 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral recognition for the complexation dynamics of β-cyclodextrin with the enantiomers of 2-naphthyl-1-ethanol†
Hao
Tang
a,
Andria S. M.
Sutherland
a,
Lana M.
Osusky
a,
Yan
Li
b,
Josef F.
Holzwarth
b and
Cornelia
Bohne
*a
aDepartment of Chemistry, University of Victoria, PO Box 3065, Victoria, BC, Canada V8W 3V6. E-mail: cornelia.bohne@gmail.com; Fax: +1-250-721-7147; Tel: +1-250-721-7151
bFritz-Haber-Institut, Department of Physical Chemistry Faradayweg 4-6, 14195 Berlin, Germany. E-mail: holzwarth@fhi-berlin.mpg.de
First published on 5th December 2013
Abstract
The focus of this study is to understand the origin of the chiral recognition for a host–guest system containing complexes with different stoichiometries. Each enantiomer of 2-naphthyl-1-ethanol forms two different 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with β-cyclodextrin, leading to the formation of three different 2:2 complexes. One of these 2:2 complexes leads to excimer emission of the guest. Fluorescence studies were employed to determine the binding isotherms for the 1:1 and 2:2 complexes. No chiral discrimination was directly observed for the formation of the 1:1 complexes, while higher equilibrium constants (29% from binding isotherms and 40% from kinetic studies) were observed for the formation of the 2:2 complexes with (R)-2-naphthyl-1-ethanol when compared to the formation of the 2:2 complexes formed from (S)-2-naphthyl-1-ethanol. The relaxation kinetics was studied using stopped-flow experiments. The formation of the 2:2 complexes was followed by detecting the excimer emission from one of the 2:2 complexes. The relaxation kinetics was faster for (S)-2-naphthyl-1-ethanol, where a higher dissociation rate constant, by 47%, was observed, suggesting that the chiral discrimination occurs because the interaction between two cyclodextrins is more favorable for the complexes containing (R)-2-naphthyl-1-ethanol when compared to (S)-2-naphthyl-1-ethanol. The same overall equilibrium constants were observed for the 1:1 complexes with both enantiomers showing that at a given cyclodextrin concentration the sum of the two types of 1:1 complexes is the same for both enantiomers. However, analysis of the binding isotherms indicates that the ratio between the two different 1:1 complexes for each enantiomer was different for (R)- and (S)-2-naphthyl-1-ethanol.
:1 complexes with β-cyclodextrin, leading to the formation of three different 2:2 complexes. One of these 2:2 complexes leads to excimer emission of the guest. Fluorescence studies were employed to determine the binding isotherms for the 1:1 and 2:2 complexes. No chiral discrimination was directly observed for the formation of the 1:1 complexes, while higher equilibrium constants (29% from binding isotherms and 40% from kinetic studies) were observed for the formation of the 2:2 complexes with (R)-2-naphthyl-1-ethanol when compared to the formation of the 2:2 complexes formed from (S)-2-naphthyl-1-ethanol. The relaxation kinetics was studied using stopped-flow experiments. The formation of the 2:2 complexes was followed by detecting the excimer emission from one of the 2:2 complexes. The relaxation kinetics was faster for (S)-2-naphthyl-1-ethanol, where a higher dissociation rate constant, by 47%, was observed, suggesting that the chiral discrimination occurs because the interaction between two cyclodextrins is more favorable for the complexes containing (R)-2-naphthyl-1-ethanol when compared to (S)-2-naphthyl-1-ethanol. The same overall equilibrium constants were observed for the 1:1 complexes with both enantiomers showing that at a given cyclodextrin concentration the sum of the two types of 1:1 complexes is the same for both enantiomers. However, analysis of the binding isotherms indicates that the ratio between the two different 1:1 complexes for each enantiomer was different for (R)- and (S)-2-naphthyl-1-ethanol.
Introduction
A change in chirality is one of the smallest perturbations that can be made to a chemical system and understanding how chiral discrimination is achieved is an ongoing quest. Supramolecular chemistry can impose chiral recognition through non-covalent interactions, which is advantageous when reversible systems are desired. Photochemical reactions in supramolecular systems provide the opportunity to study chirality control in systems where recognition is imposed through non-covalent interactions.1–3 One of the ongoing challenges is to obtain mechanistic information on how this discrimination occurs for complex supramolecular systems beyond those with 1:1 guest–host stoichiometries. An example of such complexity is the use of serum albumins, which were exploited as hosts with several binding sites with different binding affinities to which 2-anthracenecarboxylate binds as a guest. The highest chiral discrimination for the photodimerization reaction of 2-anthracenecarboxylate was obtained when pro-chiral guests were bound to protein binding sites with moderate affinity and where the guests were fairly immobile before the photodimerization reaction occurred.4–7 This example shows that the guest was bound to sites with different properties and that the desired property, chiral discrimination in this case, was obtained from a site with moderate binding affinity. Therefore, determination of systems in which one of the enantiomers binds much more strongly to a single site, such as in the case of the binding of a chiral derivative of pyrene to bovine serum albumin,8 may not be a sufficient requirement to achieve a property that requires guest release such as in a bimolecular reaction, making it necessary to investigate systems with multiple binding sites.
A different approach to complexity, described in this contribution, is to have one host and one guest where complexes with different guest–host stoichiometries are formed and where these complexes have different spectroscopic signatures.
Cyclodextrins (CDs) are cyclic glucose oligosacccharides that have a hydrophobic cavity into which guests are included.9–12 The size of the cavity depends on the number of glucose units in the ring, with the smallest being α-CD, followed by β-CD and γ-CD. The majority of CD complexes have a 1:1 stoichiometry, but higher order complexes with 1:1:1, 1:2, 2:1 and 2:2 guest–host complexes can occur depending on the relative size of the guest and the CD. Naphthalene and its derivatives were shown to form higher order complexes with all three CDs.13–20 When encapsulated by two CDs the naphthalene excited states were located in a less polar environment and were much better protected from quenching by molecules residing in the aqueous phase.19–21 The shape of the guest determined which stoichiometries were formed for the CD complexes.17 Not all complexes containing two naphthalene guests led to the observation of excimer emission because steric hindrance could impede the approach of the two guests inside the complex.18
The current work is based on previous studies of the binding of 2-naphthyl-1-ethanol (NpOH, Scheme 1) with β-cyclodextrin (β-CD).19,22 These previous studies focused on the determination of binding isotherms and the dynamics for the 1:1 complexes,19 and on theoretical calculations where predicted circular dichroism spectra were compared with experimental ones.22 The current work focuses on kinetic studies for the formation of the 2:2 complexes for which only order of magnitude estimates were reported,19 and on the possible mechanisms for chiral discrimination that previously was observed only as differences in fluorescence intensities and circular dichroism spectra but was not apparent in the determined equilibrium constants.19,22
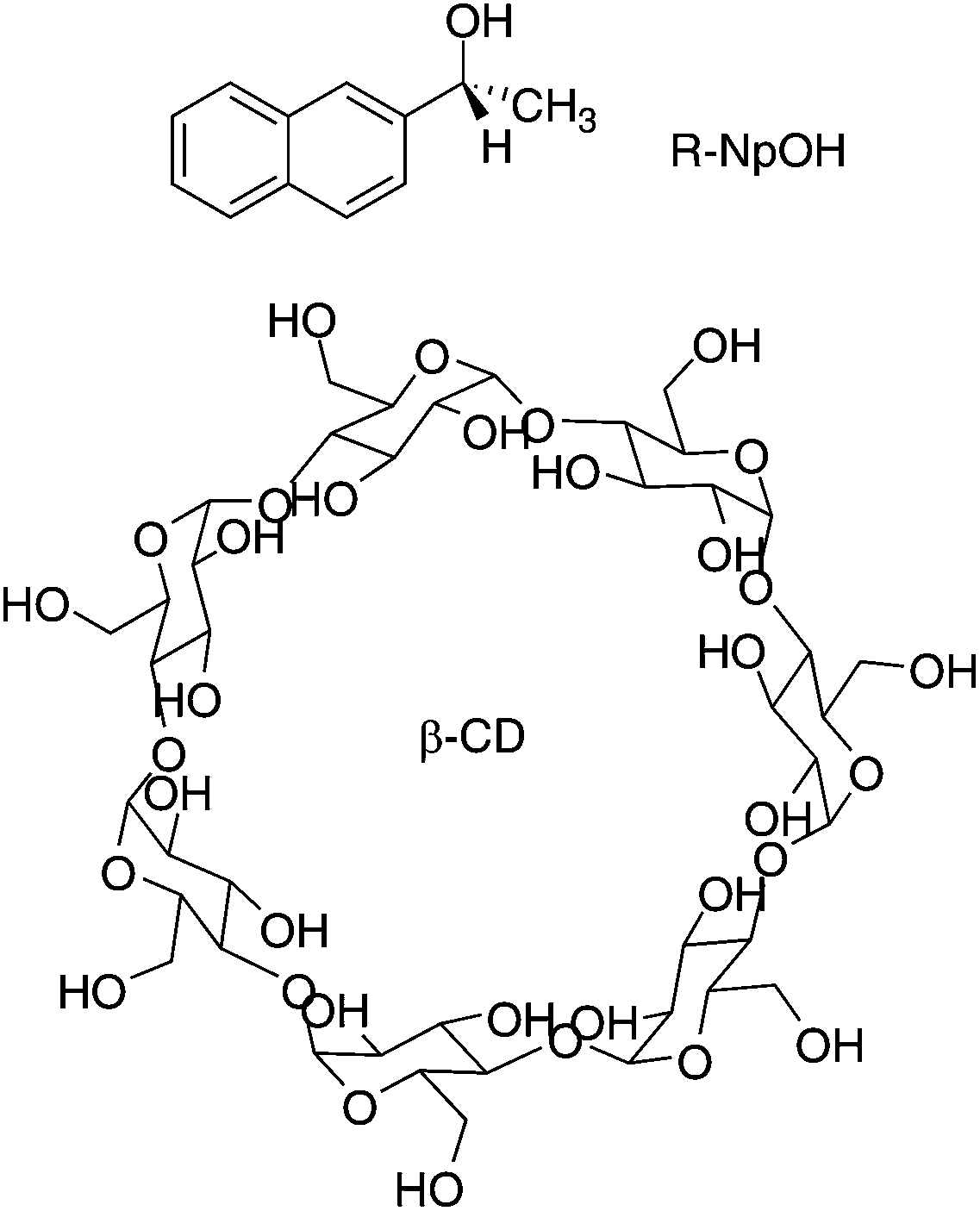 | ||
| Scheme 1 Structure of (R)-2-naphthyl-1-ethanol ((R)-NpOH) and β-cyclodextrin (β-CD). | ||
