 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fructose controlled ionophoric activity of a cholate–boronic acid†
James R. D.
Brown
,
Inmaculada C.
Pintre
and
Simon J.
Webb
*
Manchester Institute of Biotechnology (MIB) and School of Chemistry, University of Manchester, 131 Princess St., Manchester M1 7DN, UK. E-mail: S.Webb@manchester.ac.uk
First published on 27th February 2014
Abstract
Wulff-type boronic acids have been shown to act as ionophores at pH 8.2 by transporting Na+ through phospholipid bilayers. A cholate–boronic acid conjugate was synthesised and shown to be an ionophore, although the hydroxyl-lined face of the cholate moiety did not enhance ion transport. Mechanistic studies suggested a carrier mechanism for Na+ transport. The addition of fructose (>5 mM) strongly inhibited ionophoric activity of the cholate–boronic acid conjugate, mirrored by a strong decrease in the ability of this compound to partition into an organic phase. Modelling of the partitioning and ion transport data, using a fructose/boronic acid binding constant measured at pH 8.2, showed a good correlation with the extent of fructose/boronic acid complexation and suggested high polarity fructose/boronic acid complexes are poor ionophores. The sensitivity of ion transport to fructose implies that boronic acid-based antibiotic ionophores with activity modulated by polysaccharides in the surrounding environment may be accessible.
Introduction
The recognition of polyols, in particular saccharides, by boronic acids is an example of a dynamic covalent chemistry that has provided a wealth of applications in supramolecular chemistry, with recent examples in sensing,1 drug delivery,2 separation3 and hydrogel formation.4 The interaction of lipophilic or membrane-bound boronic acids with saccharides or glycolipids has risen to particular prominence in recent years,5,2b driven in part by the development of 2-(hydroxymethyl)phenylboronic acids able to bind a wider range of sugars at close to physiological conditions.6 Lipophilic boronic acids have also been known for some time to also transport saccharides, particularly fructose, across supported membranes,7 bulk organic phases8 and phospholipid bilayers.9Amphiphilic boronic acids are suggested to transport saccharides by forming complexes that are sufficiently lipophilic to diffuse across the membrane. Two mechanisms have been proposed for boronic acid-mediated diol transport across membranes (Fig. 1), with transport either occurring through trigonal planar boronate esters or tetrahedral boronate anions; the pH of the sending phase and boronic acid pKa determines which mechanism is followed.10 A cation is associated with tetrahedral boronate anions, which can be covalently linked to the boronic acid, an added lipophilic tetraalkylammonium salt, or it can be a counterion symported with the sugar from the sending phase.7,11 In the latter case a crown ether is often added to form a lipophilic complex able to pass through the apolar membrane phase.10a Despite the implication that cation transport is concomitant with saccharide transport transport,7,9b,11 boronic acid mediated M+ transport across phospholipid bilayers has not been explicitly measured and compared to the rate of concomitant saccharide transport.
 | ||
Fig. 1 Scheme showing the transport of boronic acid–diol complexes through bilayers either as (a) trigonal planar or (b) tetrahedral boron species. (c) Main 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 boronate complex formed with fructose.12 :1 boronate complex formed with fructose.12 | ||
The ability of lipophilic boronic acids embedded in phospholipid bilayers to complex sugars suggests new ways of controlling ion flux across phospholipid bilayers, with ion flow across the membrane controlled by the presence of biological catechols13 or saccharides.14 Saccharides are ideal for controlling ion transport as these highly polar molecules cannot cross the phospholipid bilayer, they will not discharge a pH gradient and unlike lipophilic polyols they will not disrupt bilayer membranes. Although the affinities of sugars for most boronic acids is generally rather weak, some sugars, such as fructose, exhibit binding constants with phenylboronic acids in buffer in the order of 100 M−1.
Previously we developed cholate-based ionophores that can be turned on or off by complexation to metal ions or proteins.15 Adding boronic acid mediated recognition/transport with mono- or bis-cholate scaffolds was expected to provide some interesting ionophores, especially as oligocholates are known to transport metal ions and dyes through bilayers, through either channel or carrier mechanisms.16,17 The high activity of cholates stems from their facial amphiphilicity, which provides hydrophobic convex surfaces that can be presented to the bilayer and concave hydrophilic surfaces that can interact with alkali metal ions.
Linking cholates with boronic acids was hoped to produce saccharide-switchable ion transport across phospholipid bilayers. Fructose forms boronic acid complexes with a variety of geometries and stoichiometries,12 including 1:2 fructose to boronic acid,12,18 and we wondered if fructose could assemble boronic acid–cholate conjugates into dimers that might form channels17 or “hairpin” carriers with a sequestered hydrophilic cavity. The compounds we investigated for cation transport ranged from phenylboronic acid 1 and Wulff-type 2-(N-methylaminomethyl)phenylboronic acid 2, to a more amphiphilic Wulff-type boronic acid conjugate 3 that has a single facially amphiphilic cholate tail (Fig. 2).
