Synthesis of novel trithiocarbonate and allyl sulfide containing monomers†
Christopher R.
Fenoli
a and
Christopher N.
Bowman
*abc
aDepartment of Chemistry and Biochemistry, University of Colorado, UCB 215, Boulder, Colorado 80309, USA. E-mail: Christopher.fenoli@colorado.edu; Christopher.Bowman@colorado.edu
bDepartment of Chemical and Biological Engineering, University of Colorado, UCB 596, Boulder, Colorado 80309, USA
cMaterial Science and Engineering Program, University of Colorado, Boulder, Colorado 80309, USA
First published on 14th August 2013
Abstract
In this study, we present a mild, efficient, and high yield synthesis of a variety of trithiocarbonates and allyl sulphide-containing AFT (addition–fragmentation termination) monomers containing terminal (meth)acrylate functional groups. The trithiocarbonate moiety core was synthesized with reaction yields often above 95%. Synthesis of monomers with allyl sulphide cores gave yields ranging from 48–92%. These monomers, designated as CRAFT (Controllable Reversible Addition Chain Transfer) monomers, offer the ability to control the mechanical and polymerization properties of the resulting thiol-Michael and chain-growth polymer networks in a previously unattainable manner. The triothiocarbonates reached 70–81% acrylate conversion in 10–200 s of light exposure while the allyl sulphide monomers reached 74–85% conversion in 10–50 s.
Introduction
The RAFT (reversible addition–fragmentation chain transfer) polymerization process, which includes AFT (addition–fragmentation chain transfer) reactions of various forms, has rapidly become one of the two most dominant controlled radical polymerization reactions and is becoming more prevalent in commercial and research practice for producing polymers of controlled structure, molecular weight and architecture. Trithiocarbonate and allyl sulphide moieties are dominant as chain transfer agents in RAFT processes and have been used for more than a decade as such. Furthermore, trithiocarbonates have also long been important intermediates in numerous industrial processes for the industrial synthesis of pesticides1 and lubricants.2 In addition to conventional RAFT polymerizations involving the addition of a conventional RAFT agent, over the past decade, there has been a growing interest in the synthesis and application of AFT compounds for numerous polymerization processes3 and of late, considerable efforts have been focused on optimizing the challenging synthesis of compounds that include the various functional moieties that facilitate RAFT polymerizations. Even more recently, both allyl sulphides and trithiocarbonates have been incorporated into multifunctional monomers for direct incorporation throughout polymer networks. The inclusion of an AFT-capable moiety within a monomer has been shown to enable stress relaxation,4 healing,5 and topographical material6 patterning by forming a network with covalent bonds that are reversible through the AFT process.The first report of addition–fragmentation chemistry was presented in the 1970's by Griese and Crich,7,8 and subsequently the use of AFT agents, such as vinyl ethers and allyl sulfides, to control molecular weight and end-group functionality of polymers was demonstrated in the 1980s.9–12 In 1998, the first RAFT polymerization process making use of trithiocarbonates was reported by Moad, Rizzardo, and Thang.12 This process has since been extensively used to affect controlled radical polymerization to form polymeric species of controlled, uniform molecular weight as well as polymers of various controlled architectures.13,14
However, one of the primary limitations of this process remains the synthesis of the trithiocarbonates themselves. Traditionally, symmetric trithiocarbonates have been synthesized in one-pot reactions using highly toxic reagents such as thiophosgene15 and chlorodithioformates.16 Symmetric trithiocarbonates have been made using a two-step approach involving grignards,17 alkyl halides, and carbon disulfide, or with carbon disulfide and alkyl halides under basic conditions.18 These approaches suffer from the use of strong bases and excess amounts of carbon disulfide, which carries a very foul and unpleasant odor. The Mitsunobu reaction has also been used to synthesize the trithiocarbonates in good yields,19 but suffers from the use of diethyl azodicarboxylate, DEAD reagent, which is costly and toxic, and these reactions are hard to purify because of the triphenylphosphine oxide by-products. Phase-transfer catalysts have been used as well but suffer from the large excess of reagents (17–20 equivalents).20 Recently, the use of mild bases and stoichiometric carbon disulfide has been employed to synthesize symmetric trithiocarbonates.21,22 This approach was limited to using non-functionalized alkyl, alkene, and benzyl halides. The allyl sulfide synthesis is straightforward when the appropriate thio–alcohols are available.23,24 The allylic sulfides are formed by a Sn2 displacement using sulphur as the nucleophile, but with limited commercially available thio–alcohols, the synthesis of these intermediate thio–alcohols makes the limited synthetic variability of the allyl sulfides noteworthy. The ability to synthesize a variety of functionalized AFT-containing moieties, including multifunctional AFT-containing monomers, is appealing for numerous polymerization and photochemical network modification strategies.5,6 By incorporating AFT moieties throughout polymer networks, radical generation triggers the bond exchange process. Despite these potential benefits, there have been only a few instances where AFT monomers have been endcapped5,25 with functional groups that enable them to be incorporated into networks via polymerization reactions.
Herein, we present a novel synthetic strategy that involves the use of a mild base, potassium carbonate, coupled with polar aprotic solvents using stoichiometric carbon disulfide with alcohol-functionalized alkyl halides. The reactions are carried out at mild temperatures, without the use of large excesses of costly or toxic reagents. The use of such mild conditions allows for the chemistry to be carried out in atmospheric conditions in the presence of oxygen and moisture. Additionally, after the formation of the AFT core, the subsequent synthetic manipulations occur using mild techniques to incorporate the (meth)acrylates or other desired polymerizable moieties without degrading the AFT core. While the core allyl sulphide monomer synthesis is straightforward, there are a limited number of commercially available thio–alcohols. The ability to synthesize these intermediate thio–alcohols for the core allyl sulphide reaction makes this synthetic approach challenging and noteworthy.