The enantiomers of NpOH form 1:1 and 2:2 (NpOH–β-CD) complexes with β-CD with a higher equilibrium constant for the 2:2 complexes compared to the equilibrium constant for the 1:1 complexes.19,22 Two types of 1:1 complexes were formed where either the naphthyl (N) or the ethanol (E) moieties were included close to the narrower rim of β-CD which contains the primary hydroxyl groups (Fig. 1). The presence of N and E complexes leads to the formation of NN, NE and EE complexes (Fig. 1).22 In previous work with naphthalene/β-CD20 and α-terthiophene/γ-CD23 complexes the excimer emission of the guest was observed for the 2:2 complexes while no triplet excited states were detected for the 2:2 complexes because the excited guest interacts readily with the second guest in the ground state leading to excimer emission. In the case of the NpOH–β-CD system, excimer emission and long-lived triplet states were observed.19 This result suggested that different 2:2 complexes were formed, one in which the naphthyl groups were in close proximity leading to the formation of the excimer and other 2:2 complexes where the naphthyl groups were kept separate leading to long-lived triplet excited states. The NN and NE complexes placed the naphthalene moieties at a distance leading to long-lived triplets that were well protected from quenchers in the aqueous phase.19 For the EE complex the naphthalene moieties were in close proximity leading to excimer emission of the guest dimer. The formation of this dimer was consistent with the observation of a broadening of the excitation spectrum for NpOH when the excimer emission was monitored and the lack of a growth kinetics for the excimer emission.22 The calculated energies for the NN, NE and EE complexes were close and a comparison of the calculated and the experimental circular dichroism spectra supported the presence of multiple 2:2 complexes.22
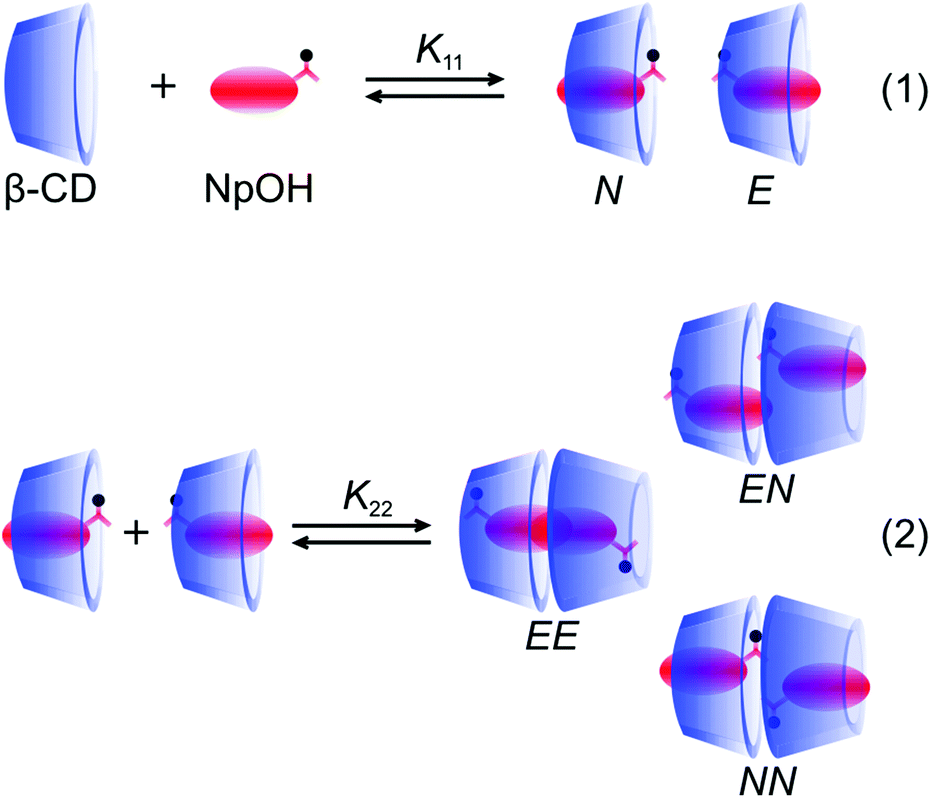 | ||
| Fig. 1 Formation of the two 1:1 complexes between NpOH and β-CD and the formation of the three 2:2 complexes from the reaction of the E and N complexes. | ||
The dynamics for the 1:1 complexation of NpOH with β-CD is fast with an association rate constant of 3 × 108 M−1 s−1 and a dissociation rate constant of 2 × 105 s−1, while the dynamics for the 2:2 complex is much slower and was estimated to occur in the millisecond time domain.19 Higher excimer intensities were observed for (R)-NpOH/β-CD than for (S)-NpOH/β-CD, suggesting that chiral discrimination occurs but was not apparent in the previous thermodynamic and kinetic studies because of the relatively large experimental errors.19,22 The objective of the current study was to investigate the origins of the chiral discrimination for a host–guest system that contains multiple complexes. The kinetics for the formation of the 2:2 complexes, for which chiral discrimination was observed in the steady-state emission intensities, was followed by the kinetics of excimer emission, which is unique for the presence of the EE complex. These kinetic data were combined with binding isotherm studies at variable guest and host concentrations. The chiral discrimination is due to the faster dissociation of the 2:2 complexes formed for (S)-NpOH, while no chiral discrimination was observed for the equilibrium constants (K11) of the 1:1 complexes.
Experimental
Materials
β-CD was a generous gift from Cerestar (lots C6034-13 and F6080-191), (R)- and (S)-NpOH (Fluka) and NaCl (BDH) were used as received. The purities of (R)- and (S)-NpOH were checked by measuring fluorescence decays of 10 μM aqueous solutions for each NpOH. The decays were mono-exponential indicating that no other emissive species were present. NaNO2 (Aldrich) was recrystallized from water. Deionized water (Barnstead NANOpure, 17.8 MΩ cm−1) was employed for all samples.Solution preparation
NpOH (1 mM) and β-CD (10 mM) stock solutions were prepared in 0.2 M NaCl aqueous solutions, since the magnitude of the equilibrium constants were shown to depend on the concentration of NaCl.21 NpOH–β-CD solutions with the highest β-CD concentration (10 mM) were prepared by dissolving β-CD in NpOH–NaCl solutions. Solutions with lower β-CD concentrations were prepared by dilution with NpOH–NaCl solutions. All solutions were shaken overnight. All samples were aerated with the exception of the solutions for laser flash photolysis experiments that were purged with N2O for 20 min. N2O is known to trap solvated electrons24 formed from the photoionization of organic molecules in aqueous solutions.25The triplet excited states of NpOH were quenched by NaNO2. NaNO2 aqueous solutions (∼0.6 M) were prepared daily and were purged with N2O for 20 min. Appropriate amounts of quencher stock solution were injected into 2.5 mL of the sample solution using a gas tight syringe.
Instrumentation
UV-Vis spectra were recorded using a Varian Cary 1 spectrometer with samples kept at room temperature. A PTI QM-2 fluorimeter was employed to measure steady-state fluorescence spectra. The excitation wavelength was set to 280 nm, and the emission spectra were recorded between 300 nm and 500 nm. The bandwidths for the excitation and emission monochromators were 2 nm. Samples contained in 10 mm × 10 mm quartz cells were kept at a constant temperature of 10.0 ± 0.1 °C using a circulating water bath. Spectra were corrected by subtracting a baseline spectrum for a solution containing all chemicals with the exception of NpOH. This baseline spectrum corresponds to the water Raman emission and a low impurity emission (300–375 nm) from β-CD.Fluorescence decays and time-resolved emission spectra (TRES) were measured using an Edinburgh Instruments OB920 single photon counter. The excitation source was a 277 nm pulsed light emitting diode (EPLED280 – Edinburgh Instruments). Decays were collected at 330 nm or 380 nm with a monochromator bandwidth of 16 nm. A Ludox (Aldrich) solution was used to collect the instrument response function (IRF) by scattering the excitation light at 277 nm. The decays were collected until at least 2000 counts were reached in the channel with maximum intensity. Samples were held in 10 mm × 10 mm quartz cells kept at 10.0 ± 0.1 °C for at least 15 min using a circulating water bath.
Time-resolved emission spectra (TRES) were collected at every 10 nm between 300 nm and 500 nm by keeping the collection time constant at 5 min, which corresponded to a collection of ca. 20000 counts in the channel of maximum intensity for the maximum emission wavelength of 330 nm. Each decay trace was integrated between specific time delays to construct the TRES.
Fluorescence decays were fit to a sum of exponentials (eqn (3)) using the Fast software (version 3, Edinburgh Instruments), where τi corresponds to the lifetime of each species and Ai is the associated pre-exponential factor. The calculated fit was reconvoluted with the IRF and was compared to the experimental data until the differences between the calculated fit and the experimental data were minimized. Fits to individual decays were deemed to be adequate when the residuals were random and the χ2 values were between 0.9 and 1.2.26 The number of exponentials was increased until an adequate fit was obtained. Global analysis (Fast software) was used as an alternate analysis method when the emission decays for the same sample were measured at two different wavelengths.
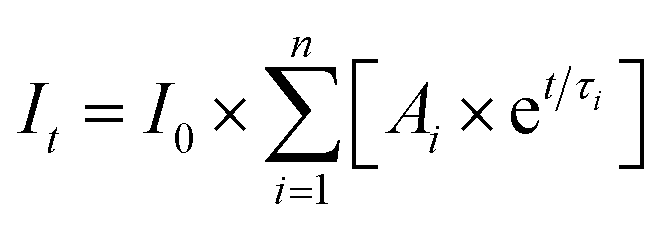 | (3) |
Laser flash photolysis (LFP) was employed to measure the kinetics for the triplet excited states of NpOH.27,28 Samples were excited with a Quanta-Ray Lab 130-4 pulsed Nd:YAG laser at 266 nm (≤20 mJ per pulse, Spectra Physics). The transient absorption changes at 420 nm were simultaneously detected on two oscilloscopes.28 This procedure was required to determine the kinetics at short and long times. The samples contained in 10 mm × 10 mm quartz cells were maintained at 10.0 ± 0.1 °C for at least 15 min using a modified cryostat (Unisoku USP-203 cryostat) with 4 windows.29
The decays were fit either to a monoexponential function (kobs2 = 0 and a2 = 0, eqn (4)) or to the sum of two exponentials (eqn (4)), where a1 and a2 are pre-exponential factors for processes with rate constants kobs1 and kobs2. It is important to note that the ai values in the case of laser flash photolysis experiments correspond to absorbance values while in the case of single photon counting experiments the Ai values correspond to fractional values.
| ΔA = a0 + a1e−kobs1t + a2e−kobs2t | (4) |
The data were fit using the Kaleidagraph software (v. 4.1, Synergy Software). The values of kobs1 and kobs2 recovered from the fit are very dependent on the value of the final offset a0. Therefore, a0 was determined from a kinetic trace collected at a sufficiently long collection time as a constant absorbance value after the decay of all triplets. The kinetic traces at two different collection times were determined simultaneously.