 | ||
| Fig. 2 Boronic acids and cholic acid. | ||
Results and discussion
Synthesis of 3
Boronate-cholate derivative 3 was synthesised in two steps from cholic acid 4. The short triethylene glycol spacer was added between the cholate body and the boronic acid to give flexibility around the complexation site and to project it away from the cholate, which will become embedded in the bilayer. Cholic acid 4 was first coupled to mono-protected 1-(benzyloxycarbonylamino)-3,6-dioxa-8-aminooctane, the product hydrogenolysed, then reductively aminated with 2-formylphenylboronic acid to give compound 3 in 59% yield from the acid.Ion transport assays
To screen the ability of these compounds to transport cations through bilayers we chose the phospholipid vesicle based 8-hydroxypyrenetrisulfonate (HPTS pKa = 7.3) assay,19 using an interior pH of 6.5 and an exterior pH of 8.5. The exterior pH was selected to be above the pKa of aminomethylboronic acids (pKa ∼ 6.5)7 and corresponding saccharide complexes (pKa < 6.5),20 a strategy designed to favour M+/OH− co-transport. EYPC/cholesterol vesicles were used as these bilayers have the right balance between fluidity and impermeability. Fluidity allows assembly in the membrane and diffusion through the membrane, but pure EYPC bilayers gave unsatisfactory levels of background leakage.The choice of buffer was crucial as boronic acid complexation of diols is strongly pH dependent, with the ideal pH between the pKa of the diol and the boronic acid.21 Buffer type and concentration can also strongly affect the binding of boronic acids to saccharides, with stronger binding at low buffer concentrations.22 To obtain precise pH control, the standard procedure for the HPTS assay was modified, with HPTS-loaded vesicles directly added to buffer at pH 8.5 rather than addition of an external aliquot of NaOH (the base pulse). The ionophore was then added after 180 s to initiate transport, which also eliminates the jump in fluorescence observed if the base pulse is applied after ionophore addition.
Initially cholate–boronic acid 3 was assayed for its ability to allow sodium ions to cross a bilayer. Addition of cholate–boronic acid 3 (10 μM) to give a membrane loading of 5% mol/mol gave an immediate increase in fluorescence that suggested Na+/H+ antiport or Na+/OH− symport across the bilayer of these EYPC/chol vesicles. Cholic acid 4 (pKa ∼ 4.6)23 did not conduct sodium ions across the membrane and gave little transport above the background methanol control (Fig. 3).
 | ||
Fig. 3 Transport of Na+ across EYPC/chol bilayers by boronic acid 2 ( ), boronate-cholate 3 ( ), boronate-cholate 3 ( ) and cholate 4 ( ) and cholate 4 ( ). The methanol control has been subtracted from the data. First order curve fits are to guide the eye. ). The methanol control has been subtracted from the data. First order curve fits are to guide the eye. | ||
To confirm that the boronic acid is the active moiety in mediating ion transport, phenylboronic acid 1 and 2-(N-methylaminomethyl)phenylboronic acid 2 were also assessed for their ability to transport Na+ through an EYPC/chol bilayer. Phenylboronic acid (10 μM, 5% mol/mol) did not transport sodium ions above the background rate, but the 2-(N-methylaminomethyl) derivative 2 (10 μM, 5% mol/mol) gave good transport of Na+ at a rate above that observed for cholate boronic acid 3. This observation suggests that the cholate hydroxyls play little role in assisting ion transport and the sodium ion is co-transported in close proximity to an anionic boronate headgroup. The pKa of 2 is ∼6.5,7 and at pH 8.5 the major species in solution will be the tetrahedral boronate anion, but phenylboronic acid 1 has a higher pKa of 8.8,21a,22 suggesting that at pH 8.5 the major boron species in solution will be uncharged; this trigonal planar boronic acid would be unable to antiport Na+/H+ or symport Na+/OH− across the membrane.
Effect of fructose on ion transport
Based upon literature binding constants (K from 100 to 500 M−1) in methanol–buffer mixtures at near neutral pHs,22,24,25 we hoped that 100 mM fructose would be sufficient to give extensive complexation of the boronic acid headgroups in these ionophores. The effect of this concentration of fructose on ion transport by ionophore 3 was assessed using the HPTS assay. During formation and purification of the EYPC/chol vesicles, all pH 6.5 buffer solutions also contained 100 mM fructose, maintaining the osmotic balance across the vesicle membrane. As previously, an aliquot of this vesicle solution was transferred to iso-osmotic buffer with 100 mM fructose at pH 8.5 before ionophore 3 was added at 180 s. Under these conditions, little Na+ transport was observed, with discharge of the pH gradient only marginally above the methanol control (Fig. 4a). As expected the low level of transport exhibited by cholate control 4 was found to be little affected by the addition of fructose (ESI Fig. S3†). A similar switching off of ion transport was observed for boronate 2 in the presence of 100 mM fructose, showing that complexation to fructose diminished the ability of these Wulff-type boronic acids to act as ionophores. | ||
Fig. 4 (a) Transport of Na+ across EYPC/chol bilayers by 2 ( ), 2 with 100 mM fructose ( ), 2 with 100 mM fructose ( ), 3 ( ), 3 ( ) and 3 with 100 mM fructose ( ) and 3 with 100 mM fructose ( ). First order curve fits are to guide the eye. (b) Plot of Na+ transport by 3 after 1200 s at different concentrations of fructose. Curve fitted using eqn (2) with K = 400 M−1. ). First order curve fits are to guide the eye. (b) Plot of Na+ transport by 3 after 1200 s at different concentrations of fructose. Curve fitted using eqn (2) with K = 400 M−1. | ||
If the complexation of the boronic acid group to fructose blocks the activity of these ionophores, then the rate of Na+ transport should depend on the fructose concentration. Ion transport by 3 was monitored in the presence of 2.5 mM, 5 mM, 10 mM and 100 mM fructose, which revealed a steady decrease in activity as the concentration of fructose increased. A plot of the extent of transport after 1200 seconds against the concentration of fructose showed an inverse dependence, a relationship that mirrors the fraction of free ionophore 3 expected at different concentrations of fructose (Fig. 4b).