Discussion
The synthetic strategy is based on the design of AFT synthons that contain the general structure illustrated needed for incorporation within a polymer network (Fig. 1).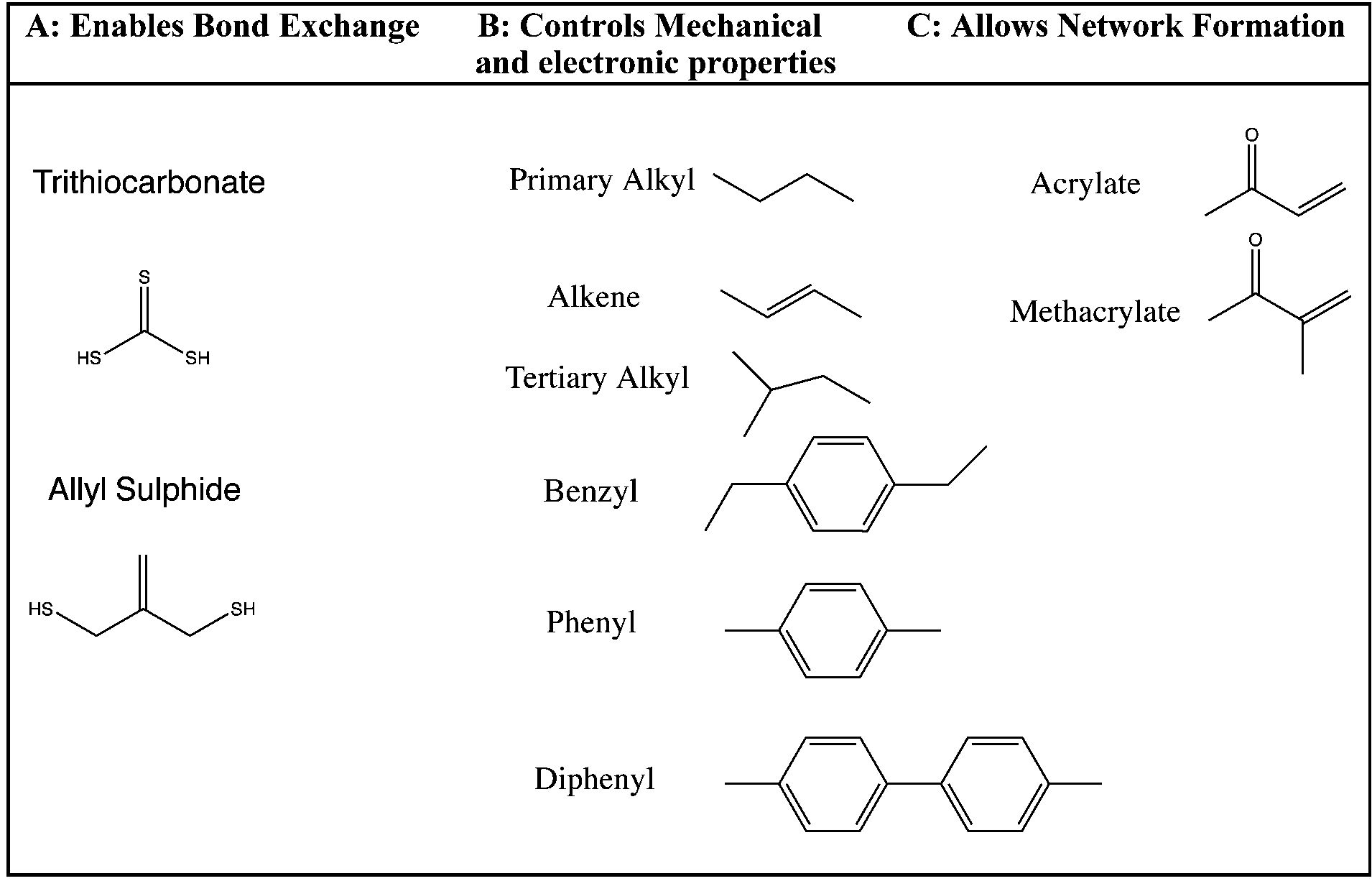 | ||
| Fig. 1 General monomer structure for monomers with the generalized structure C–B–A–B–C with each monomer containing A (bond exchange core), B (linkers), and C (end groups that are necessary for addition–fragmentation) moieties. | ||
The synthons consist of an AFT core (A) that is either an allyl sulfide or trithiocarbonate moiety. The linker (B) is synthetically connected to the AFT core (A) and capped with an end group (C) that is polymerizable in one or more types of polymerization reactions. The linkers included into the AFT monomers were chosen to elicit specific mechanical behavior from the final networks and also to induce various electronic effects in the AFT process. Tailored at the end (C), segments are incorporated that consist generally of a polymerizable subunit. Here, we focus specifically on acrylates that can either be reacted via conventional radical polymerization or be formed into a network by reaction with a multifunctional thiol via thiol-Michael addition, among other possible reactions. The A, B, and C subunits that were systematically evaluated in each monomer (Fig. 2).