A SX20 stopped-flow system from Applied Photophysics was employed to measure the kinetics for the formation of the 2:2 NpOH–β-CD complexes. The dead time for mixing was 1 ms. Samples were excited at 280 nm (9.3 nm bandwidth) and fluorescence was collected with an interference filter with the centre wavelength of 405 nm (FWHM of 38 nm, Melles Griot ANDV7248, lot 13637). Two solutions were contained in separate syringes; one syringe with the NpOH solution and the second one with the β-CD solution. These solutions were mixed in a 1:1 ratio. Samples were kept at 10.0 ± 0.1 °C using a circulating water bath for at least 20 min before experiments were performed. The following control experiments were performed: (i) mixing of a 0.2 M NaCl solution with a 0.2 M NaCl solution to determine the reading of the “zero intensity” for the equipment, (ii) mixing of β-CD solution with 0.2 M NaCl solution to determine the baseline value for the system in the absence of fluorophore. The values for control experiments (i) and (ii) are very similar and were less than 2% of the lowest offset intensity measured for the system containing the lowest concentrations of NpOH (50 μM) and β-CD (50 μM) employed.
The kinetic data were fit to a monoexponential function or to a sum of exponentials using the Pro-data Viewer software from Applied Photophysics. This fitting procedure was used to determine how many relaxation processes were observed for a particular set of data. A global analysis method using the Pro-Kineticists II software from Applied Photophysics was employed to simultaneously fit a set of data where an experimental condition, e.g. the β-CD concentration was systematically varied. The global analysis method requires the definition of a model for the sequence of reactions, which in this study included a fast equilibrium for the formation of the 1:1 complexes followed by a slow reaction forming the 2:2 complexes.
Results
The various complexes in solution need to be known in order to interpret the kinetic data. The experiments were performed at 10 °C in order to sufficiently slow down the reaction for kinetic experiments to be feasible with the stopped-flow system. The changes in the fluorescence of NpOH in the presence of β-CD (Fig. 2) were qualitatively the same as observed previously at 20 °C.19,22 At a low NpOH concentration (5 μM) only the monomer emission of NpOH was observed in the presence of β-CD and no differences in the emission intensities were observed for both enantiomers (inset Fig. 2). At this concentration of NpOH only 1:1 NpOH–β-CD complexes are formed. The same intensities observed for both enantiomers suggest that either no chiral discrimination occurs for the formation of the 1:1 complexes or that all 1:1 complexes formed have the same emission efficiency for NpOH.
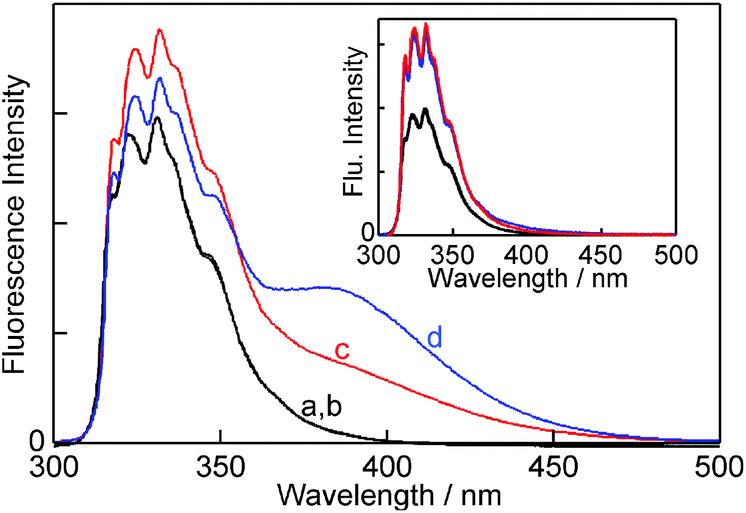 | ||
| Fig. 2 Fluorescence spectra for (R)- and (S)-NpOH (150 μM) in the absence (a, b – black; both spectra are superimposed) and presence of 10 mM β-CD (c, red (S)-NpOH; d, blue (R)-NpOH). The inset shows the fluorescence spectra for (R)- and (S)-NpOH (5 μM) in the absence (black; both spectra are superimposed) and presence of 10 mM β-CD (red (S)-NpOH and blue (R)-NpOH; both spectra are superimposed). | ||
At a high NpOH concentration (150 μM) the formation of the 2:2 complex was observed as a broad red-shifted excimer emission (Fig. 2). The excimer emission intensity is related to the formation of the EE complex, whereas the monomer emission includes the emissions from free NpOH, N, E, NN and NE complexes. At a NpOH concentration of 150 μM the excimer emission is higher for (R)-NpOH than for (S)-NpOH, while the reverse trend was observed for the fluorescence intensities for the emission from NpOH–β-CD complexes other than EE all of which emit as a NpOH monomer. This result indicates a chiral discrimination for the formation of the 2:2 complexes.
Characterization of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 NpOH–β-CD complexes
:1 NpOH–β-CD complexes
Excited-state lifetime measurements can be used to differentiate fluorophores located in different environments. The lifetime for (R)- and (S)-NpOH in water measured at 330 nm was 25.4 ± 0.3 ns, which was similar to the lifetimes measured at 20 °C (25 ns).19 The ratio between the NpOH and β-CD concentrations was low and an impurity emission from β-CD was detectable in the lifetime measurements at the NpOH concentration (5 μM) where only monomer emission was observed. The lifetime for this impurity emission (4.6 ± 0.7 ns) was shorter than the lifetimes for NpOH (>20 ns). The emission decays in the presence of β-CD were adequately fit to a sum of three exponentials where the values for the impurity emission of β-CD (4.6 ns) and NpOH in water (25.4 ns) were fixed. The third lifetime recovered was 37.6 ± 0.1 ns for (R)-NpOH and 38.1 ± 0.1 ns for (S)-NpOH. These values were similar to the 34–38 ns lifetimes previously determined for the 1:1 NpOH–β-CD complex at 20 °C.19,22 The longer lifetimes for the NpOH complexes are consistent with the increase in the steady-state fluorescence intensities observed for both NpOHs in the presence of β-CD (inset Fig. 2).
The pre-exponential factors for the fluorescence decays (Ai, eqn (3)) are related to the relative concentrations of the fluorophores when the singlet excited state lifetimes are sufficiently short so that interconversion between the excited species does not occur during their lifetimes. The lifetimes for NpOH in 1:1 complexes with β-CD were shorter (<50 ns) than the relaxation time for complex formation (310 ns), i.e. sum of the association and dissociation processes, at the highest β-CD concentration employed.19 Therefore, the Ai values can be related to the relative concentrations for free and β-CD bound NpOH. The same pre-exponential values were measured for both enantiomers. These values were 0.14 ± 0.03 and 0.13 ± 0.03 for free (R)- and (S)-NpOH, respectively and 0.86 ± 0.03 and 0.87 ± 0.03 for (R)- and (S)-NpOH in the 1:1 complexes.
The binding isotherms based on the changes in the fluorescence intensity were fit numerically (see ESI† for details) assuming a 1:1 binding stoichiometry between NpOH and β-CD (Fig. 3). The parameters for the fit were the equilibrium constant for the 1:1 complex (K11, eqn (1)) and the relative emission intensity of the 1:1 complex (C11) compared to the intensity in water. The recovered K11 values from three independent experiments were (1.10 ± 0.03) × 103 M−1 and (1.08 ± 0.03) × 103 M−1 for (R)- and (S)-NpOH, respectively. The C11 values were 1.74 ± 0.01 and 1.78 ± 0.02 for (R)- and (S)-NpOH, respectively.