Mechanistic investigations
Smith and co-workers have shown that boronic acids transport saccharides across bilayers via a carrier mechanism, which implies a similar mechanism for Na+ transport by 2 and 3. A classical test for an ion carrier mechanism is the ability to transport ions through a bulk organic phase in a U-tube experiment.19 The boronic acids 2 and 3 were assessed for their ability to transport sodium picrate across a chloroform phase, using 0.01% wt/vol sodium picrate in sodium phosphate buffer (2.5 mL, 20 mM phosphate, 100 mM NaCl, pH 8.5) in the sending phase and sodium phosphate buffer (2.5 mL, 20 mM phosphate, 100 mM NaCl, pH 6.5) in the receiving phase. In addition, cholic acid 4 and dibenzo-18-crown-6 were also assessed for their ability to transport sodium picrate. Boronic acid 3 (1 mM) transported sodium picrate through the chloroform layer at a rate comparable to dibenzo-18-crown-6 (respectively 1% and 3% picrate transport after 6 h, ESI Fig. S5†).26 As expected, cholate 4, lacking the boronic acid, was unable to transport sodium picrate through the organic phase (∼0.07% after 6 h). Surprisingly, boronic acid 2 was also relatively ineffective (0.2% after 6 h), which was ascribed to its fairly high solubility in the aqueous phase.Given that lipophilic boronate complexes are known to transport fructose through organic phases,7–9 the relationship between fructose transport and Na+ transport was also assessed. The ability of ionophore 3 to transport fructose/Na+ through the organic CHCl3 phase was assessed using a U-tube and a combination of 1H-NMR analysis (fructose transport) with UV-visible spectroscopy (Na+ transport). A CDCl3 (99.8 atom % D) solution of 3 (1 mM) was transferred to a glass U-tube. A receiving phase of sodium phosphate buffer (D2O containing 0.75% sodium 3-(trimethylsilyl)-2,2,3,3-d4-propionate salt as an internal standard, 20 mM phosphate, 100 mM NaCl, pH 6.5) was added to one side of the U-tube, and to the other side was added a source phase of fructose (1 M) and 0.01% wt/vol sodium picrate in sodium phosphate buffer (D2O containing 0.75% internal standard, 20 mM phosphate, 100 mM NaCl, pH 8.5). Aliquots were taken at intervals from the receiving phase and analysed by 1H NMR spectroscopy for the presence of fructose, but no fructose transport was detected over 10 h. Similarly, UV-visible measurements of the receiving phase showed a concomitant lack of picrate in the receiving phase, confirming that little Na+ transport was occurring. Both observations suggest that the fructose complex of 3 is too polar to efficiently partition into the apolar phase. The lack of fructose transport by 3 is similar to that observed by Karpa et al. for a cholesterol–phenylboronic acid conjugate, which they ascribed to slow diffusion across the aqueous–organic interface for very lipophilic boronic acids.27
To measure changes in lipophilicity caused by complexation to fructose, the distribution constant (KD) for cholate boronic acid 3 and cholate 4 in the presence of fructose was measured. Solutions of 3 or 4 in 1-octanol (10 mM) were prepared and an equal volume of pH 8.5 buffer was added. The aqueous–organic mixture was vigorously mixed for 180 s before standing for 1 h. To calculate the concentration of 3 or 4 in the organic layer, the UV-visible absorbances (260 nm) of aliquots from the organic layer were measured. The same procedure was repeated for 0.01, 0.1 and 0.5 M fructose in pH 8.5 phosphate buffer. A plot of the buffer/octanol distribution constant (KD) at pH 8.5 for ionophore 3 and cholic acid 4 as a function of changing fructose concentration revealed a strong decrease in the lipophilicity of 3 in the presence of fructose, decreasing from 2.0 in the absence of fructose to 1.2 at concentrations of fructose above 0.1 M. In contrast the pH 8.5 buffer/octanol distribution constant for cholic acid 4 was relatively unaffected by the presence of fructose, and was between 2.2 and 2.6 at all concentrations of fructose tested (Fig. 5a). These observations are consistent with ionophore 3 binding to fructose to produce a complex that is less able to partition into the membrane and mediate transport.