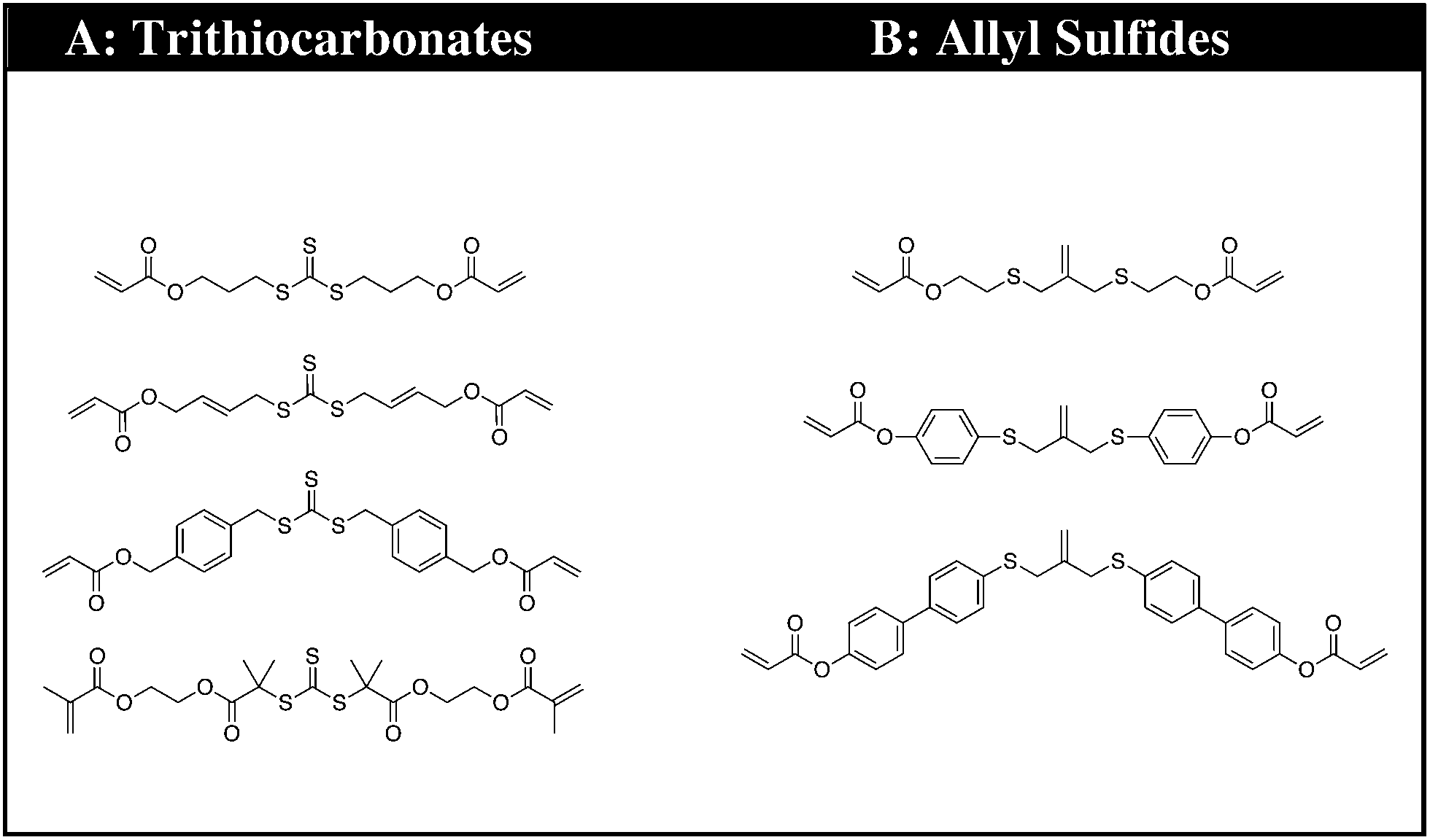 | ||
| Fig. 2 The CRAFT monomers consisting of four trithiocarbonate and three allyl sulphide monomers each containing the different functionality incorporated into the core of the monomer structure. | ||
The premise of the synthesis of these compounds is based on the desire to design a tailor made controllable addition–fragmentation chain transfer (CRAFT) monomer that is polymerizable into a network and contains functionality(ies) that are designed to elicit specific mechanochemical behavior from the network, including both static behavior in the absence of radicals and dynamic adaptability in the presence of radicals. Copolymerizations of these monomers with other functional materials may also readily be used to alter these behaviors as needed. Depending on whether the (C) subunit is an acrylate or methacrylate group, the CRAFT monomers can undergo photoinitiated radical polymerization, thiol-(meth)acrylate radical polymerization, and/or base or nucleohile-catalyzed thiol-Michael addition polymerizations. This approach facilitates a wide range of spatial, temporal, and overall polymer property behavior control in the network.
The key reaction to build the trithiocarbonate backbone followed a modified procedure from the literature for making the trithiocarbonate core.22 This synthesis used the reaction of carbon disulfide with a mild base, potassium carbonate, in a polar aprotic solvent with an alkyl, vinyl or benzylic halide, which contained a silyl protected alcohol at one end (Scheme 1).
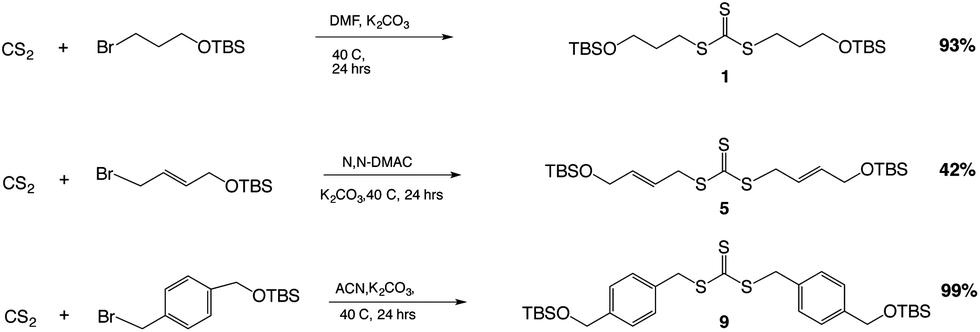 | ||
| Scheme 1 Reaction of trithiocarbonate cores using CS2 with various alkyl halides, reaction conditions, and overall reaction yields. Reaction conditions: (a) K2CO3, polar aprotic solvent, carbon disulfide, 25 °C. (b) TBSO-R-X, 25–40 °C, 24 hours. | ||
The alcohol is subsequently deprotected using tetrabutyl ammonium fluoride and acetic acid, TBAF/AcOH, which allows the use of fluorine to deprotect the silyl group without allylation of the AFT core by TBAF.24 The deprotection is followed by a Mitsunobu coupling to add the (meth)acrylate end groups (Scheme 2).
 | ||
| Scheme 2 Reaction conditions for the removal of the TBDMS protecting group for the Mitsunobu coupling with overall reaction yields. Reaction conditions: (a) TBAF/ACOH, DCM or THF, 25 °C, 16 h, (b) diethyl azodicarboxylate, triphenylphosphine, THF, acrylic acid, 0–25 °C, 16 h. | ||
The allyl sulfides were synthesized following a modified procedure from the literature.6 To 3-chloro-2-chloromethyl-1-propene was added the thio–alcohol in a solution of sodium methoxide. The thio–alcohols used were mercaptoethanol, thiophenol, and 4-hydroxyl-4′-mercaptobiphenyl. 4-Hydroxyl-4′-mercaptobiphenyl was synthesized from 4-iodoanisol and 4-bromothioanisol via a Kumada coupling. The product was subsequently reduced with sodium ethanethiolate to yield the 4-hydroxyl-4′-mercaptobiphenyl (Scheme 3). Once all the thio–alcohols were in hand, the core allyl disulfide scaffold was formed. The acrylate or methacrylate was added to the pendent alcohol using modified Schotten–Baumann conditions, where the acyl chloride is reacted with the alcohol using triethylamine as an acid scavenger (Scheme 4).
