, where the model applied. Indeed, in this case the ring correction has a limited impact on the specific viscosity. In contrast, in toluene, taking into account the presence of rings significantly lowers the specific viscosity, so the fit was better but not perfect, especially at medium concentrations (around 0.05 g cm−3, Fig. 6b). An explanation for the differences observed in chloroform and toluene could be that not only KThy–DAT is higher in toluene than in chloroform, but also there are different aggregation mechanisms in toluene and in chloroform. More specifically, an isodesmic mechanism could take place in chloroform and a nucleation-elongation mechanism in toluene, which means then that there are secondary interactions that take place in addition to hydrogen bonding. One possible interaction between the Thy and DAT associative groups, besides hydrogen bonding, is aromatic stacking, commonly encountered for DNA bases and biological recognition.41 In fact, π-stacking of Thy in toluene could be evidenced by comparing the 1H NMR chemical shifts of Thy CH3 (A) and CH (C) protons in different solvents. Indeed, aromatic electron circulation induces a local magnetic field that shields protons lying above aromatic rings.42Methyl (A) and ethylene (C) signals were shielded by around 0.4 ppm in toluene-d8 compared to CDCl3. This suggested that these protons laid above aromatic cycles in toluene, but not in chloroform. Thus, in toluene Thy was believed to π-stack with other aromatic rings (such as: toluene,43 Thy,44 and/or DAT45) in an off-centered parallel displaced manner.46
However, small Thy and DAT derivatives (such as thymine-1-acetic acid, thymine, 4,6-diamino-2-methyl-1,3,5-triazine [DAT-Me], 2-chloro-4,6-diamino-1,3,5-triazine [DAT-Cl], as well as 1:1 thymine and 4,6-diamino-2-methyl-1,3,5-triazine mixture) were quasi-insoluble in toluene. Thy-PPO-460-Thy 6a and 50/50-M-460 8a were also quasi-insoluble in toluene (tested down to 0.002 mol L−1), but slightly soluble in chloroform (soluble under 0.03 mol L−1), although the PPO chain does not have more affinity with chloroform than with toluene (Hildebrand solubility parameters δPPO ≅ 18 (MPa)1/2, δCHCl3 = 18.9 (MPa)1/2 and δtoluene = 18.2 (MPa)1/2).47 This indicated that Thy and DAT groups had very little affinity with toluene, even when they were associated together via hydrogen bonds, despite the fact that Thy and DAT could a priori π-stack with toluene. Indeed, Thy and DAT are polar groups, while toluene is mostly apolar. Therefore, it seems likely that, in toluene, Thy and DAT preferably π-stack with Thy or DAT, rather than toluene. As a result, objects consisting of a PPO shell and a Thy and DAT core, with hydrogen bonds and π-stacking interactions between Thy and DAT, should form in toluene (Fig. 7C). Such inversed micelles (“flowers”) could connect to each other,48 especially at a high concentration (C > C*), resulting in an increase of viscosity.