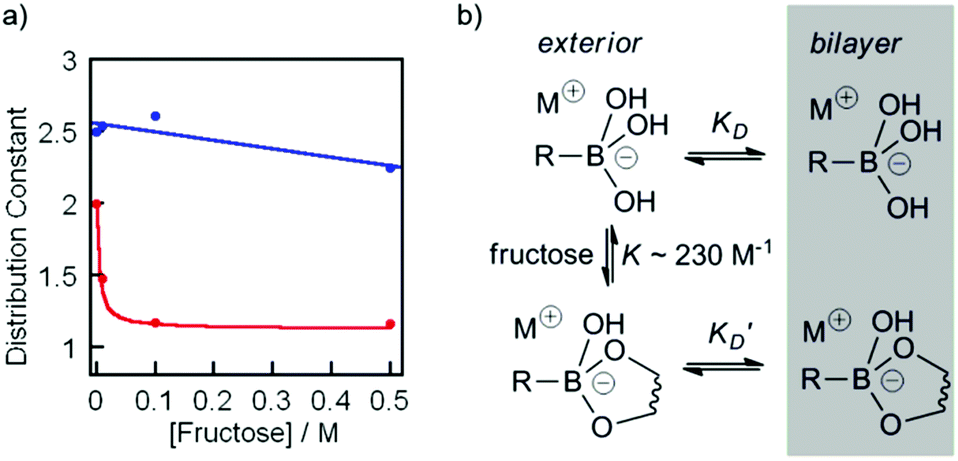 | ||
Fig. 5 (a) Changes in the distribution constant KD for ionophore 3 (red,  ), and cholate 4 (blue, ), and cholate 4 (blue,  ) in the presence of 0 M, 0.01 M, 0.1 M, and 0.5 M fructose. Curve fit added for 3 using eqn (1) and K = 230 M−1, with linear curve fit for 4 to guide the eye. (b) Model mechanism for the partition of 3 into the bilayer in the presence of fructose. ) in the presence of 0 M, 0.01 M, 0.1 M, and 0.5 M fructose. Curve fit added for 3 using eqn (1) and K = 230 M−1, with linear curve fit for 4 to guide the eye. (b) Model mechanism for the partition of 3 into the bilayer in the presence of fructose. | ||
To allow us to model these changes in KD, the binding affinity (K) of compound 2 for fructose was determined under conditions similar to those used for the HPTS assays (pH 8.5). By using pyrocatechol violet in a competitive displacement assay28 and following the method developed by Wang,22 the affinity of 2 for fructose in phosphate buffer (20 mM NanHmPO4, 100 mM NaCl, pH 8.5) was determined as K ∼ 230 M−1. The partition data for fructose + 3 were then fitted to a simple model, where the measured value of the distribution constant KD(obs) will be an average of KD and K′D weighted according to the fraction of free boronate and fructose-boronate complex respectively (Fig. 5b). With the concentration of fructose much greater than the concentration of boronate, this gives the relationship below:
 | (1) |
By approximating the values of KD and K′D from the distribution coefficients of 3 with and without 1 M fructose, combined with the approximate binding constant for 2 to fructose in pH 8.5 buffer, a good fit was found to the distribution constant data using eqn (1) (Fig. 5a).
This analogy was extended by fitting the HPTS responses for 3 after 1200 s (ESI Fig. S4†) to eqn (2). In this relationship, 1 is the normalised response in the absence of fructose, and 0.17 is the estimated normalised response at saturating fructose concentrations.
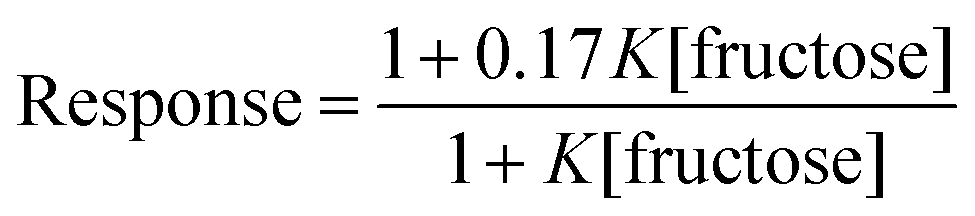 | (2) |
This equation gave a reasonable fit (Fig. 4b) with values of K between 300 and 400 M−1, similar to the measured affinity of 2 for fructose and consistent with complexation of fructose inhibiting of ion transport. This agreement is remarkable given that ion transport recorded by an HPTS assay is a combination of factors, including the rate of ionophore distribution across the population of vesicles.29a
The similarity between the decline in ionophoric activity of 3 with increasing fructose and the decrease in KD with increasing fructose suggests that ionophoric activity is modulated by the ability of the boronate to partition into the bilayer. Support for this suggestion comes from models of carrier-mediated ion transport across bulk organic phases.30 By assuming that transport across a bilayer will be broadly analogous to transport through a bulk organic phase and that low saturation conditions exist at both bilayer–aqueous interfaces, diffusion of the Na+/boronate complex through the buffer–bilayer interface would be the rate limiting step. The rate of transport across the bilayer would then be proportional to the ability of the ionophore to partition into the membrane.