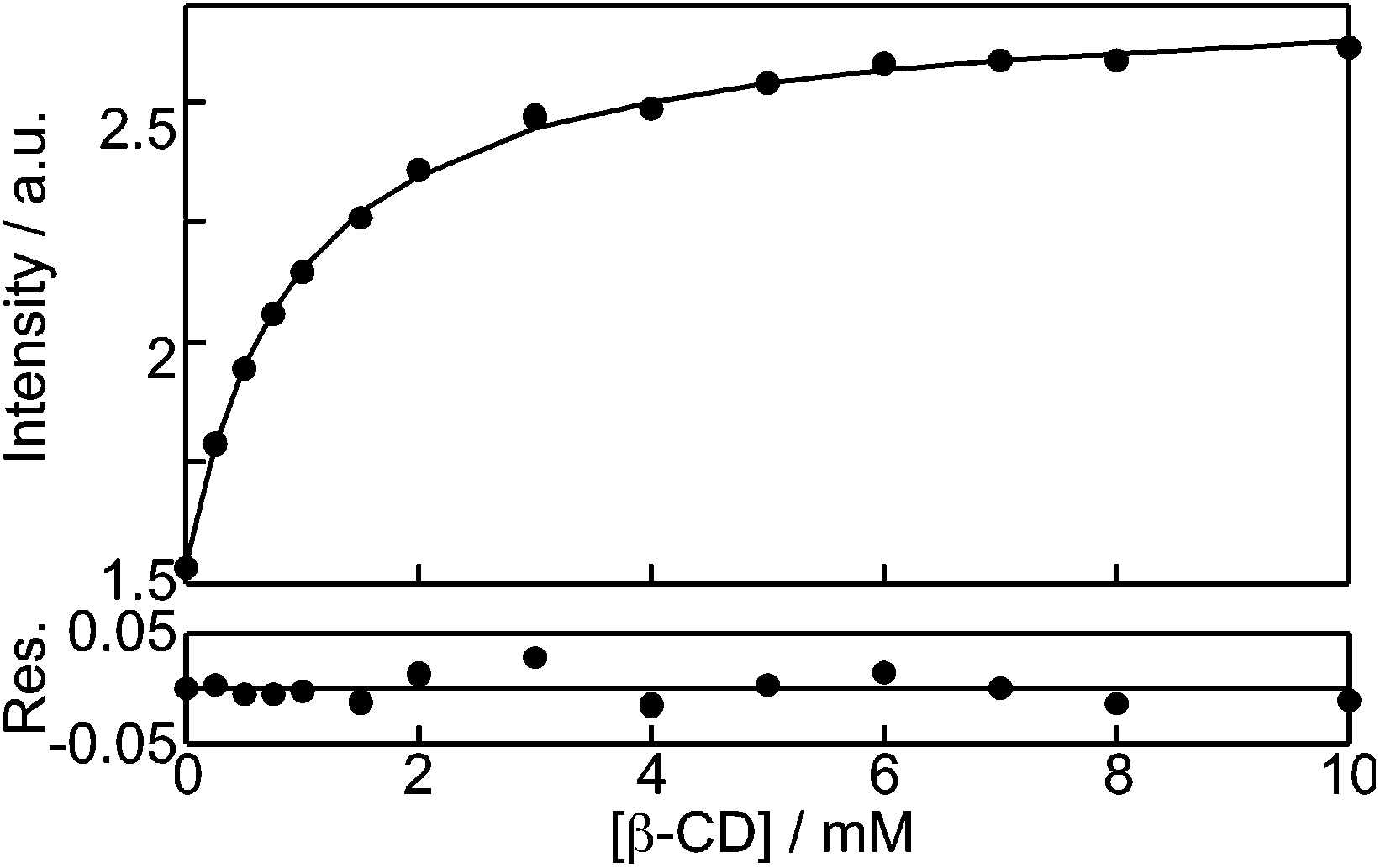 | ||
| Fig. 3 Numerical fit for the dependence of the (R)-NpOH (5 μM) monomer fluorescence intensity with the β-CD concentration assuming the formation of a 1:1 complex. | ||
The observation of the same K11 values for both enantiomers is in line with the same pre-exponential values observed for bound NpOH in the time-resolved emission studies. The fluorescence studies for the 1:1 NpOH complex showed that fluorescence cannot differentiate between the formation of the E and N complexes (Fig. 1). The presence of both these complexes had previously been established in circular dichroism experiments combined with theoretical calculations.22 Unfortunately circular dichroism experiments were not possible for the determination of the equilibrium constant of E and N because of the poor signal-to-noise ratio at the low NpOH concentration required for these experiments. The lack of discrimination between the E and N complexes in the binding studies suggests that their fluorescence properties, such as excited state lifetimes and emission quantum yields, are similar. In this respect, it is important to note that time-resolved experiments are quite sensitive to differentiate between similar lifetimes and the observation of only one lifetime for the 1:1 complexes supports the suggestion that the complexes E and N cannot be differentiated by fluorescence. Therefore, experimentally it was only possible to determine the value for K11, which corresponds to the sum of the individual equilibrium constants for the formation of the E (KE) and N (KN) complexes.
Characterization of the 2:2 NpOH–β-CD complexes
The emission of the NpOH excimer was observed at high NpOH concentrations (150 μM) indicating that the 2:2 EE complex was formed. The kinetics for the decay of the single excited states of NpOH were measured at 330 nm and 380 nm. The emission from NpOH free in water and from NpOH in E, N, EN and NN complexes predominate at 330 nm, while the emission from the excimer emission due to the EE complex predominates at 380 nm. The impurity emission from β-CD was negligible at this NpOH concentration. No growth kinetics was observed at 380 nm indicating that the excimer emission was due to a NpOH dimer present before excitation, indicating that both guest molecules are in close proximity in the EE complex.
The emission from the monomers and the excimer contribute to the intensities at 330 nm and 380 nm. For this reason, global analysis was employed in which the decays at both wavelengths were simultaneously fit to the sum of three exponentials. No further lifetimes could be resolved. The shortest lifetime, τ1, was fixed to 25.4 ns assigned to NpOH in water, while the second lifetime, τ2, was fixed to 38 ns for the monomer emission of NpOH complexed to β-CD. The third lifetime recovered corresponds to the excimer emission and was 72.7 ± 0.3 ns for (R)-NpOH and 72.0 ± 0.3 ns for (S)-NpOH. The A3 values were significantly higher for (R)-NpOH than for (S)-NpOH (Table 1) in line with the higher steady-state emission intensity for the excimer emission observed for the (R)-enantiomer (Fig. 2). This result shows that for (R)-NpOH a larger amount of the EE complex was formed when compared to the 2:2 complexes for (S)-NpOH. The different intensities could also have been related to different lifetimes for the excimer emission of the two enantiomers, which was not the case. In contrast, there is a higher fraction of the E, N, EN and NN complexes formed for (S)-NpOH (see A2 values at both wavelengths, Table 1). The relative concentrations of the E, N, EN and NN complexes could not be determined from the time-resolved fluorescence studies because all these complexes had lifetimes close to 38 ns.
| NpOH | λ em/nm | A 1 | A 2 | A 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The errors for the Ai values are 0.03. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R- | 380 | 0.17 | 0.11 | 0.72 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S- | 380 | 0.21 | 0.38 | 0.42 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R- | 330 | 0.22 | 0.70 | 0.08 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S- | 330 | 0.09 | 0.86 | 0.05 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Complex EE is expected not to form triplet excited states based on previous studies where the excimer emission from guests in 2:2 CD complexes were observed.20,23 The formation of excimer precludes intersystem crossing of the singlet excited state to the triplet state in line with the lack of a growth kinetics for the excimer emission. For this reason, studies on the absorption of the triplet excited states of NpOH in the presence of β-CD is selective to differentiate between the E, N and EN, NN complexes without interfering signals from the EE complex. In the EN and NN complexes the naphthalene moieties are too far apart to form excimers and excitation leads to the formation of one of the NpOHs in the triplet state.22 Previous work19 also showed that triplet NpOH in the 2:2 complexes was much better protected from quenchers in the aqueous phase than triplet NpOH in 1:1 complexes. Laser flash photolysis was employed where the samples were excited at 266 nm to measure the kinetics of triplet NpOH at 420 nm in the absence and presence of β-CD. Quenching experiments were performed at 10 °C using nitrite anions as a quencher (Q).30 The quenching plots for NpOH in water (eqn (5), where kobs is the observed decay rate constant and k0 is the rate constant in the absence of quencher) were linear leading to quenching rate constants (kq) of (2.5 ± 0.1) × 109 M−1 s−1 and (2.6 ± 0.1) × 109 M−1 s−1 for (R)- and (S)-NpOH, respectively. These values were similar to the quenching rate constant for triplet NpOH by nitrite ions of 3.1 × 109 M−1 s−1 determined in water at 20 °C.31
| kobs = k0 + kq[Q] | (5) |
The decay for triplet NpOH in the presence of 10 mM β-CD was adequately fit to the sum of two exponentials. This result indicated that two populations of triplets were present that did not interconvert during the lifetime of these excited states (<100 μs). The short-lived triplet was previously assigned to triplet NpOH in the 1:1 complex (E and N), while the long-lived triplet was assigned to triplet NpOH in the 2:2 complexes (EN and NN).19,22 No triplet for NpOH free in water was observed because the contribution from free NpOH was negligible in the presence of 10 mM β-CD. Decays were collected simultaneously for short and long time windows to ensure the precise determination of the final absorbance values (a0) and of the rate constants, kobs1 and kobs2 (Fig. S1 and S2, see ESI† for details). The quenching plots (eqn (5)) for the short- and long-lived species were linear (Fig. S3†), and the same quenching rate constants were obtained for (R)- and (S)-NpOH. The quenching rate constants for the short-lived species assigned to the 1:1 complex ((8.8 ± 0.2) × 108 M−1 s−1 for (R)-NpOH and (8.4 ± 0.2) × 108 M−1 s−1 for (S)-NpOH) were lower than for the quenching in water, but were higher than the kq values for the 2:2 complexes ((2.9 ± 0.1) × 107 M−1 s−1 for (R)-NpOH and (3.1 ± 0.2) × 107 M−1 s−1 for (S)-NpOH). For each type of β-CD complex, that is 1:1 or 2:2 complexes, the quenching rate constants were the same for both enantiomers of NpOH, showing that the accessibility of nitrite anions to NpOH inside one or two β-CDs is not determined by the chirality of the guest.
The pre-exponential factors for the slow decay (eqn (4), a2) were 0.020 ± 0.001 for (R)-NpOH and 0.023 ± 0.004 for (S)-NpOH when solutions were excited that had the same absorbance values at 266 nm. The a2 values correspond to the sum of the concentrations of EN and NN complexes. The fact that the same a2 values, within experimental error, were obtained for (R)- and (S)-NpOH at a given β-CD concentration showed that the amount of EN and NN complexes formed were similar for both enantiomers of NpOH.
The dependence of the EE emission at 390 nm with the concentration of β-CD was used to determine the equilibrium constant for the 2:2 complexes. The monomer and excimer emissions contribute to the intensity at 390 nm. The spectrum for the excimer emission needs to be known in order to calculate the contribution of the monomer emission at 390 nm. TRES were collected for the emission of (S)- and (R)-NpOH (Fig. S4 in the ESI†) in the presence of 10 mM β-CD. The spectra at long delays correspond to the emission from the EE complex because the lifetime for the excimer emission is longer than the lifetime for the NpOH monomer emission. The same spectra were obtained for (R)- and (S)-NpOH. The fraction of the emission intensity from the monomer at 390 nm with respect to the intensity at 330 nm was calculated using the NpOH emission spectrum in water, while the fraction of the excimer emission at 330 nm with respect to the intensity at 390 nm was calculated from the TRES. From these values operational equations were derived for the corrected monomer and excimer intensities at 330 nm and 390 nm, respectively (eqn (S14)–(15) in the ESI†).
The equilibria for the formation of the 1:1 and 2:2 complexes are coupled (Scheme 2) and all equilibria need to be considered when fitting the binding isotherm for the formation of one of the products, which corresponds to the EE complex in this study.