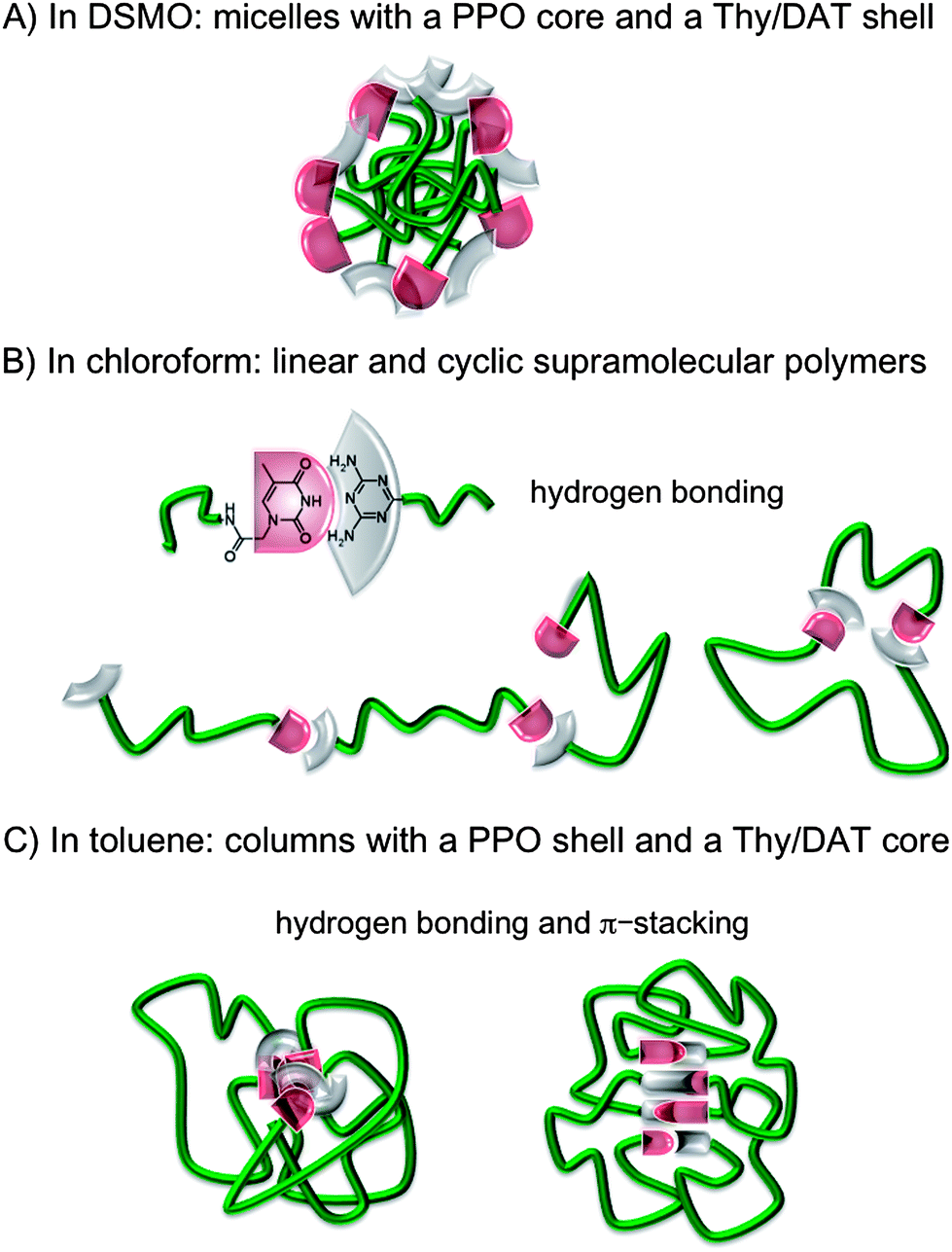 | ||
| Fig. 7 Heterotelechelic oligomer Thy-PPO-2200-DAT 5b solution mesoscopic organization: (A) in DMSO, (B) in chloroform, (C) in toluene. | ||
In contrast, no or much less π-stacking of Thy and DAT seemed to occur in chloroform. The reason could be that chloroform solvates better than toluene, not only the hydrogen bonds donors and acceptors as mentioned above, but also the Thy and DAT aromatic cycles, illustrating the connection between solvation and π-stacking. Therefore, the classical vision of linear supramolecular polymers in equilibrium with cycles seemed to manifest in chloroform (Fig. 7B). The higher KThy–DAT value in toluene, compared to chloroform, may be attributed to poor solvation of the hydrogen bond donors and acceptors in toluene. However, π-stacking may also be held responsible for the high KThy–DAT in toluene. Indeed, hydrogen bonds involving its CO groups cause an increase of thymine's aromatic character.49 Reciprocally, when the thymine cycle interacts by π-stacking with an aromatic ring, the thymine cycle's aromaticity increases, resulting in a more basic CO, and a more acidic NH. Moreover, π-stacking may induce a parallel alignment of Thy and DAT, with an optimal NH2 and CO relative orientation.1b Therefore, the supramolecular Thy–DAT hydrogen bonding association should be reinforced when Thy and DAT interact by π-stacking with an aromatic cycle. Furthermore, π-stacking and solvation are connected. π-stacking may occur between Thy and DAT in toluene and not in chloroform because chloroform better solvates Thy and DAT rings.