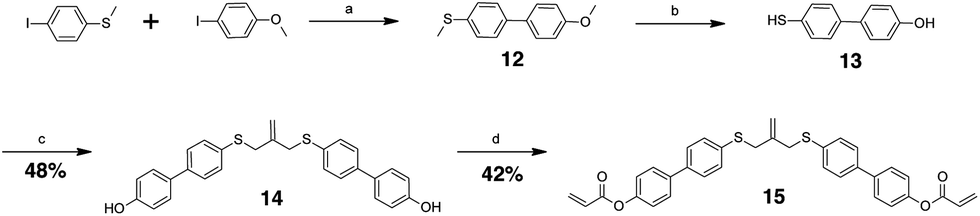 | ||
| Scheme 3 Reaction diagram for the synthesis of the biphenyl allyl sulphide diacrylate with overall reaction yields as indicated. Reaction conditions: (a) Mg, THF, I2; Pd(Ph3P)4, THF, Δ (heat), (b) sodium thiosulfate, DMF, Δ, (c) 3-chloro-2-chloromethyl propene, NaOMe, Δ, (d) acryloyl chloride, Et3N, THF, 0 °C. | ||
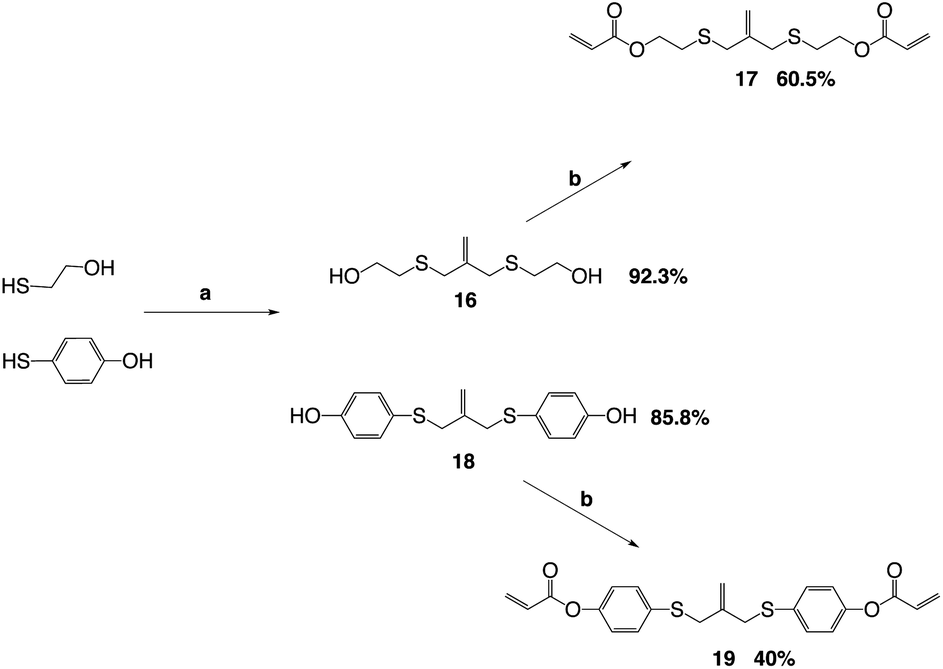 | ||
| Scheme 4 Reaction diagram for the synthesis of alkyl and phenyl allyl sulphides with overall reaction yields. Reagents and conditions: (a) sodium methoxide in MeOH, 3-chloro-2-chloromethyl-1-propene, 16 h, Δ, (b) acryloyl chloride, Et3N, 0 °C to 25 °C. | ||
The acid chloride reaction to produce the diester acrylate functionality gives yields from 40–60% depending on the thiol–alcohol in which the diacrylate is coupled onto. The reaction is used because the ease of the reaction conditions and the cost of acryloyl chloride. The reaction can also be done using Mitsunobu conditions, as well. The Mitsunobu reaction gives yields in the 80–90%, but the purification of the final product is difficult. The DEAD and triphenylphosphine bi-products are hard to separate from the final product.
With the newly synthesized monomers in hand, the monomers were incorporated into polymer networks to evaluate the effects of each monomer on the polymer network. In particular, for the acrylate functionalized AFT synthons, a polymer network can be formed either by a photocatalyzed radical polymerization or by a base (or nucleophile) catalyzed thiol-Michael addition reaction (Fig. 3 for the base-catalyzed mechanism).
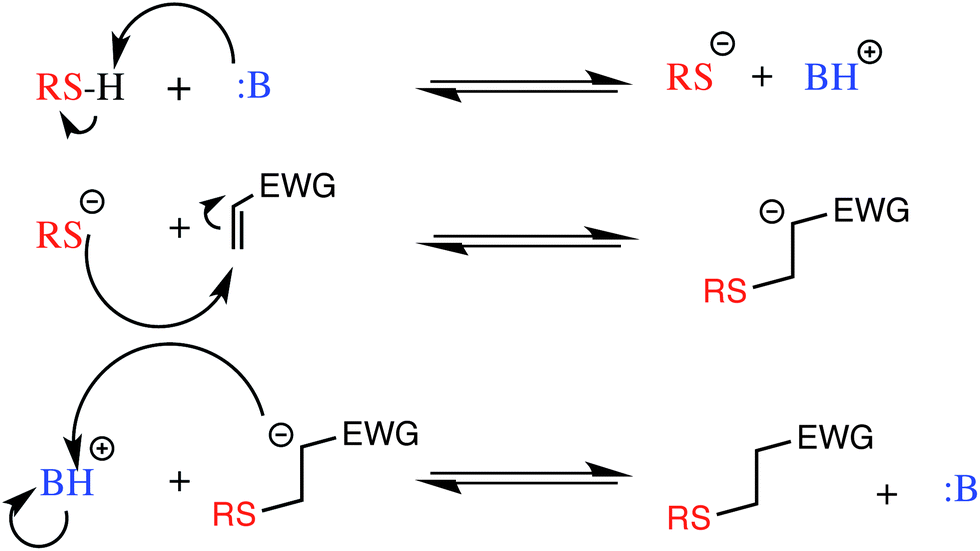 | ||
| Fig. 3 The base catalyzed thiol-Michael addition reaction mechanism. EWG represents an electron withdrawing group that activates the vinyl for Michael addition. | ||
When the network is formed by a photocatalyzed radical polymerization, AFT reactions may occur both concurrently with and subsequent to polymerization. In contrast, when the thiol-Michael addition reaction is used, the AFT mechanism occurs only during post-polymerization rearrangement of the network as no radicals are generated during the thiol-Michael addition reaction to activate the AFT subunits. Uniquely, the differences in when the AFT rearrangement of the network occurs relative to polymerization, as well as differences in the resulting network structure, optical characteristics of the material, and other physical properties of the material are readily adjusted simply by changing the polymerization mechanism.