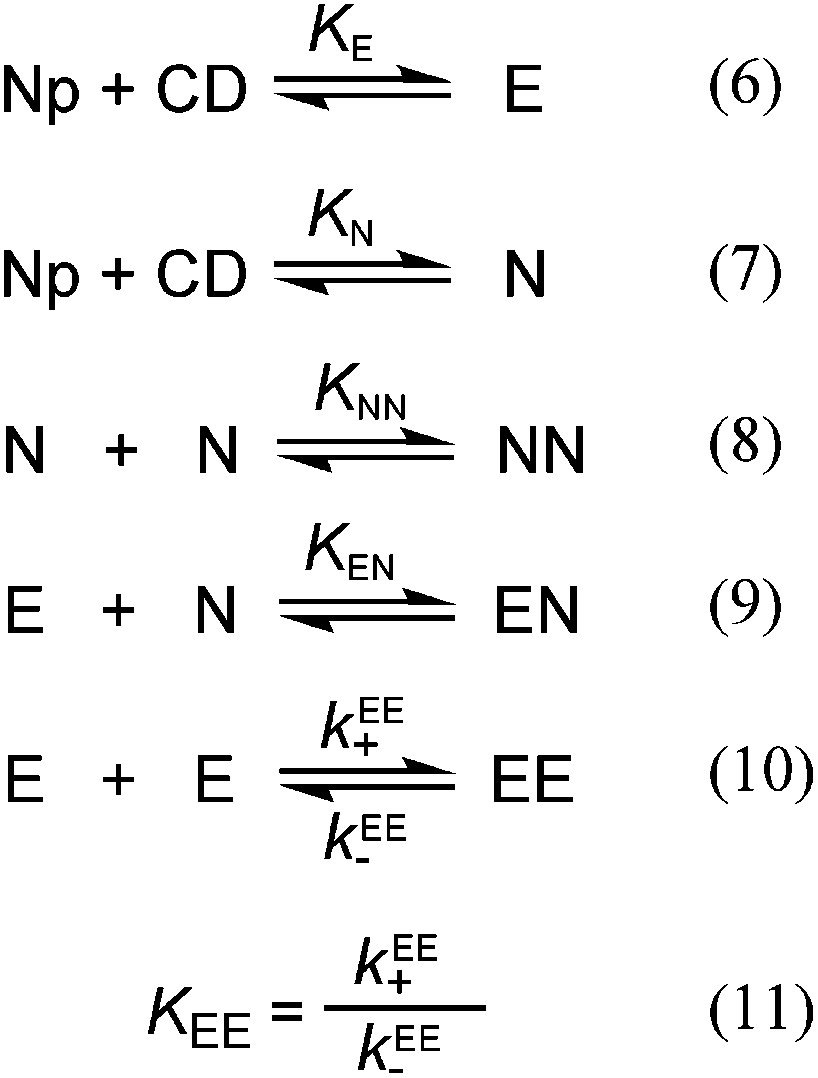 | ||
| Scheme 2 | ||
The binding isotherm was numerically fit considering the equilibria shown in Scheme 2 by making the following assumptions (see ESI† for details): (i) the value for KE was fixed for each fit to a value between 0 and K11 (1100 M−1), which corresponds to the theoretical limits for this parameter, and (ii) the values for KEE, KEN and KNN were considered to be equal based on the analysis of the kinetic data for the formation of the EE complex where only one relaxation process was observed (see details below). The binding isotherm fit well to this model (Fig. 4) and the quality of the fit was the same irrespective of the KE values used. The KEE (inset Fig. 4) and the relative emission efficiency (CEE, Fig. S5†) values varied in a systematic manner as the KE values were changed showing that these parameters were correlated. In all cases, the values for KEE and CEE were higher for (R)-NpOH than for (S)-NpOH, showing that a chiral discrimination occurs for the formation of the 2:2 complexes.
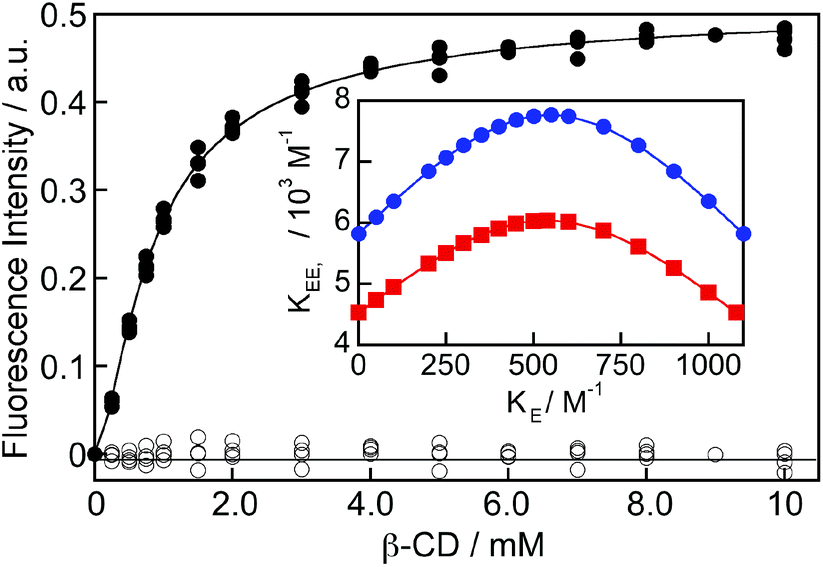 | ||
| Fig. 4 Dependence of the corrected excimer emission intensity at 390 nm for (R)-NpOH (150 μM) with the β-CD concentration (solid symbols; three independent experiments). The solid line corresponds to the numerical fit of the data using fixed KE values between 0 and 1100 M−1. The residuals between the data and the fit are shown as open symbols. The inset shows the variation of the KEE value as the fixed value for KE was changed in the numerical fit for (R)-NpOH (blue circles) and (S)-NpOH (red squares). The lines in the inset were included to guide the eye. | ||
The limiting values for KE are when this parameter is equal to zero, equal to KN, or equal to K11. The KEE value for (R)-NpOH was (5.8 ± 0.2) × 103 M−1, while it was (4.5 ± 0.2) × 103 M−1 for (S)-NpOH when KE was either fixed at zero or equal to K11. The increase in the emission efficiency at 390 nm (CEE) corresponds to the ratio of the intensity of the EE complex with respect to the intensity for the monomer of NpOH in water at the detection wavelength. The CEE values were infinity when KE was zero and (1.43 ± 0.03) × 104 and (0.72 ± 0.01) × 104 for the (R)- and (S)-enantiomers when KE was equal to K11. Fits were attempted by fixing the CEE and KE for both enantiomers to the same values, but the fits were not adequate suggesting that either CEE or KE values were different for the two enantiomers. The singlet-state lifetimes for EE complex were the same for both enantiomers, suggesting that the CEE values for both enantiomers were similar. Therefore, the KE values were different for (R)- and (S)-NpOH. The CEE value of infinity has no physical meaning. KE cannot be zero since EE complexes were observed. However, this limit was included to show the continuous variation of KEE and CEE when KE was varied. The maximum values of KEE of (7.8 ± 0.3) × 103 M−1 for (R)-NpOH and (6.0 ± 0.3) × 103 M−1 for (S)-NpOH were obtained when KE and KN were fixed to be equal. It is important to note that the dependencies for KEE and CEE with KE for (R)-NpOH never cross with the dependencies for (S)-NpOH.
Kinetics for EE complex formation
The kinetics for the formation of the EE complex was measured using a stopped-flow set-up by mixing a solution of NpOH with a solution containing β-CD. The fluorescence intensity levelled off within 0.1 seconds (Fig. 5) indicating that the reaction was completed within this timeframe. All kinetic traces were adequately fit to a mono-exponential function (Fig. S6†). At given NpOH and β-CD concentrations the observed rate constant for the relaxation kinetics of (S)-NpOH was always higher than for (R)-NpOH, showing that the relaxation kinetics for the (S)-enantiomer were faster.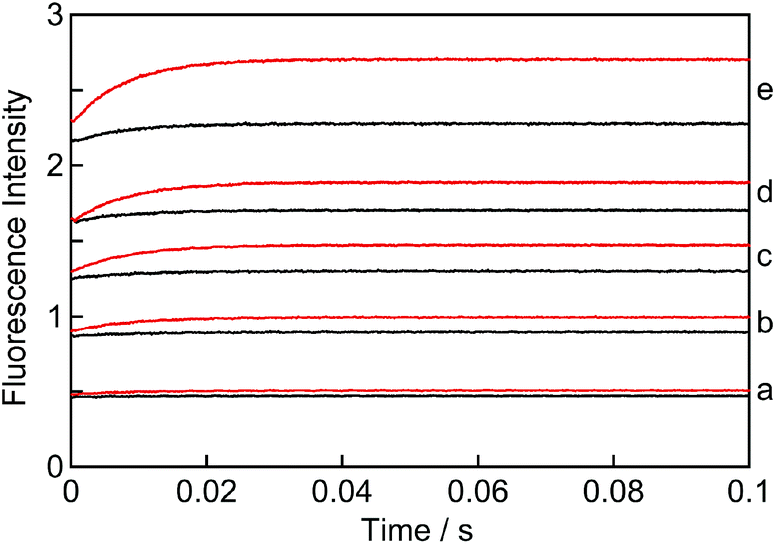 | ||
| Fig. 5 Kinetics for the formation of the EE complex of (R)-NpOH when mixed with β-CD (black: 50 μM; red: 100 μM) at various concentrations of (R)-NpOH: a, 50 μM; b, 100 μM; c, 150 μM; d, 200 μM and e, 300 μM. | ||
The kinetic traces showed an initial offset value (Fig. 5). Most of the offset is due to the presence of NpOH as a fluorophore and therefore the offset was higher as the NpOH concentration was raised. However, at a constant NpOH concentration, the offset was higher at the higher β-CD concentration (for clarity see traces “e” in Fig. 5). This additional increase in the presence of β-CD indicates that a process occurred involving β-CD with a time constant faster than the 1 ms time-resolution of the stopped-flow system. This process corresponds to the formation of the 1:1 NpOH–β-CD complex, which was previously shown to occur in the microsecond time domain.19 This process is observable because the NpOH monomer emits to some extent at the detection wavelengths (405 nm, FWHM = 38 nm) and the intensity for NpOH in the 1:1 complex is higher than in water (see above).
The amplitude of the kinetic traces (final offset intensities) corresponds to the amount of EE complex formed. This amplitude increases with the concentration of β-CD at constant NpOH concentrations (Fig. 5: compare red and black traces for each set) and also increases with the NpOH concentrations at constant β-CD concentrations (Fig. 5: compare all black or all red traces). These trends are consistent with the formation of a 2:2 complex where the concentration of NpOH and β-CD needs to be high for the formation of the complex to occur.