If our supramolecular polymers form colloidal objects in toluene as suggested by NMR and as described above, then we should be able to fit their viscosities with a model for colloids. Eqn (5) is a commonly used semi-empirical relationship for colloidal suspensions,50 with Φmax the maximum value of the particle volume fraction and Φ the particle volume fraction. Φ can be expressed as a function of C the supramolecular polymer concentration (in g cm−3), V the volume of colloidal object, M the molecular weight of one unit (here, M = 2500 g.mol−1) and p the number of units in one colloid object. Na is the Avogadro number.
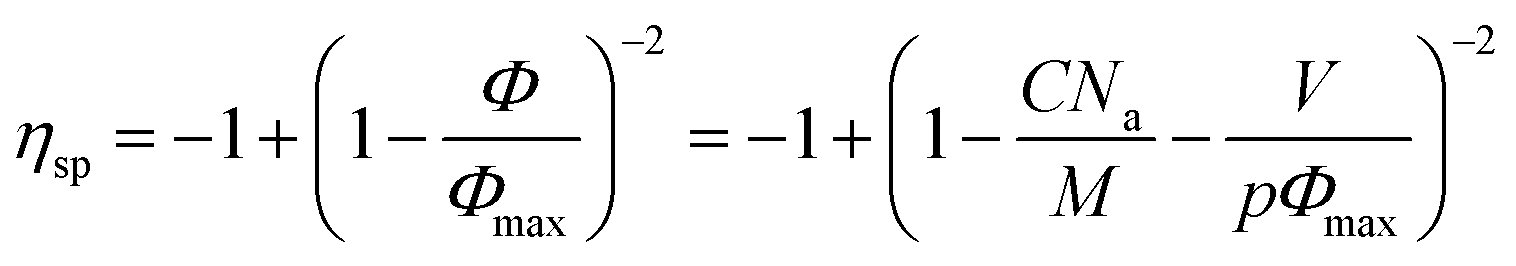 | (5) |
The specific viscosity of 50/50-M-2200 8b could thus be expressed as a function of the concentration C through eqn (5) which comprises only one fitting parameter:
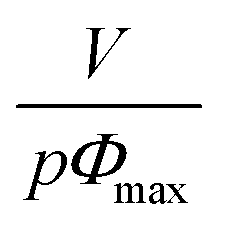
The best fit was plotted in Fig. 8 and was obtained for
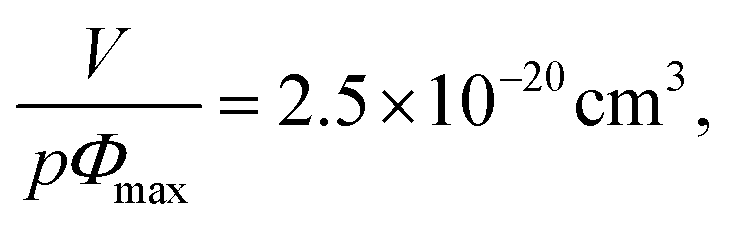
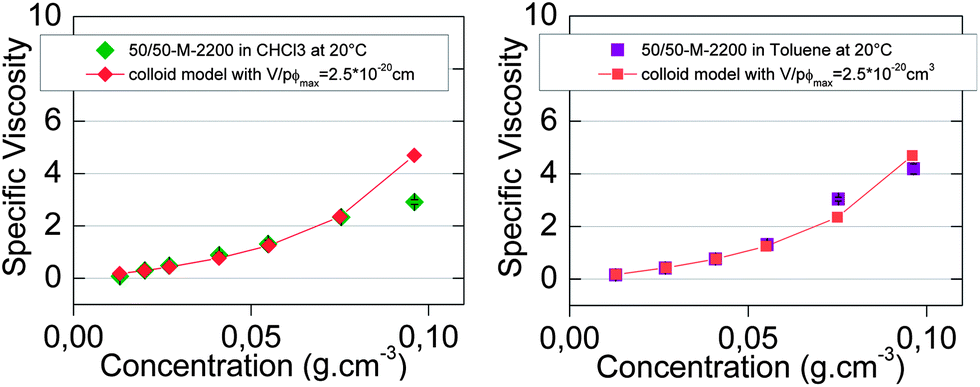 | ||
| Fig. 8 Concentration-dependent specific viscosity of 50/50-M-2200 8b in (a) chloroform (◆) and (b) toluene (■) at 20 °C. Viscosity model for colloidal supramolecular polymers (◆) (■). | ||
Finally, the colloidal model with these reasonable parameters fit relatively well with the specific viscosity of 50/50-M-2200 8b from 0 to 0.1 g cm−3 in toluene (Fig. 8b) but not in chloroform (Fig. 8a).