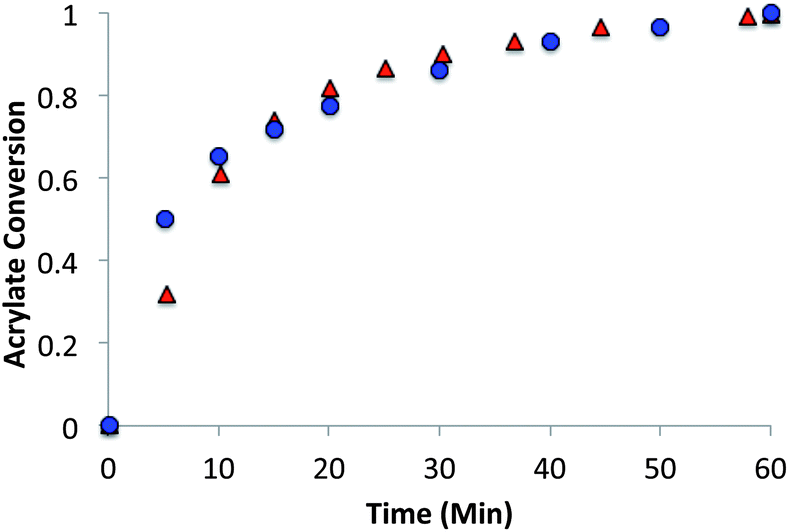
Trithiocarbonate networks were formed using a base catalyzed thiol-Michael “click” reaction by reacting stoichiometric mixtures (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acrylate–thiol functional group ratio 1.025 equivalents of pentaerytheritol tetrakis(3-mercaptopropionate) (PETMP) and 0.9 equivalents of tetraethylene glycol diacrylate (TEGDA) and 0.9 equivalents of the RAFT trithiocarbonate diacrylate. The CRAFT monomer containing networks conversion and kinetics were evaluated for 60 minutes by infrared (IR) spectroscopy after the addition of 1% triethylamine as the thiol-Michael catalyst (Fig. 4).
:1 acrylate–thiol functional group ratio 1.025 equivalents of pentaerytheritol tetrakis(3-mercaptopropionate) (PETMP) and 0.9 equivalents of tetraethylene glycol diacrylate (TEGDA) and 0.9 equivalents of the RAFT trithiocarbonate diacrylate. The CRAFT monomer containing networks conversion and kinetics were evaluated for 60 minutes by infrared (IR) spectroscopy after the addition of 1% triethylamine as the thiol-Michael catalyst (Fig. 4).
 | ||
| Fig. 4 Thiol-Michael polymerization of trithiocarbonate CRAFT resins. Thiol-Michael addition reaction kinetics monitoring the acrylate conversion of the alkyl (○), alkene (■), and benzyl (Δ) substituted trithiocarbonates at ambient temperature. 1% triethylamine was used as a catalyst for all resins. Resins consisted of a 1:1 stoichiometric mixture of the acrylate and thiol functional groups where the acrylate was comprised of a 1:1 stoichiometric ratio of the CRAFT monomer and TEGDA. | ||
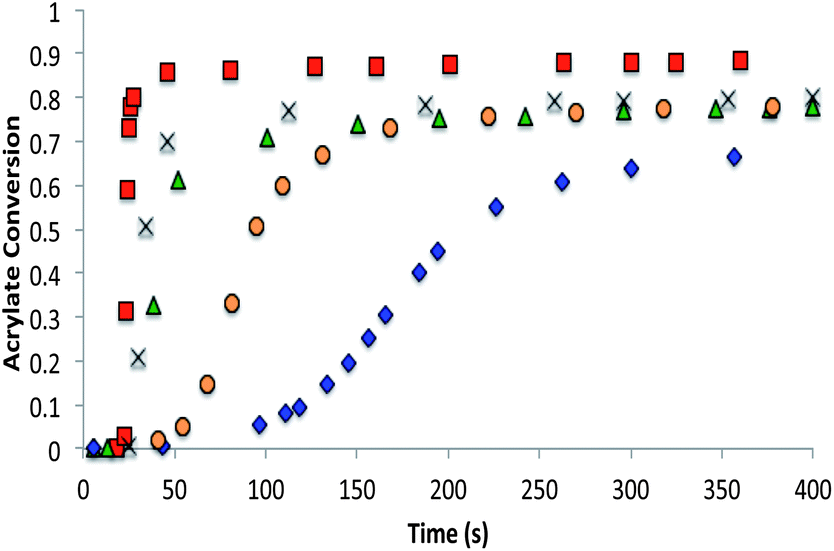
The allyl sulfide networks were constructed using the same base catalyzed thiol-Michael “click” reaction by reacting stoichiometric mixtures (1:1 acrylate–thiol functional group ratio) of 1.025 equivalents of pentaerytheritol tetrakis(3-mercaptopropionate) (PETMP) and 0.9 equivalents of tetraethylene glycol diacrylate (TEGDA) and 0.9 equivalents of the allyl sulfide The CRAFT monomer containing network acrylate conversion and kinetics were evaluated for 60 minutes by IR spectroscopy after the addition of 1% triethylamine or 0.8% imidazole as the thiol-Michael catalyst (Fig. 5). In particular, the phenyl allyl sulphide CRAFT monomer achieved 100% acrylate conversion much more rapidly and was slowed to a comparable level here by using 0.8% imidazole in place of the triethyl amine. In comparison to the alkyl allyl sulphide, which is catalysed by 1% triethylamine, the phenyl allyl sulphide uses a base that is 10000 times weaker and at a lower concentration, 0.8% to achieve similar overall rates.