Conclusions
2-(Methylaminomethyl)phenylboronic acids have been shown to transport Na+ across phospholipid bilayers. Boronic acid–cholate conjugate 3 transported Na+ through carrier rather than channel mechanisms despite cholate derivatives being well known to form channels in phospholipid bilayers.16,17 The conjugate 3 was less effective than the smaller 2-(methylaminomethyl)phenylboronic acid 2 at transporting Na+ across bilayers, perhaps due to slower diffusion through the membrane or slower redistribution across the vesicle population,29 but 3 was more effective at transporting across a bulk organic phase. We have also shown that Na+ transport by 2-(methylaminomethyl)phenylboronic acids 2 and 3 is not coupled to saccharide transport, and that fructose complexation instead switches off ion flow. Mechanistic studies suggest that complexation to fructose shuts down ionophoric activity due to the lower lipophilicity of the Na+/saccharide/boronate complexes inhibiting partitioning into the bilayer.The studies presented here may open a pathway towards saccharide-sensitive ionophores, provided that high selectivity for targeted saccharides under physiological conditions6 can be engineered into the boronic acid moiety. Several design rules can be elucidated for the construction of such saccharide sensitive boronic acid ionophores. The boronic acid must be designed so that the pKa lies below the pH of the sending (exterior) solution. It may not be necessary to have a crown ether or other Na+ complexing site appended to the boronic acid for sodium ion transport,11 as the hydroxyl-lined cholate on 3 did not provide greater activity than simple boronic acid 2. Other simple saccharides should produce a different effect on ionophoric activity, depending upon binding constant and complex lipophilicity. Covalently linking together cholates into bis(cholate) dimers17 with boronic acids at both termini did not give any improvement in Na+ transport activity, and these compounds also followed a carrier mechanism with similar activity.31 Transport of other cation/anion combinations are yet to be investigated, although preliminary studies indicate K+ is transported in much the same way as Na+ (ESI Fig. S6†).
Given recently reported biological applications of a new generation of boronates able to complex to polysaccharides under physiological conditions,2b,5e,32 work is ongoing to generate new types of boronic acid-based antibiotics that are responsive to the concentration of targeted polysaccharides in the surrounding environment.
Experimental section
Benzeneboronic acid 1 and cholic acid 4 were purchased from Sigma-Aldrich and used as received. Phospholipids were obtained from Avanti Polar Lipids. Unless otherwise stated, all other reagents and solvents were obtained from commercial suppliers (Sigma-Aldrich, Fluka, Acros and Alfa Aesar). Anhydrous dichloromethane was obtained by distillation from calcium hydride, and anhydrous tetrahydrofuran was obtained by distillation from sodium. Thin layer chromatography (TLC) was carried out using Merck aluminum-backed F254 silica gel plates and visualised with either UV light (256 nm or 365 nm), alkaline aqueous KMnO4, ninhydrin, 2,4-dinitrophenylhydrazine, or cerium molybdate. Preparative column (flash) chromatography was carried out using commercially available normal-phase silica gel. 1-(Benzyloxy carbonylamino)-3,6-dioxa-8-aminooctane and 2-(methylamino-methyl)phenylboronic acid 2 were synthesised according to literature procedures.33,34NMR spectra were measured on Bruker DPX300, AV400, or AMX500 instruments and were assigned using COSY, HMBC, HMQC and DEPT spectra where appropriate. Coupling constants J are given in hertz (Hz); multiplicities are given as singlet (s), doublet (d), triplet (t), quartet (q), quintet (qn), sextet (sxt), septet (spt), or multiplet (m); broad signals are indicated by (br). Due to quadrupolar relaxation, the aryl carbon atoms attached directly to boron are not always observed by 13C NMR.24 Electrospray mass spectra were measured on Micromass Prospec and Micromass Platform spectrometers; samples were prepared using 50:50:0.1 acetonitrile–water–formic acid solution. High resolution mass spectra (HRMS) were made on a Thermo Finnegan MAT95 XP instrument. Infrared spectra were recorded on a Bruker Alpha-P spectrometer and analysed using OPUS 6.5 software package. Elemental analysis was performed using a Thermo Scientific FLASH 2000 Series CHNS/O Analyzer. UV-Visible absorption measurements used Sigma Spectrophotometer Silica (Quartz) cuvettes (10 mm pathlength) and were recorded on a Jasco V660 spectrophotometer with Jasco EHC-716 Peltier temperature control. Fluorescence spectroscopy was carried out on a Perkin-Elmer LS55 fluorimeter, with an attached Julabo F25-HE water circulator for temperature control. All pH measurements were recorded on a HANNA pH 212 microprocessor pH meter using a Hamilton MINITRODE pH electrode that was calibrated using Fisher Chemicals standard buffer solutions (pH 4.0 – phthalate, 7.0 – phosphate, and 10.0 – borate). Unless otherwise stated all binding constants and pKa values were determined at 20 °C.
Synthesis of compound 3
:1 v/v) of the residue provided the title compound as a white foam (1.06 g, 66%). 1H-NMR (400 MHz, CD3OD, 25 °C): δH ppm 0.58 (3H, s, C18/19 CH3), 0.79 (3H, s, C18/19 CH3), 0.91 (3H, d, J = 4.7 Hz, C21H3), 1.15–2.22 (24H, steroid CH/CH2), 3.33 (5H, t, J = 5.1 Hz, 2 × NCH2 + C3H), 3.43–3.53 (8H, m, CH2OCH2CH2OCH2), 3.73 (1H, m, C7H), 3.86 (4H, m, C12H + OH), 5.02 (2H, s, PhCH2), 5.68 (1H, br t, J = 5.2 Hz, amide NH), 6.65 (1H, br, carbamate NH), 7.27 (5H, m, CHAr). 13C-NMR (100 MHz, CD3OD, 25 °C): δC ppm 12.5, 17.4, 22.5, 23.3, 26.3, 27.6, 28.1, 30.4, 31.7, 33.1, 34.7, 34.8, 35.4, 35.5, 39.2, 39.4, 39.5, 40.8, 41.6, 46.4, 46.5, 66.7, 68.4, 69.9, 70.0, 70.1, 70.2, 71.8, 73.1, 128.1, 128.2, 128.5, 136.6, 156.7, 174.5. MS (ES+): m/z 673.5 [M + H]+, 695.6 [M + Na]+, 1346.3 [2M + H]+.