Therefore, the model of colloidal inversed micelles in toluene was consistent with the viscosity results as well as the NMR results, unlike the ring-chain equilibrium model in toluene (Fig. 6b). Still, the ring-chain equilibrium specific viscosity model in toluene was relatively good because rings had a size comparable to the colloidal inversed micelles. In fact, the specific viscosity models by themselves could not distinguish between the two interpretations, but the NMR and viscosity results together pointed toward the colloidal model in toluene and ring-chain equilibrium model in chloroform.
Conclusions
To sum up, we synthesized heterotelechelic oligomers of poly(propylene oxide) oligomeric spacers capped with a thymine group (Thy) on one end and a 2,6-diamino-1,3,5-triazine (DAT) on the other (5a-b), in a four step procedure. We showed that, in a chosen solvent, as expected, viscosity of solutions depends on the association constant (KThy–Thy, KDAT–DAT ≪ KThy–DAT). Viscosities of homotelechics blends were slightly lower than those of corresponding heterotelechelic oligomers, which could be attributed to a Thy–DAT ratio closer to 1 in the latter case and thus no chain-stopper effect takes place. Finally, we can conclude that the solvent has an important and subtle impact on the size and supramolecular organization of telechelic oligomers. In the polar dissociating DMSO, micelles with a PPO core and a Thy, DAT shell could form. In the less-polar non-dissociating chloroform, linear supramolecular polymers form through hydrogen bonding between Thy and DAT, in equilibrium with ring supramolecular polymers. In the apolar non-dissociating toluene, by combining hydrogen bonding and π–π interactions, small π-stacked objects with a PPO shell and a Thy, DAT core could form. Such solvent-dependent organization and driving force of the assembly had already been observed, notably by Bouteiller and his coworkers,51 as well as Meijer and his coworkers on a self-associative homotelechelic system.6a It is interesting to find the same solvent effect for a complementary heterotelechelic system, Thy-PPO-2200-DAT 5b.Finally, one might expect that when the materials are obtained by solvent evaporation, the organization in the bulk is solvent dependent as this is often the case for ABC block copolymers, for example.52
Notes and references
- (a) J.-M. Lehn, Supramolecular chemistry – Concepts and perspectives, VCH, Weinhein, Germany, 1995 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular chemistry, 2nd Edition, Wiley, Chippenham, UK, 2009 Search PubMed.
- (a) B. Rybtchinski, ACS Nano, 2011, 5, 6791 CrossRef CAS PubMed; (b) M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 4, 3591 CrossRef CAS PubMed.
- (a) A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39 CrossRef CAS; (b) P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- (a) J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823 CrossRef CAS; (b) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- J. Cortese, C. Soulié-Ziakovic, M. Cloitre, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2011, 133, 19672 CrossRef CAS PubMed.