The fast formation of the E and N complexes can be treated as being in pre-equilibrium. The kinetics for the formation of the EE complex is coupled to the kinetics for the formation of the other 2:2 complexes (EN and NN). All kinetic traces fit adequately to a mono-exponential function indicating that only one relaxation process was observed. This result suggests that the kinetics for the formation of all 2:2 complexes have similar relaxation times. If one of the complexes was formed faster, then it would transiently accumulate before redistribution to the other complexes occurred. Such a scenario would lead to kinetics with more than one relaxation process. Simulations were performed in which the association or dissociation rate constants for the EE and the EN/NN complexes were varied in order to establish how different the rate constants had to be for the simulated data to lead to non-random residuals when fit with the model used to analyze the experimental data (Fig. S7, see ESI† for details). Association and/or dissociation rate constants that differed by 20–25% would lead to non-random residuals and to the detection of more than one relaxation time. This simulation set the upper limit for the difference in the association and dissociation rate constants for the EE, EN and NN complexes, and consequently for differences in their equilibrium constants. Differences of ca. 25% in the rate constants and consequently in the equilibrium constants would not be discernable in the experiments.
The kinetic data were fit using global analysis to a model in which a pre-equilibrium is followed by the formation of 2:2 complexes (Scheme 3). Global analysis was employed to fit simultaneously the kinetic data at different NpOH and β-CD concentrations. The K11 value was fixed to the value determined in the fluorescence binding isotherm studies. All residuals (Fig. S8†) were random showing that the model is consistent with the experimental data. The k22+ values were the same for both enantiomers of NpOH, while a larger k22− value was observed for (S)-NpOH when compared to (R)-NpOH (Table 2). The faster dissociation of the 2:2 complexes for (S)-NpOH is responsible for the faster relaxation kinetics and the lower equilibrium constants observed for this enantiomer.
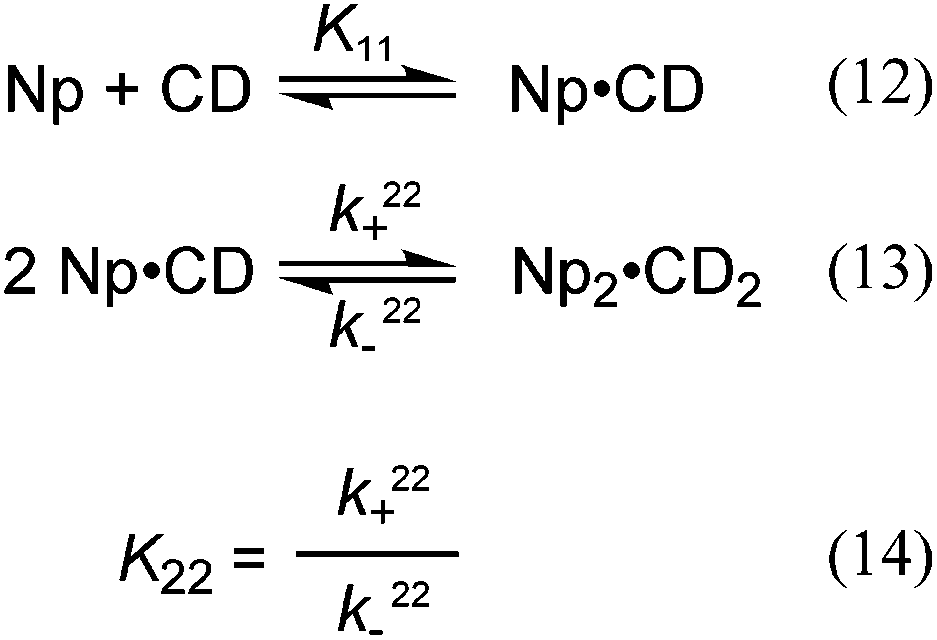 | ||
| Scheme 3 | ||
:2 complexes of the enantiomers
| NpOH | k 22+/105 M−1 s−1 | k 22−/102 s−1 |
|---|---|---|
| (R)- | 6.9 ± 0.3 | 1.11 ± 0.05 |
| (S)- | 7.2 ± 0.4 | 1.63 ± 0.02 |
A relationship can be obtained between the rate constants in Table 2 and the rate constants for the formation of the EE complex (Scheme 2) when it is assumed that the equilibrium constants for the formation of the three 2:2 complexes are the same (eqn (15)–(18), see ESI† for derivation).
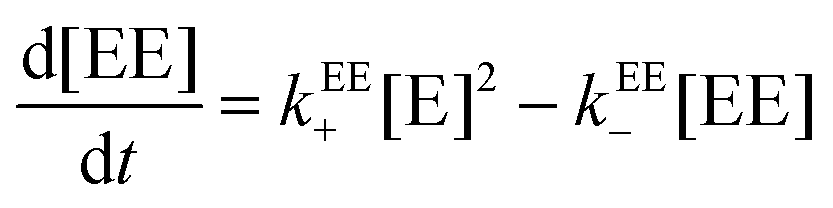 | (15) |
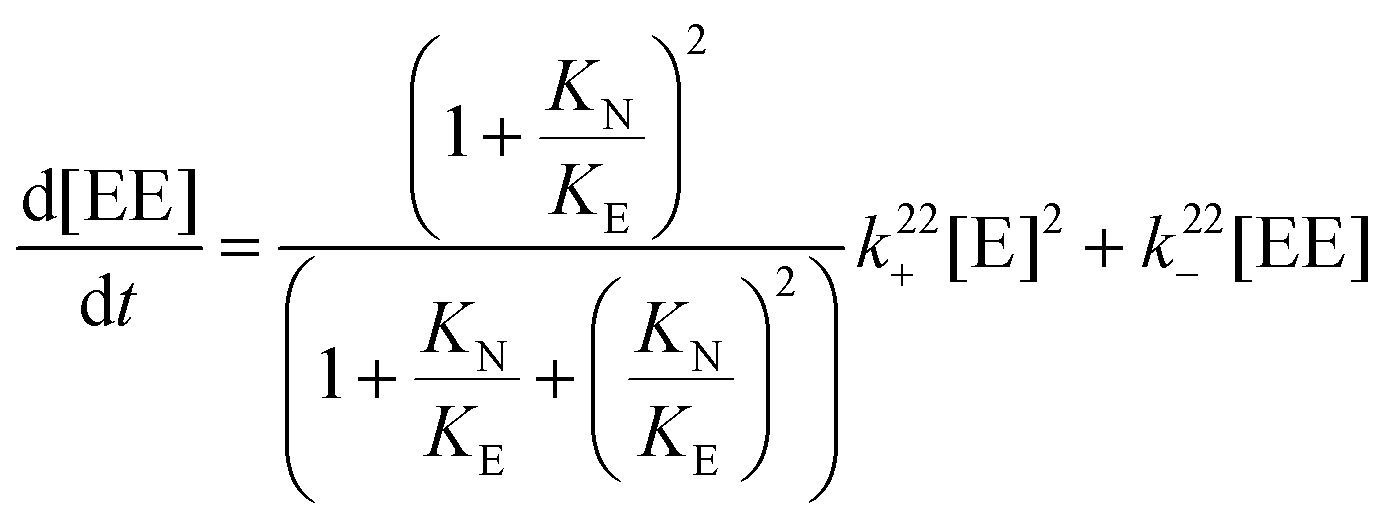 | (16) |
The relationship between the association and dissociation rate constants for the EE complex and the experimentally determined parameters k22+ and k22− are:
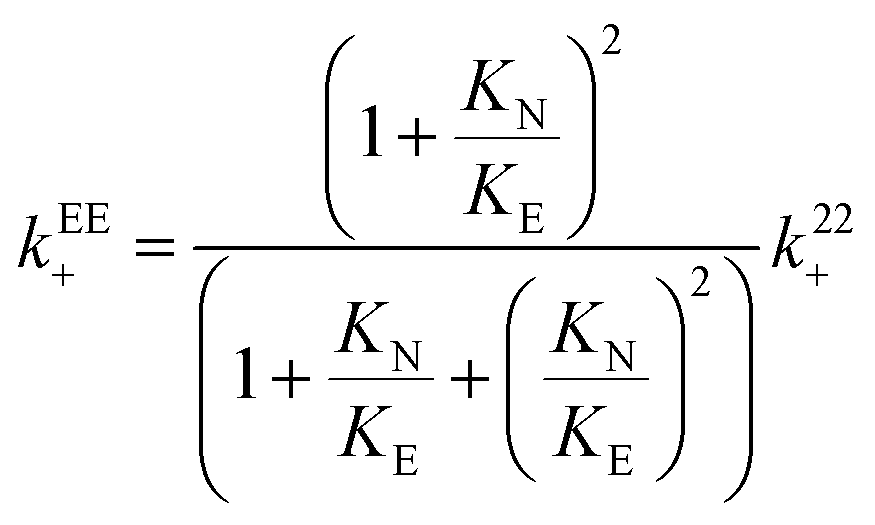 | (17) |
| kEE− = k22− | (18) |
The value for the dissociation rate constant is independent of the magnitude of KE with respect to KN for the two enantiomers. Therefore, the higher dissociation rate constant for (S)-NpOH does not depend on the exact value for KE. The value for kEE+ is minimum when KE is equal to K11 ((6.9 ± 0.3) × 105 M−1 s−1 for (R)-NpOH and (7.2 ± 0.4) × 105 M−1 s−1 for (S)-NpOH), while the upper limit is reached when KE and KN are equal ((9.2 ± 0.4) × 105 M−1 s−1 for (R)-NpOH and (9.6 ± 0.5) × 105 M−1 s−1 for (S)-NpOH). The values for kEE+ are the same within experimental errors for both enantiomers for the same KE value assumed. The values for the equilibrium constant for the EE complex can be calculated from the association and dissociation rate constants (eqn (11), Scheme 2) and these calculated KEE values are the same as those obtained from the binding isotherms based on the variation of the steady-state emission intensities (Table S1 in the ESI†). This equality shows that the analysis of the kinetic study and the binding studies are self-consistent.