Procedure for HPTS fluorescent assay of metal ion transport for boronocholates with and without fructose present
A lipid thin film was first prepared by the addition of spectroscopic grade chloroform to egg yolk phosphatidylcholine (EYPC, 40 mg, 52 μmol) and cholesterol (5 mg, 13 μmol) in a round bottomed flask (5 mL). Chloroform was slowly evaporated in vacuo at room temperature to leave a lipid thin film on the interior of the flask. HPTS phosphate buffer (1.2 mL, 100 μM HPTS, 20 mM MnHmPO4, 100 mM MCl, pH 6.5 where M+ = Na+ or K+ as appropriate) was added, followed by detachment of the lipid film from the interior of the flask by vortex agitation, and extruded through a polycarbonate membrane (200 nm pore size, 19×). To remove unencapsulated HPTS, an aliquot of the suspension (1 mL) was diluted with phosphate buffer (1.5 mL, pH 6.5) and purified by gel permeation chromatography (GPC) on a PD10 desalting column (eluted with 3.5 mL phosphate buffer). This provided a purified suspension of HPTS-encapsulated vesicles (15.5 mM total lipid concentration).An aliquot (25 μL) of the vesicle suspension was transferred to a fluorescence cuvette containing phosphate buffer (1.975 mL, 20 mM MnHnPO4, 100 mM MCl, pH 8.5 where M+ = Na+ or K+ as appropriate) with stirring (0.19 mM total lipid concentration) and fluorescence measurements were immediately started (ex. 405 nm and 460 nm, em. 510 nm, time interval 8 s) as a function of time. After 180 s the relevant ionophore was added (20 μL, 0.01 mM total ionophore concentration, 5 mol%) and vesicles were lysed after 1200 s by the addition of Triton X-100 (25 μL, 25% v/v in distilled water). The change in the normalized ratio I460/I405 as a function of time gave the rate of M+/OH− antiport across the phospholipid bilayer. The above procedure was repeated at fructose concentrations of 0.25 mM, 0.5 mM, 10 mM, 0.1 M, 0.5 M, achieved by ensuring each buffer composition contained the relevant concentration of fructose.
U-tube metal picrate transport experiments
A chloroform solution of the potentially transporting species (1 mM, 5 mL) was transferred to a glass U-tube (internal diameter 10 mm) and incubated at 25 °C. To one side of the U-tube was added a receiving phase of phosphate buffer (2.5 mL, 20 mM NanHmPO4, 100 mM NaCl, pH 6.5), and to the other side was added a source phase of 0.01% sodium picrate in phosphate buffer (2.5 mL, 20 mM NanHmPO4, 100 mM NaCl, pH 8.5). Addition of the source phase marked the start of the experiment; aliquots (1 mL) were taken at intervals from the receiving phase and analysed for the presence of picrate by UV spectroscopy (356 nm); after each measurement the sample was immediately replaced back in the receiving phase of the U-tube. The chloroform was stirred (300 rpm) for the entire experiment to aid diffusion through to the receiving phase.U-tube fructose transport experiments
A chloroform solution of the potentially transporting species (1 mM, 5 mL) was transferred to a glass U-tube (internal diameter 10 mm) incubated at 25 °C. To one side of the U-tube was added a receiving phase of phosphate buffer (2.5 mL D2O containing 0.75% % 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid as an internal standard, 20 mM NanHmPO4, 100 mM NaCl, pH 6.5), and to the other side was added a source phase of fructose (1 M) and 0.01% sodium picrate in phosphate buffer (2.5 mL D2O containing 0.75% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid as an internal standard, 20 mM NanHmPO4, 100 mM NaCl, pH 8.5). Addition of the source phase marked the start of the experiment; aliquots (0.8 mL) were taken at intervals from the receiving phase and analysed for the presence of D-fructose by 1H NMR spectroscopy, after each measurement the sample was immediately replaced back to the receiving phase of the U-tube. The chloroform was stirred (300 rpm) for the entire experiment to aid diffusion through to the receiving phase.Distribution coefficient determinations