- (a) J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS PubMed; (b) L. Brunsveld, J. A. J. M. Vekemans, J. H. K. K. Hirschberg, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4977 CrossRef CAS PubMed; (c) E. Kolomiets, E. Buhler, S. J. Candau and J.-M. Lehn, Macromolecules, 2006, 39, 1173 CrossRef CAS; (d) R. Iwaura, F. J. M. Hoeben, M. Masuda, A. P. H. J. Schenning, E. W. Meijer and T. Shimizu, J. Am. Chem. Soc., 2006, 128, 13298 CrossRef CAS PubMed; (e) R. Iwaura, T. Iizawa, H. Minamikawa, M. Ohnishi-Kameyama and T. Shimizu, Small, 2010, 6, 1131 CrossRef CAS PubMed.
- (a) F. Ouhib, M. Raynal, B. Jouvelet, B. Isare and L. Bouteiller, Chem. Commun., 2011, 47, 10683 RSC; (b) E. Sabadini, K. R. Francisco and L. Bouteiller, Langmuir, 2010, 26, 1482 CrossRef CAS PubMed; (c) L. Bouteiller, Adv. Polym. Sci., 2007, 207, 79 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761 CrossRef CAS.
- D. Montarnal, P. Cordier, C. Soulié-Ziakovic, F. Tournilhac and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7925 CrossRef CAS.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC.
- (a) H. M. Keizer, R. van Kessel, R. P. Sijbesma and E. W. Meijer, Polymer, 2003, 44, 5505 CrossRef CAS; (b) D. J. M. van Beek, M. A. J. Gillissen, B. A. C. van As, A. R. A. Palmans and R. P. Sijbesma, Macromolecules, 2007, 40, 6340 CrossRef CAS; (c) W. H. Binder, M. J. Kuntz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749 CrossRef CAS; (d) U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950 CAS; (e) S. K. Yang, A. V. Ambade and M. Weck, Chem. Soc. Rev., 2011, 40, 129 RSC.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2002, 8, 1227 CrossRef CAS.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 14236 CrossRef CAS PubMed.
- T. F. A. de Greef, G. Ercolani, G. B. W. L. Ligthart, E. W. Meijer and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 13755 CrossRef CAS PubMed.
- (a) A. Ambade, S. Yang and M. Weck, Angew. Chem., Int. Ed., 2009, 48, 2894 CrossRef CAS PubMed; (b) S. K. Yang, A. V. Ambade and M. Weck, J. Am. Chem. Soc., 2010, 132, 1637 CrossRef CAS PubMed.
- U. Mansfeld, A. Winter, M. D. Hager, R. Hoogenboom, W. Gunther and U. S. Schubert, Polym. Chem., 2013, 4, 113 RSC.
- A. Bertrand, S. Chen, G. Souharce, C. Lavadière, E. Fleury and J. Bernard, Macromolecules, 2011, 44, 3694 CrossRef CAS.
- O. Altintas, T. Rudolph and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2566–2576 CrossRef CAS.
- J. Xu, E. A. Fogleman and S. L. Craig, Macromolecules, 2004, 37, 1863 CrossRef CAS.
- O. A. Scherman, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 2072 CrossRef CAS PubMed.
- G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Org. Chem., 2006, 71, 375 CrossRef CAS PubMed.
- T. Shimizu, R. Iwaura, M. Masuda, T. Hanada and K. Yase, J. Am. Chem. Soc., 2001, 123, 5947 CrossRef CAS PubMed.
- J. Cortese, C. Soulié-Ziakovic, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2012, 134, 3671 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, New York, 3rd edn, 1999 Search PubMed.
- (a) D. W. Lee, H.-J. Ha and W. K. Lee, Synth. Commun., 2007, 37, 737 CrossRef CAS; (b) M. Pittelkow, R. Lewinsky and J. B. Christensen, Synthesis, 2002, 15, 2195 Search PubMed.
- A. P. Krapcho, M. J. Maresch and J. Lunn, Synth. Commun., 1993, 23, 2443 CrossRef CAS.
- J. N. Spencer and T. A. Judge, J. Solution Chem., 1983, 12, 847 CrossRef CAS.