Discussion
CDs have been used as chiral selectors in a variety of analytical techniques.32–34 The potential of CDs as chiral selectors was realized early on when these hosts were used for the partial chiral discrimination of racemates.35,36 This discrimination was attributed to differences in the efficiencies for the formation of the enantiomer–CD complexes. Preferential precipitation of one enantiomer through formation of a 1:2 (guest–CD) complex was shown to be due to a combination of differential binding constants and differential inherent solubilities of the enantiomers.37 The chiral discrimination of natural CDs is in general low,12,38 and the ability to discriminate between enantiomers of guests is attributed to binding of the guest within the cavity and its interactions with the rim of the CDs.39,40 Chiral recognition is improved for modified CDs because these modifications introduce asymmetries to the host.3,41 Chiral discrimination is also improved when higher order CD complexes are formed.42 In general, CDs are poor in discriminating for guests with central chirality, and better discrimination is observed when the stereocenters are located axially or the guests are helical.43 Hydrogen bonding is not a pre-requisite, as chirality was achieved by asymmetric “twisting” of the guests within the chiral CD. For example, circularly polarized luminescence was observed for the excimer of pyrene in γ-CD.44 There is no direct relationship between the thermodynamic parameters for formation of 1:1 complexes and the degree of chiral recognition observed for different chiral guests.38
Asymmetric photoreactions were achieved for various systems in CDs.2,3 Most examples are related to unimolecular reactions in 1:1 complexes. Different outcomes were observed when benzaldehyde was photolyzed in the solid state with different CDs. For the 1:1 complex with β-CD an enantiomeric excess for the chiral product was observed, while no chiral discrimination was observed for the 2:1 (guest–CD) complex with γ-CD.45 The rate of bimolecular photocyclizations reactions for CD complexes containing two guests is frequently enhanced.2 In the case of anthracene derivatives the enhancement was observed for the 2:2 complexes with β-CD and 2:1 complexes with γ-CD.46–48 Photodimerization of 2-anthracenecarboxylate in γ-CD occurred immediately upon excitation leading to moderate enantiomeric excesses.49 These examples show that higher order complexes containing more than one CD play a role in the chiral discrimination achieved with CDs.
The objective of this study was to determine the origin for the chiral discrimination of a system with different host–guest stoichiometries (1:1 vs. 2:2) and with different geometries for the 1:1 and 2:2 complexes (E vs. N and EE vs. EN and NN). The chiral discrimination observed for this system is modest, but the importance of this study is to understand the mechanism of the chiral discrimination for a complex system where several host–guest complexes co-exist. The formation of these five complexes was established previously.19,22 The chiral discrimination for the NpOH–β-CD system appears as a higher excimer emission intensity for (R)-NpOH and a faster relaxation kinetics for (S)-NpOH. The higher excimer intensity indicates that for (R)-NpOH a higher concentration of EE was formed than for (S)-NpOH. It is important to note that the observed rate constant of a relaxation process corresponds to the sum of the association and dissociation processes and either the association or the dissociation process (or both) could be faster for (S)-NpOH when compared to (R)-NpOH.
No chiral discrimination was observed for the values of K11, which corresponds to the sum of KE and KN. Only one singlet excited state lifetime was observed for the NpOHs in the 1:1 complexes and the intensity increase for the fluorescence and the lengthening of the lifetimes for the 1:1 complexes were the same for both enantiomers. A similar result was previously observed for the binding of the enantiomers of 1-naphthyl-1-ethylamine with β-CD as a 1:1 complex.50 The observation of one lifetime for the 1:1 complexes indicates that E and N cannot be differentiated spectroscopically and that the environment for the chromophore in E and N is similar for (R)-NpOH and (S)-NpOH. Therefore, the binding isotherms do not provide information on the ratio between E and N for each one of the enantiomers of NpOH. Analysis of the binding isotherm for the formation of the EE complex (Fig. 4) showed that parameters KE and CEE are correlated (Fig. S5†). CEE corresponds to the relative emission efficiency for the EE complex. CEE can be assumed to be the same for both enantiomers because the lifetimes for the excimer emission of (R)-NpOH and (S)-NpOH in the 2:2 complexes were the same. For a constant CEE value, the value of KE for (R)-NpOH is always higher than for (S)-NpOH (Fig. S5). Since the K11 values are the same for both enantiomers (1100 M−1) the values of KE can be estimated to be between 600 and 700 M−1 for (R)-NpOH and 400 and 450 M−1 for (S)-NpOH. This analysis shows that for (R)-NpOH there is a preference for the formation of the E complex over the N complex.
The kinetics for the formation of EE probed the formation of all 2:2 complexes because all equilibria are coupled. No growth kinetics was observed for the excimer emission suggesting that its formation occurred instantly upon excitation in the same fashion as for the photodimerization reaction of 2-anthracenecarboxylate in γ-CD.49 The structure of the EE complexes are likely to be similar for both NpOH enantiomers, since the same lifetimes were observed for their excimer emissions. In addition, the structure for the EN and NN complexes are also similar because the same quenching rate constants were observed for the triplet excited states of the NpOHs in these complexes. A comparable observation was reported for the binding of the enantiomers of ketoprofen with β-CD in a 1:1 (guest–CD) complex where the binding constants and the decarboxylation kinetics of the excited states were the same and subtle structural changes were only observed by circular dichroism and NMR experiments.51 These results suggest that subtle changes in the structure of the complexes are not always detectable in photophysical experiments.
The concentration of EE was higher for (R)-NpOH than for the (S)-enantiomer as the pre-exponential factor for the excimer emission was higher for the former guest. Consequently, the pre-exponential factor for the monomer emission, which includes the EN and NN complexes, was higher for the (S)-enantiomer. The same concentrations within experimental errors were observed for the sum of EN and NN for both enantiomers from the analysis of the triplet excited state decays, but the errors in these experiments were too large, ca. 25%, to be conclusive. The kinetics for EE formation showed only one relaxation time for both NpOH enantiomers. This result is consistent with similar dynamics for the formation of the three 2:2 complexes (EE, EN and NN). Simulations showed that the differences in the association and dissociation rate constants for the various 2:2 complexes would have to be larger than 25% for more than one relaxation process to be observed in the kinetics for the formation of EE. Based on this analysis we assumed that the equilibrium for all 2:2 complexes were the same. Therefore, the higher excimer intensity observed for (R)-NpOH when compared to (S)-NpOH is because of a higher concentration of all 2:2 complexes.
The dissociation rate constant for the 2:2 complexes of (S)-NpOH was higher than for (R)-NpOH. It is important to note that this result is independent of the KE value assumed. The latter parameter only affects the magnitude of the association rate constants. The values for the overall association rate constants (k22+) were the same for (R)-NpOH and (S)-NpOH, suggesting that the association of the E and N complexes leading to the formation of the 2:2 complexes is not responsible for the chiral discrimination observed. The faster dissociation rate constants for the 2:2 complexes with (S)-NpOH suggest that these complexes are less stable than the ones formed with (R)-NpOH. This difference could be related to the alignment of the secondary hydroxyl groups at the rim of the CDs that interact in the 2:2 complex. This alignment is more favorable in the case of (R)-NpOH. The chiral recognition is not primary related to the difference in the E and N complexes because the overall equilibrium constant for the formation of the 1:1 complexes is the same for both enantiomers. E is preferred for (R)-NpOH, while N is the preferred complex for (S)-NpOH, but the sum of both is the same for each enantiomer. This result maybe a reflection of the similar energies for these two complexes.22
Conclusions
The origin for the chiral discrimination in a system containing two 1:1 and three 2:2 guest–host complexes was investigated with the same guest and host for the NpOH–β-CD system. The insights for this work is that for complex systems an interplay of factors affects the chiral discrimination and that kinetic studies can be instrumental to understand such systems. No chiral discrimination was observed for equilibrium constants of the 1:1 complexes between (R)- or (S)-NpOH with β-CD. However, the analysis of the binding isotherm for the 1:1 and 2:2 complexes suggests that the formation of one of the 1:1 complexes is higher for each one of the enantiomers (E complex for (R)-NpOH and N complex for (S)-NpOH), while the sum of both complexes is the same. The dynamics for the formation of the three 2:2 complexes (EE, EN and NN) were similar leading to one relaxation process being observed in the kinetic studies. The faster relaxation kinetics observed for (S)-NpOH is because the 2:2 complexes for this enantiomer dissociate faster. This result shows that the chiral discrimination observed is primarily dictated by the stability of the 2:2 complexes and not by the structural diversity of the 1:1 complexes. This system is an example where the formation of higher order complexes is essential for the observation of the chiral discrimination of enantiomers.
Acknowledgements
The Natural Sciences and Engineering Council of Canada in the form of Discovery (DG) and Research Tools and Instruments (RTI) grants funded this work.Notes and references
- C. Müller and T. Bach, Chirality Control in Photochemical Reactions: Enantioselective Formation of Complex Photoproducts in Solution, Aust. J. Chem., 2008, 61, 557–564 CrossRef.
- T. Wada and Y. Inoue, in Chiral Photochemistry, ed. Y. Inoue and V. Ramamurthy, Marcel Dekker, New York, 2004, pp. 341–384 Search PubMed.
- C. Yang and Y. Inoue, in Supramolecular Photochemistry: Controlling Photochemical Processes, ed. V. Ramamurthy and Y. Inoue, John Wiley & Sons, Singapore, 2011, pp. 115–153 Search PubMed.
- D. Fuentealba, H. Kato, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Bohne, Explaining the Highly Enantiomeric Photocyclodimerization of 2-Anthracenecarboxylate Bound to Human Serum Albumin Using Time-Resolved Anisotropy Studies, J. Am. Chem. Soc., 2013, 135, 203–209 CrossRef CAS PubMed.
- M. Nishijima, T. C. S. Pace, A. Nakamura, T. Mori, T. Wada, C. Bohne and Y. Inoue, Supramolecular Photochirogenesis with Biomolecules. Mechanistic Studies on the Enantiodifferentiation for the Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Bovine Serum Albumin, J. Org. Chem., 2007, 72, 2707–2715 CrossRef CAS PubMed.
- M. Nishijima, T. Wada, T. Mori, T. C. S. Pace, C. Bohne and Y. Inoue, Highly Enantiomeric Supramolecular [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Human Serum Albumin, J. Am. Chem. Soc., 2007, 129, 3478–3479 CrossRef CAS PubMed.
- T. C. S. Pace, M. Nishijima, T. Wada, Y. Inoue and C. Bohne, Photophysical Studies on the Supramolecular Photochirogenesis for the Photocyclodimerization of 2-Anthracenecarboxylate within Human Serum Albumin, J. Phys. Chem. B, 2009, 113, 10445–10453 CrossRef CAS PubMed.
- C. V. Kumar, A. Buranaprapuk and H. C. Sze, Large Chiral Discrimination of a Molecular Probe by Bovine Serum Albumin, Chem. Commun., 2001, 297–298 RSC.
- P. Bortolus and S. Monti, Photochemistry in Cyclodextrin Cavities, Adv. Photochem., 1996, 21, 1–133 CrossRef CAS.
- K. A. Connors, The Stability of Cyclodextrin Complexes in Solution, Chem. Rev., 1997, 97, 1325–1357 CrossRef CAS PubMed.