 | ||
| Fig. 5 Thiol-Michael polymerization of allyl sulphide CRAFT resins. Thiol-Michael addition reaction kinetics monitoring the acrylate conversion of the alkyl (○) and phenyl (Δ) allyl sulphide at ambient temperature. 1% Triethylamine was used as a catalyst for the alkyl allyl sulphide and 0.8% imidazole was used for the phenyl allyl sulfide to slow the kinetic rate to a comparable level. Resins consisted of a 1:1 stoichiometric mixture of the acrylate and thiol functional groups where the acrylate was comprised of a 1:1 stoichiometric ratio of the CRAFT monomer and TEGDA. | ||
The thiol-Michael addition networks were evaluated on a dynamic mechanical analyzer (DMA) to analyze the how the different monomers affected the glass transition and rubbery moduli (as an indicator of crosslinking density) of the networks (Table 1).
| RAFT monomer used in the resin formulation (thiol–acrylate ratio) | T g (°C) | Rubbery E′ at 40 °C (MPa) |
|---|---|---|
| Alkyl trithiocarbonate | −9.7 ± 0.2 | 10 ± 1 |
| S,S′-Bis(isobutyric acid)-trithiocarbonate | −13.6 ± 0.6 | 2 ± 2 |
| Alkene trithiocarbonate | −3.1 ± 0.5 | 19.8 ± 0.1 |
| Benzyl trithiocarbonate | 9.0 ± 0.6 | 18.0 ± 0.3 |
| Alkyl allyl sulfide | −8 ± 1 | 24 ± 5 |
| Phenyl allyl sulfide | 13.3 ± 0.8 | 15 ± 2 |
| Bi-phenyl allyl sulfide | 29.8 ± 0.7 | 24 ± 2 |
The crosslinking densities are within approximately a factor of 2 with the exception of the S,S′-bis(isobutyric acid) system, which contains a small impurity and must be catalyzed by a strong phosphine nucleophilic catalyst. The lower crosslink density of this resin arises from the lower conversion achieved under these circumstances as the incomplete reactions causes fewer crosslinks to form. The phenyl systems showed higher Tg's, which can be ascribed to the greater rigidity of the polymer backbone and an increase in the amount of pi–pi stacking within the systems. Finally, the CRAFT monomers were formulated into a chain-growth polymerization network. The monomers were evaluated based on their ability to affect the polymerization rate and conversion in a radically polymerized acrylate photopolymerization, as RAFT monomers. The monomers were specifically designed to affect the RAFT mechanism and thereby their ability to control the polymerization (Fig. 6).
 | ||
| Fig. 6 Generic RAFT mechanism with the addition and B-cleavage components. | ||
The chain-growth system contained 70% bisphenol A ethoxylated diacrylate, 28.5–20% tetraethylene glycol diacrylate, and 10–1.5 wt% of the trithiocarbonate CRAFT monomers. The CRAFT monomer concentrations were altered so that the rate retardation observed due to the CRAFT monomers did not prevent the overall conversion of the polymerization from reaching 70% (Fig. 7).
 | ||
| Fig. 7 Radical polymerization of trithiocarbonate CRAFT resins. The acrylate conversion of the nonfunctional diacrylate control composed of 70% bisphenol A ethoxylated diacrylate and 30% tetraethylene glycol diacrylate (■) as a function of time as well as when a portion of the TEGDA is replaced with the trithiocarbonate diacrylates as follows: 10% alkyl (○), 5% S,S′-bis(isobutyric acid)-trithiocarbonate (Δ), 1.5% alkene (◆), and 2.0% benzyl (X's). The samples were irradiated at 5 mW cm−2 with 365 nm light for 10 minutes. | ||
The allyl sulfide CRAFT monomers were formulated in networks and were photoinitiated via a radical-mediated reaction in the presence of PETMP, in a manner analogous to the network formation with the trithiocarbonate monomers. It is well known that the allyl sulfide containing compounds do not react reversibly in radical polymerizations of (meth)acrylic monomers.26 The allyl sulfide monomers polymerize much more rapidly than the trithiocarbonate monomers because of the different reactivity and reversibility of the allyl sulfide (Fig. 8). Polymerizations for the allyl sulphides are generally complete in less than 10 seconds of exposure while the analogous trithiocarbonate systems require upwards of 100 s to achieve their maximum conversion. When considering the radical polymerizations of these CRAFT monomers, it is interesting to note that the kinetic results are strongly correlated to the behavior of analogous RAFT agents as indicated by their ability to lead to controlled radical polymerizations. As noted, the trithiocarbonate CRAFT monomer react more slowly, likely due to their improved ability to sequester the radicals. Furthermore, the phenyl allyl sulphide reacts more slowly than the alkyl ally sulphide (Fig. 8). This observation is in agreement with the work by Thang and co-workers,26 where the phenyl allyl sulphide has a higher transfer rate constant. Similarly, within the trithiocarbonate monomers, the reaction rates and acrylate conversion match nicely with the trends described previously by Thang and co-workers26 with the exception of the alkene trithiocarbonate. Generally, RAFT moieties that function well to control radical polymerization of (meth)acrylates tend to inhibit the radical polymerization of CRAFT monomers when those moieties are included within the monomer structure. This outcome occurs because of the enhanced ability of those moieties to sequester the radicals that would otherwise increase the polymerization rate.