Solutions of relevant ionophores were prepared in 1-octanol (10 mM, 0.5 mL) and sodium phosphate buffer (20 mM NanHmPO4, 100 mM NaCl, pH 8.5) was added (0.5 mL). The aqueous–organic mixture was vigorously mixed using a vortex mixer for 180 s before standing for 1 h to allow separation of the organic and aqueous layers. Aliquots (100 μL) of the 1-octanol layer were transferred to a UV-visible cuvette containing HPLC-grade methanol (1.9 mL) and mixed before measurement (260 nm). The procedure was repeated for different fructose concentrations in sodium phosphate buffer (20 mM NanHmPO4, 100 mM NaCl, pH 8.5, [fructose] = 0.01, 0.1, 0.5 mol L−1). To provide a maximum absorbance reading solutions of relevant ionophores were prepared in 1-octanol (5 mM, 100 μL) and transferred to a UV-visible cuvette containing HPLC-grade methanol (1.9 mL, 0.25 mM total ionophore concentration) and mixed before measurement (260 nm).Binding constant estimation with (2-methylamino methylphenyl)boronic acid 2
Following the protocol of Wang et al.,22 a three-component competitive assay with pyrocatechol violet28 was used to determine the binding constant between 2 and fructose. Two experiments were conducted to determine the equilibrium constants of the competitive system. First, the association for 2 with pyrocatechol violet (PV) was determined (Keq1). A solution of PV (2.74 mM, 2 mL) in phosphate buffer (20 mM NanHmPO4, 100 mM NaCl, pH 8.5) was transferred to a UV-visible cuvette and a solution of 2 in PV-phosphate buffer (2.74 mM PV, 20 mM NanHmPO4, 100 mM NaCl, pH 8.5) was titrated into the cuvette. A plot of changes in absorbance (596 nm) as a function of the concentration of 2, and subsequent iterative non-linear curve fitting (Dynafit35) of the data provided the binding constant for the PV-2 complex (Keq1).The binding constant for the 2-fructose complex (Keq2) was found by titrating a PV-2 solution with fructose. The binding constant Keq2 was determined by plotting 1/P as a function of Q, where P is defined as:
 | (3) |
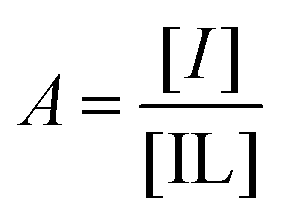 | (4) |
Acknowledgements
J.R.D.B. and S.J.W. thank the EPSRC for funding (EP/F055315/1). I.C.P. would like to thank the EU Seventh Framework Programme for a Marie Curie Intra-European Fellowship (Grant PIEF-GA-2012-328025 MagNanoVes).References
- (a) R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1106–1123 RSC; (b) S. D. Bull, M. G. Davidson, J. M. H. Van Den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed; (c) R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS PubMed.
- (a) H. Kim, Y. J. Kang, S. Kang and K. T. Kim, J. Am. Chem. Soc., 2012, 134, 4030–4033 CrossRef CAS PubMed; (b) G. A. Ellis, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 3631–3634 CrossRef CAS PubMed; (c) Y. Li, W. Xiao, K. Xiao, L. Berti, J. Luo, H. P. Tseng, G. Fung and K. S. Lam, Angew. Chem., Int. Ed., 2012, 51, 2864–2869 CrossRef CAS PubMed.
- (a) C. Lu, H. Li, H. Wang and Z. Liu, Anal. Chem., 2013, 85, 2361–2369 CrossRef CAS PubMed; (b) Y. Liu, L. Ren and Z. Liu, Chem. Commun., 2011, 47, 5067–5069 RSC.
- (a) L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC; (b) Y. Kotsuchibashi, R. V. C. Agustin, J.-Y. Lu, D. G. Hall and R. Narain, ACS Macro Lett., 2013, 2, 260–264 CrossRef CAS.
- (a) O. Savsunenko, H. Matondo, S. Franceschi-Messant, E. Perez, A. F. Popov, I. Rico-Lattes, A. Lattes and Y. Karpichev, Langmuir, 2013, 29, 3207–3213 CrossRef CAS PubMed; (b) A. Kashiwada, M. Tsuboi, N. Takamura, E. Brandenburg, K. Matsuda and B. Koksch, Chem.–Eur. J., 2011, 17, 6179–6186 CrossRef CAS PubMed; (c) A. Kashiwada, M. Tsuboi, T. Mizuno, T. Nagasaki and K. Matsuda, Soft Matter, 2009, 5, 4719–4725 RSC; (d) E. Han, L. Ding and H. Ju, Anal. Chem., 2011, 83, 7006–7012 CrossRef CAS PubMed; (e) A. Mahalingam, A. R. Geonnotti, J. Balzarini and P. F. Kiser, Mol. Pharm., 2011, 8, 2465–2475 CrossRef CAS PubMed.
- (a) M. Berube, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471–6479 CrossRef CAS PubMed; (b) M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS PubMed.
- P. J. Duggan, T. A. Houston, M. J. Kiefel, S. M. Levonis, B. D. Smith and M. L. Szydzik, Tetrahedron, 2008, 64, 7122–7126 CrossRef CAS PubMed.
- (a) T. Shinbo, K. Nishimura, T. Yamaguchi and M. Sugiura, J. Chem. Soc., Chem. Commun., 1986, 349–351 RSC; (b) M.-F. Paugam, L. S. Valencia, B. Boggess and B. D. Smith, J. Am. Chem. Soc., 1994, 116, 11203–11204 CrossRef CAS; (c) L. K. Mohler and A. W. Czarnik, J. Am. Chem. Soc., 1993, 115, 7037–7038 CrossRef CAS.