- J. Szejtli, Introduction and General Overview of Cyclodextrin Chemistry, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Complexation Thermodynamics of Cyclodextrins, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- A. Ueno, K. Takahashi and T. Osa, One Host-Two Guests Complexation Between γ-Cyclodextrin and Sodium α-Naphthylacetate as shown by Excimer Fluorescence, J. Chem. Soc., Chem. Commun., 1980, 921–922 RSC.
- S. Hamai, Association of Inclusion Compounds of β-Cyclodextrin in Aqueous Solution, Bull. Chem. Soc. Jpn., 1982, 55, 2721–2729 CrossRef CAS.
- S. Hamai, The Excimer Fluorescence of 2-Methylnaphthalene in β- and γ-Cyclodextrin Aqueous Solution, Bull. Chem. Soc. Jpn., 1996, 69, 543–549 CrossRef CAS.
- S. Hamai and N. Mononobe, The Room-Temperature Phosphoresence of 2-Chloronaphthalene Induced by Inclusion Complexation with 6-Iodo-6-deoxy-β-cyclodextrin and the Excimer Fluorescence of a β-Cyclodextrin-2-chloronaphthalene Inclusion Complex in Aqueous Solution, J. Photochem. Photobiol., A, 1995, 91, 217–221 CrossRef CAS.
- S. Hamai, Inclusion of Methyl 2-Naphthalenecarboxylate and Dimethyl 2,3-, 2,6-, and 2,7-Naphthalenedicarboxylates by Cyclodextrins in Aqueous Solution, Bull. Chem. Soc. Jpn., 2010, 83, 1489–1500 CrossRef CAS.
- S. Hamai and A. Hatamiya, Excimer Formation in Inclusion Complexes of β-Cyclodextrin with 1-Alkylnaphthalenes in Aqueous Solutions, Bull. Chem. Soc. Jpn., 1996, 69, 2469–2476 CrossRef CAS.
- T. C. Barros, K. Stefaniak, J. F. Holzwarth and C. Bohne, Complexation of Naphthylethanols with β-Cyclodextrin, J. Phys. Chem. A, 1998, 102, 5639–5651 CrossRef CAS.
- G. Grabner, K. Rechthalter, B. Mayer and G. Köhler, Solvent Influences on the Photophysics of Naphthalene: Fluorescence and Triplet State Properties in Aqueous Solutions and in Cyclodextrin Complexes, J. Phys. Chem. A, 2000, 104, 1365–1376 CrossRef CAS.
- S. Sau, B. Solanki, R. Orprecio, J. van Stam and C. H. Evans, Higher-order Cyclodextrin Complexes: The Naphthalene System, J. Inclusion Phenom. Macrocyclic Chem., 2004, 48, 173–180 CrossRef CAS.
- R. S. Murphy, T. C. Barros, B. Mayer, G. Marconi and C. Bohne, Photophysical and Theoretical Studies on the Stereoselective Complexation of Naphthylethanols with β-Cyclodextrin, Langmuir, 2000, 16, 8780–8788 CrossRef CAS.
- S. De Feyter, J. van Stam, F. Imans, L. Viaene, F. C. De Schryver and C. H. Evans, Observation of α-Terthiophene Excited Dimer Fluorescence in Aqueous Solutions of γ-Cyclodextrin, Chem. Phys. Lett., 1997, 277, 44–50 CrossRef CAS.
- E. Janata and R. H. Schuler, Rate Constant for Scavenging e–aq in N2O-Saturated Solutions, J. Phys. Chem., 1982, 86, 2078–2084 CrossRef CAS.
- L. T. Okano, R. Ovans, V. Zunic, J. N. Moorthy and C. Bohne, Effect of Cyclodextrin Complexation on the Photochemistry of the Lignin Model α-Guaiacoxyacetoveratrone, Can. J. Chem., 1999, 77, 1356–1365 CrossRef CAS.
- C. Bohne, R. W. Redmond and J. C. Scaiano, in Photochemistry in Organized and Constrained Media, ed. V. Ramamurthy, VCH Publishers, New York, 1991, pp. 79–132 Search PubMed.
- Y. Liao and C. Bohne, Alcohol Effect on Equilibrium Constants and Dissociation Dynamics of Xanthone-Cyclodextrin Complexes, J. Phys. Chem., 1996, 100, 734–743 CrossRef CAS.
- L. T. Okano, T. C. Barros, D. T. H. Chou, A. J. Bennet and C. Bohne, Complexation Dynamics of Xanthone and Thioxanthone to β-Cyclodextrin Derivatives, J. Phys. Chem. B, 2001, 105, 2122–2128 CrossRef CAS.
- T. C. S. Pace and C. Bohne, Temperature Effect on Xanthone/β-cyclodextrin Binding Dynamics, Can. J. Chem., 2011, 89, 395–401 CrossRef CAS PubMed.
- A. Treinin and E. Hayon, Quenching of Triplet States by Inorganic Ions. Energy Transfer and Charge Transfer Mechanisms, J. Am. Chem. Soc., 1976, 98, 3884–3891 CrossRef CAS.
- O. Rinco, M.-C. Nolet, R. Ovans and C. Bohne, Probing the Binding Dynamics to Sodium Cholate Aggregates using Naphthalene Derivatives as Guests, Photochem. Photobiol. Sci., 2003, 2, 1140–1151 CAS.
- S. Li and W. C. Purdy, Cyclodextrins and Their Applications in Analytical Chemistry, Chem. Rev., 1992, 92, 1457–1470 CrossRef CAS.
- S. Fanali, Enantioselective determination by capillary electrophoresis with cyclodextrins as chiral selectors, J. Chromatogr., A, 2000, 875, 89–122 CrossRef CAS.
- Z. Juvancz, R. B. Kendrovics, R. Ivanyi and L. Szente, The role of cyclodextrins in chiral capillary electrophoresis, Electrophoresis, 2008, 29, 1701–1712 CrossRef CAS PubMed.
- F. Cramer and W. Dietrsche, Spaltung von Racematen mit Cyclodextrinen, Chem. Ber., 1959, 92, 378–384 CrossRef CAS.
- M. Mikolacczyk and J. Drabowicz, Optical Resolution of Chiral Sulfinyl Compounds via β-Cyclodextrin Inclusion complexes, J. Am. Chem. Soc., 1978, 100, 2510–2515 CrossRef.
- H. Bakirc and W. M. Nau, Chiral Resolution through Precipitation of Diastereomeric Capsules in the Form of 2:1 β-Cyclodextrin-Guest Complexes, J. Org. Chem., 2005, 70, 4506–4509 CrossRef PubMed.
- M. Rekharsky and Y. Inoue, Chiral Recognition Thermodynamics of β-Cyclodextrin: The Thermodynamic Origin of Enantioselectivity and the Enthalpy-Entropy Compensation Effect, J. Am. Chem. Soc., 2000, 122, 4418–4435 CrossRef CAS.
- W. L. Hinze and T. E. Riehl, Liquid Chromatographic Separation of Enantiomers Using a Chiral β-Cyclodextrin Bonded Stationary Phase and Conventional Aqueous-Organic Mobile Phase, Anal. Chem., 1985, 57, 237–242 CrossRef CAS.
- D. W. Armstrong, T. J. Ward, R. Armstrong and T. E. Beeley, Separation of Drug Stereoisomers by the formation of β-Cyclodextrin Inclusion Complexes, Science, 1986, 232, 1132–1135 CAS.
- C. J. Easton and S. F. Lincoln, Chiral Discrimination by Modified Cyclodextrins, Chem. Soc. Rev., 1996, 25, 163–170 RSC.
- P. Bortolus, G. Marconi, S. Monti and B. Mayer, Chiral Discrimination of Camphorquinone Enantiomers by Cyclodextrins: A Spectroscopic and Photophysical Study, J. Phys. Chem. A, 2002, 106, 1686–1694 CrossRef CAS.
- K. Kano, Mechanism for Chiral Recognition by Cyclodextrins, J. Phys. Org. Chem., 1997, 10, 286–291 CrossRef CAS.
- K. Kano, H. Matsumoto, S. Hashimoto, M. Sisido and Y. Imanishi, Chiral Pyrene Excimer in the γ-Cyclodextrin Cavity, J. Am. Chem. Soc., 1985, 107, 6117–6118 CrossRef CAS.
- V. P. Rao and N. J. Turro, Asymmetric Induction in Benzoin by Photolysis of Benzaldehyde Adsorbed in Cyclodextrin Cavities, Tetrahedron Lett., 1989, 30, 4641–4644 CrossRef CAS.
- T. Tamaki and T. Kokubu, Acceleration of the Photodimerization of Water-Soluble Anthracenes Included in β- and γ-Cyclodextrins, J. Inclusion Phenom., 1984, 2, 815–822 CrossRef CAS.
- T. Tamaki, T. Kokubu and K. Ichimura, Regio- and Stereoselective Photodimerization of Anthracene Derivatives Included by Cyclodextrins, Tetrahedron, 1987, 43, 1485–1494 CrossRef CAS.
- T. Tamaki, Reversible Photodimerization of Water-soluble Anthracenes include in γ-Cyclodextrin, Chem. Lett., 1984, 53–56 CrossRef CAS.
- A. Nakamura and Y. Inoue, Supramolecular Catalysis of the Enantiodifferentiating [4 + 4] Photocyclodimerization of 2-Anthracene by γ-Cyclodextrin, J. Am. Chem. Soc., 2003, 125, 966–972 CrossRef CAS PubMed.
- C. D. Tran and J. H. Fendler, Photophysical Investigations of Chiral Amine Guest-Cyclodextrin Host Interaction and Diastereomeric Recognition, J. Phys. Chem., 1984, 88, 2167–2173 CrossRef CAS.
- G. Marconi, E. Mezzina, I. Manet, F. Manoli, B. Zambelli and S. Monti, Stereoselective interaction of ketoprofen enantiomers with β-cyclodextrin: ground state binding and photochemistry, Photochem. Photobiol. Sci., 2011, 10, 48–59 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of 1:1 equilibrium constants, decays for the triplet excited states of NpOH, analysis of the binding isotherms for the 2:2 complexes, dependence of CEE with KEE, stopped-flow kinetics for the formation of EE, simulation of the stopped-flow experiments, global analysis for the 2:2 formation kinetics, derivation of the dependence of the overall 2:2 kinetics and the kinetics for EE formation. See DOI: 10.1039/c3pp50298h |
| This journal is © The Royal Society of Chemistry and Owner Societies 2014 |