- S. Zimmerman and P. Corbin, Struct. Bonding, 2000, 96, 63 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chippenham, UK, 2nd edn, 2009 Search PubMed.
- L. Fielding, Tetrahedron, 2000, 56, 6151 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 2, 311 RSC.
- F. H. Beijer, R. P. Sijbesma, J. A. J. M. Vekemans, E. W. Meijer, H. Kooijman and A. L. Spek, J. Org. Chem., 1996, 61, 6371 CrossRef CAS PubMed.
- F. Herbst, K. Schröter, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Publishers, 3rd edn, 2003 Search PubMed.
- M. Salas, B. Gordillo and F. Gonzalez, ARKIVOC, 2003, 11, 72 Search PubMed.
- F. Herbst and W. H. Binder, Polym. Chem., 2013, 4, 3602 RSC.
- W. W. Graessley, Polymer liquids & networks: dynamics and rheology, Taylor & Francis Group, New York, 2008 Search PubMed.
- M. Rubinstein and R. Colby, Polymer Physics, Oxford University Press, USA, 2003 Search PubMed.
- H. Jacobson, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607 CrossRef CAS.
- H. Jacobson, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607 CrossRef CAS.
- A. K. Tewari and R. Dubey, Bioorg. Med. Chem., 2008, 16, 126 CrossRef CAS PubMed.
- J. A. N. F. Gomes and R. B. Mallion, Chem. Rev., 2001, 101, 1349 CrossRef CAS PubMed.
- M. H. Rahman, S.-C. Liao, H.-L. Chen, J.-H. Chen, V. A. Ivanov, P. P. J. Chu and S.-A. Chen, Langmuir, 2009, 25, 1667 CrossRef CAS PubMed.
- (a) R. Iwaura, F. J. M. Hoeben, M. Masuda, A. P. H. J. Schenning, E. W. Meijer and T. Shimizu, J. Am. Chem. Soc., 2006, 128, 13298 CrossRef CAS PubMed; (b) M. Surin, P. G. A. Janssen, R. Lazzaroni, P. Leclère, E. W. Meijer and A. P. H. J. Schenning, Adv. Mater., 2009, 21, 1126 CrossRef CAS; (c) Y. Umezawa and M. Nishio, Nucleic Acids Res., 2002, 30, 2183 CrossRef CAS PubMed.
- T. J. Mooibroek and P. Gamez, Inorg. Chim. Acta, 2007, 360, 381 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed.
- A. F. M. Barton, Handbook of solubility parameters and other cohesion parameters, CRC Press, Boca Raton, FL, 1991 Search PubMed.
- A. N. Semenov, J.-F. Joanny and A. R. Khokhlov, Macromolecules, 1995, 28, 1066 CrossRef CAS.
- M. K. Cyranski, M. Gilski, M. Jaskolski and T. M. Krygowski, J. Org. Chem., 2003, 68, 8607 CrossRef CAS PubMed.
- J. Mewis and N. J. Wagner, Colloidal suspension rheology, Cambrigde University Press, Cambridge, 2012 Search PubMed.
- E. Obert, M. Bellot, L. Bouteiller, F. Andrioletti, C. Lehen-Ferrenbach and F. Boué, J. Am. Chem. Soc., 2007, 129, 15601 CrossRef CAS PubMed.
- (a) L. Corte, K. Yamaguchi, F. Court, M. Cloitre, T. Hashimoto and L. Leibler, Macromolecules, 2003, 36, 7695 CrossRef CAS; (b) D. Yamaguchi, M. Cloitre, P. Panine and L. Leibler, Macromolecules, 2005, 38, 7798 CrossRef CAS; (c) S. Brinkmann-Rengel, V. Abetz, R. Stadler and E. L. Thomas, Kautsch. Gummi Kunstst., 1999, 12, 806 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and NMR studies of heterotelechelic oligomers 5a-b; determination of association constants by 1H NMR, viscosity models. See DOI: 10.1039/c3py00638g |
| This journal is © The Royal Society of Chemistry 2014 |