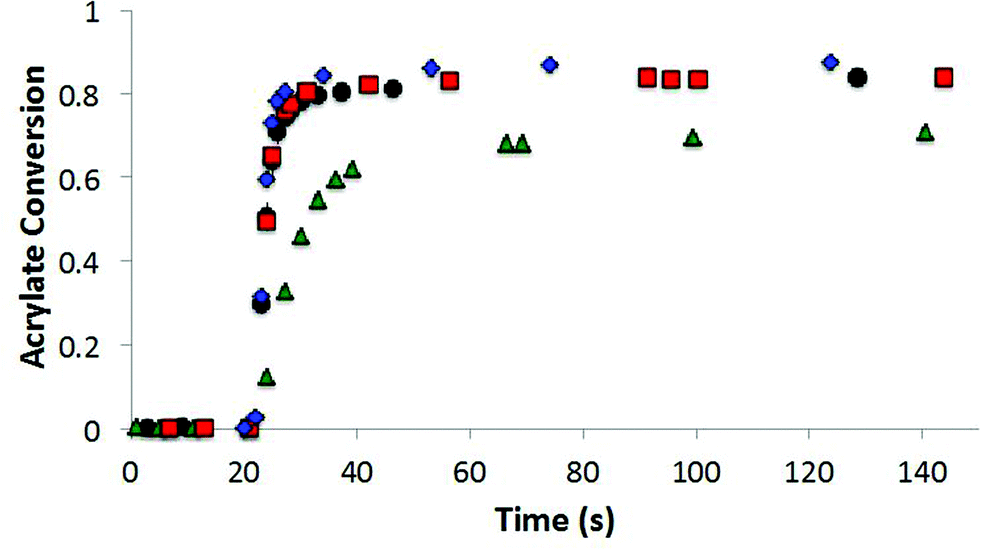 | ||
| Fig. 8 Radical polymerization of allyl sulphide CRAFT resins. The acrylate conversion of the nonfunctional diacrylate control composed of 70% bisphenol A ethoxylated diacrylate and 30% tetraethylene glycol diacrylate (■) as a function of time as well as diluted with the allyl sulphides as follows: 10% alkyl (◆), 10% phenyl (Δ), and 10% biphenyl (○). The samples were irradiated at 5 mW cm−2 at 365 nm for 10 minutes using 0.25% DMPA. | ||
Procedures
Methods and materials
The functional group conversion was observed during polymerization using a FTIR (Nicolet 670), which was equipped with near-infrared transmitting optical fiber patch cables and an indium gallium arsenide (InGaAs) detector. The formulated resin was injected between the two glass rods, which results in a specimen geometry of 6 mm diameter and 1 mm thickness. Samples were irradiated with 365 nm filtered UV light at 5 mW cm−2 (Acticure 4000, EXPO) for 10 min. Evolution of the acrylate and methcrylate double bond concentrations was determined by monitoring the infrared absorption peaks centered at 6200 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching, overtone). The elastic moduli (E′) and glass transition temperatures (Tg's) of polymerized samples were measured by dynamic mechanical analysis (DMA, TA Instruments Q800). NMR spectra were acquired on a Bruker AVANCE-III 400 NMR Spectrometer system operating at 400.13 MHz for 1H observation. Solvent chemical shifts were referenced through Mesternova from J. Org. Chem., 1997, 62, 7512–7515. The UV-Vis was employed using a diode array double-beam spectrophotometer with Ebert monochromator (Evolution 300, Thermo-Scientific, West Palm Beach, FL). Absorption spectra were collected in quartz cuvettes with a 1 cm path length. Mass Spectrometry was done on a Synapt G2 High Definition Mass Spectrometer (Waters Company). The ESI-MS settings are as follows: capillary voltage: 2.8 kV (positive mode), 2.2 kV (negative mode); sampling cone voltage: 20–40 V depending on the molecular weight. (The higher the mw, the higher the sampling cone.) Extraction cone: 4.0 V; source temperature: 80 °C; desolvation temperature: 150 °C; desolvation gas (N2): 600 L h−1 2. The samples were scanned for molecular ion analysis using ESI by dissolving or diluting the sample in MeOH or MeOH/DCM (50:50), and infusing the sample in Q-TOF MS with 5 μL min−1 flow rate to acquire the data.
C–H stretching, overtone). The elastic moduli (E′) and glass transition temperatures (Tg's) of polymerized samples were measured by dynamic mechanical analysis (DMA, TA Instruments Q800). NMR spectra were acquired on a Bruker AVANCE-III 400 NMR Spectrometer system operating at 400.13 MHz for 1H observation. Solvent chemical shifts were referenced through Mesternova from J. Org. Chem., 1997, 62, 7512–7515. The UV-Vis was employed using a diode array double-beam spectrophotometer with Ebert monochromator (Evolution 300, Thermo-Scientific, West Palm Beach, FL). Absorption spectra were collected in quartz cuvettes with a 1 cm path length. Mass Spectrometry was done on a Synapt G2 High Definition Mass Spectrometer (Waters Company). The ESI-MS settings are as follows: capillary voltage: 2.8 kV (positive mode), 2.2 kV (negative mode); sampling cone voltage: 20–40 V depending on the molecular weight. (The higher the mw, the higher the sampling cone.) Extraction cone: 4.0 V; source temperature: 80 °C; desolvation temperature: 150 °C; desolvation gas (N2): 600 L h−1 2. The samples were scanned for molecular ion analysis using ESI by dissolving or diluting the sample in MeOH or MeOH/DCM (50:50), and infusing the sample in Q-TOF MS with 5 μL min−1 flow rate to acquire the data.
1-Chloro-2-chloromethyl-1-propene, mercaptoethanol (99%), 3-(tert-butyl) dimethyl silyl-1-bromopropane, 4-bromocrotonoate, DIBAL-H, diethyl azodicarboxylate, triphenylphosphine, TBDMSCl, imidazole, tetrabutyl ammonium fluoride, glacial acetic acid, 4-hydroxythiophenol, 4-iodoanisol, 4-bromothioanisole, triethylamine (>99%), acryloyl chloride, sodium metal (99%) and ethylene glycol dimercaptopropoinate (EGDMP), tetraethylene glycol diacrylate, (TEGDA), carbon disulfide, potassium carbonate, N,N-di-methylformamide, acetonitrile, N,N-dimethyl acetamide, hexanes, ethyl acetate, methanol, dry tetrahydrofuran (99.5%), dry dichloromethane (99.5%), p-chlorobenzyl chloride, and all other chemicals unless specified were obtained from Sigma Aldrich. Pentaerythritol tetrakis (3-mercaptopropionate), (PETMP), was obtained from Evans Chemetics. Irgacure 651 (2,2-dimethoxy-2-phenylacetophenone–DMPA was obtained from BASF (formerly Ciba)). All chemicals were used without further purification unless noted.
General procedure for trithiocarbonate synthesis
In a polar aprotic solvent (DMF, Acetonitrile, or N,N-dimethylacetamide) was added 1 equivalent of carbon disulfide and 2 equivalents of potassium carbonate (K2CO3). The reaction was heated to 40 °C for thirty minutes as the solution turned a deep red color. To the solution was added 2 equivalents of the appropriate TBDMS-protected alkyl halide. The reaction was stirred for 24 hours at 40 °C. The reaction was cooled, filtered, and purified. See ESI† for detailed synthetic procedures.General procedure for Allyl sulphide synthesis
In a freshly prepared solution of sodium methoxide (NaOMe), was added the thio–alcohol. The reaction was heated to a gentle reflux. To the solution was added dropwise and carefully 1-chloro-2-chloromethyl-1-propene over 30 minutes to 1 hour. The reaction is allowed to reflux overnight. The reaction is cooled, filtered and purified elicit the title compound of interest. See ESI† for detailed synthetic procedures.Conclusions
In this study, we have synthetically designed a set of monomers with exquisite ability to control the polymerization kinetics of the resulting networks in a controlled and predictable manner. Additionally, we have detailed a high yielding, mild, and efficient synthesis of (meth)acrylate capped trithiocarbonate and allyl sulfide monomers for incorporation into a variety of networks. By incorporating different CRAFT monomers, the ability to formulate polymers across a range of glass transition temperatures from the thiol-Michael resins has been demonstrated.In the acrylate chain-growth polymerizations, the CRAFT monomers showed a tremendous propensity to control the polymerization rate and the overall acrylate conversion. In the thiol-Michael step-growth network formation, all allyl sulphides and trithiocarbonate monomers were both highly reactive and able to achieve nearly complete conversion in less than an hour under relatively mild conditions. The novel monomers synthesized and characterized in this work enable the optimization of CRAFT-based polymer systems for numerous potential applications such adhesives, material substrates for mechanopatterning and as novel polymer networks with capability for stress relaxation under constant strain.
Acknowledgements
The author would like to thank the Industry-University Cooperative Research Center for Fundamentals and Applications of Photopolymerizations and NSF CBET 1264298Notes and references
- F. Duus, in Comprehensive Organic Chemistry, ed. D. Barton and W. D. Ollis, Pergamon, New York, 1979, vol. 3, p. 342 Search PubMed; M. Bogemann, S. Peterson, O. E. Schultz and H. Soll, in Methoden der organischen chemie, ed. E. Muller, Houben-Weyl, Berlin, 1955, vol. 9, p. 804 Search PubMed.
- H. C. Godt and A. E. Wanns, J. Org. Chem., 1961, 26, 4047 CrossRef CAS , and references therein.
- G. Moad, E. Rizzardo and S. H. Thang, Radical addition–fragmentation chemistry in polymer synthesis, Polymer, 2008, 49(5), 1079–1131 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Photoinduced plasticity in cross-linked polymers, Science, 2005, 308(5728), 1615–1617 CrossRef CAS PubMed.
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Responsive Gels Based on a Dynamic Covalent Trithiocarbonate Cross-Linker, Macromolecules, 2010, 43(9), 4355–4361 CrossRef.
- C. J. Kloxin, T. F. Scott, H. Y. Park and C. N. Bowman, Mechanophotopatterning on a photoresponsive elastomer, Adv. Mater., 2011, 23(17), 1977–1981 CrossRef CAS PubMed.
- B. Griese, Radicals in organic synthesis: formation of carbon–carbon bonds, Pergamon Press, Oxford, 1986 Search PubMed.
- W. B. Motherwell and D. Crich, Free radical chain reactions in organic synthesis, Academic Press, London, 1992 Search PubMed.
- G. F. Meijs and E. Rizzardo, Makromol. Chem. Rapid Commun., 1988, 9, 547–551 CrossRef CAS.
- G. F. Meijs, E. Rizzardo and S. H. Thang, Macromolecules, 1988, 21, 3122–3124 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A: Pure Appl.Chem., 1986, 23, 839–852 CrossRef.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and H. Thang, Living Free-Radical Polymerization by Reversible Addition–Fragmentation Chain Transfer: The RAFT Process, Macromolecules, 1998, 31(16), 5559–5562 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539 CrossRef CAS.
- H. Y. Park, C. J. Kloxin, A. S. Abuelyaman, J. D. Oxman and C. N. Bowman, Stress Relaxation via Addition–Fragmentation Chain Transfer in High T, Macromolecules, 2012, 45, 5640–5646 CrossRef CAS PubMed.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- Comprehensive Organic Chemistry, ed. D. Barton and W. D. Ollis, Pergamon PressLtd, Oxford, UK, 1979, vol. 3, pp. 434–436 Search PubMed.
- A. Sugawara, M. Shirahata, S. Sato and R. Sato, Bull. Chem. Soc. Jpn., 1984, 57, 3353 CrossRef CAS.
- M. K. Leung, D. T. Hsieh, K. H. Lee and J. C. Liou, J. Chem. Res., 1995, 478–480 CAS.
- D. Chaturvedi, A. K. Chaturvedi, N. Mishra and V. Mishra, An efficient, one-pot synthesis of trithiocarbonates from the corresponding thiols using the Mitsunobu reagent, Tetrahedron Lett., 2008, 49(33), 4886–4888 CrossRef CAS PubMed.
- A. W. M. Lee, W. H. Chan and H. C. Wong, Synth. Commun., 1988, 18, 1531 CrossRef CAS.
- N. Aoyagi, B. Ochiai, H. Mori and T. Endo, Mild and Efficient One-Step Synthesis of Trithiocarbonates Using Minimum Amount of CS 2, Synlett, 2006, 4, 0636–0638 Search PubMed.
- N. Aoyagi and T. Endo, Functional RAFT agents for radical – controlled polymerization: quantitative synthesis of trithiocarbonates containing functional groups as RAFT agents using equivalent amount of CS2, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(14), 3702–37093 CrossRef CAS.
- G. F. Meijis, E. Rizzardo and S. H. Thang, Preparation of Controlled Molecular Weight, Olefin-Terminated Polymers by Free Radical Methods. Chain Transfer Using Allylic Sulfides, Macromolecules, 1988, 21, 3122–3124 CrossRef.
- D. H. R. Barton and D. Crich, Tetrahedron Lett., 1984, 25, 2787 CrossRef CAS.
- W. D. Cook, T. L. Schiller, F. Chen, C. Moorhoff, S. H. Thang, C. N. Bowman and T. F. Scott, Effect of Cross-Link Density on Photoplasticity of Epoxide Networks Containing Allylic Dithioether Moieties, Macromolecules, 2012, 45(24), 9734–9741 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Radical addition–fragmentation chemistry in polymer synthesis, Polymer, 2008, 49(5), 1079–1131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures containing NMR and MS spectral data. See DOI: 10.1039/c3py00709j |
| This journal is © The Royal Society of Chemistry 2014 |