- (a) P. R. Westmark and B. D. Smith, J. Pharm. Sci., 1996, 85, 266–269 CrossRef CAS PubMed; (b) P. R. Westmark, S. J. Gardiner and B. D. Smith, J. Am. Chem. Soc., 1996, 118, 11093–11100 CrossRef CAS.
- (a) M.-F. Paugam, G. T. Morin and B. D. Smith, Tetrahedron Lett., 1993, 34, 7841–7844 CrossRef CAS; (b) G. T. Morin, M. P. Hughes, M.-F. Paugam and B. D. Smith, J. Am. Chem. Soc., 1994, 116, 8895–8901 CrossRef CAS; (c) G. T. Morin, M.-F. Paugam, M. P. Hughes and B. D. Smith, J. Org. Chem., 1994, 59, 2724–2128 CrossRef CAS.
- J. T. Bien, M. Shang and B. D. Smith, J. Org. Chem., 1995, 60, 2147–2152 CrossRef CAS.
- J. C. Norrild and H. Eggert, J. Chem. Soc., Perkin Trans. 2, 1996, 2583–2588 RSC.
- S. Hagihara, H. Tanaka and S. Matile, J. Am. Chem. Soc., 2008, 130, 5656–5657 CrossRef CAS PubMed.
- S. Litvinchuk, N. Sordé and S. Matile, J. Am. Chem. Soc., 2005, 127, 9316–9317 CrossRef CAS PubMed.
- (a) C. P. Wilson and S. J. Webb, Chem. Commun., 2008, 4007–4009 RSC; (b) C. P. Wilson, C. Boglio, L. Ma, S. L. Cockroft and S. J. Webb, Chem.–Eur. J., 2011, 17, 3465–3473 CrossRef CAS PubMed.
- (a) P. Bandyopadhyay, V. Janout, L. Zhang, J. A. Sawko and S. L. Regen, J. Am. Chem. Soc., 2000, 122, 12888–12889 CrossRef CAS; (b) P. Bandyopadhyay, V. Janout, L. Zhang and S. L. Regen, J. Am. Chem. Soc., 2001, 123, 7691–7696 CrossRef CAS PubMed; (c) M. Mehiri, W.-H. Chen, V. Janout and S. L. Regen, J. Am. Chem. Soc., 2009, 131, 1338–1339 CrossRef CAS PubMed; (d) L. Ma, M. Melegari, M. Colombini and J. T. Davis, J. Am. Chem. Soc., 2008, 130, 2938–2939 CrossRef CAS PubMed; (e) Y. Zhao, H. Cho, L. Widanapathirana and S. Zhang, Acc. Chem. Res., 2013, 46, 2763–2772 CrossRef CAS PubMed.
- M. Yoshii, M. Yamamura, A. Satake and Y. Kobuke, Org. Biomol. Chem., 2004, 2, 2619–2623 CAS.
- S. J. Gardiner, B. D. Smith, P. J. Duggan, M. J. Karpa and G. J. Griffin, Tetrahedron, 1999, 55, 2857–2864 CrossRef CAS.
- S. Matile, N. Sakai and A. Hennig, Transport Experiments in Membranes published in Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd., 2012 Search PubMed.
- J. Yan, H. Fang and B. Wang, Med. Res. Rev., 2005, 25, 490–520 CrossRef CAS PubMed.
- (a) J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS PubMed; (b) M. Vanduin, J. A. Peters, A. P. G. Kieboom and H. Vanbekkum, Tetrahedron, 1984, 40, 2901–2911 CrossRef CAS; (c) P. A. Sienkiewicz and D. C. Roberts, J. Inorg. Nucl. Chem., 1980, 42, 1559–1575 CrossRef CAS.
- G. Springsteen and B. H. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- D. J. Cabral, J. A. Hamilton and D. M. Small, J. Lipid Res., 1986, 27, 334–343 CAS.
- C. J. Ward, P. Patel and T. D. James, J. Chem. Soc., Perkin Trans. 1, 2002, 462–470 RSC.
- A. Adamczyk-Woźniak, K. M. Borys, I. D. Madura, A. Pawełko, E. Tomecka and K. Żukowski, New J. Chem., 2013, 37, 188–194 RSC.
- Both compounds exhibited a significant lag period due to slow diffusion though the chloroform phase under our experimental conditions. See A. Giannetto, S. Lanza, F. Puntoriero, M. Cordaroa and S. Campagna, Chem. Commun., 2013, 49, 7611–7613 RSC.
- M. J. Karpa, P. J. Duggan, G. J. Griffin and S. J. Freudigmann, Tetrahedron, 1997, 53, 3669–3678 CrossRef CAS.
- Z. Zhong and E. V. Anslyn, J. Am. Chem. Soc., 2002, 124, 9014–9015 CrossRef CAS PubMed.
- (a) V. Gorteau, G. Bollot, J. Mareda and S. Matile, Org. Biomol. Chem., 2007, 5, 3000–3012 RSC; (b) B. D. Smith, J. P. Davis, S. P. Draffin and P. J. Duggan, Supramol. Chem., 2004, 16, 87–90 CrossRef CAS.
- J.-P. Behr, M. Kirch and J.-M. Lehn, J. Am. Chem. Soc., 1985, 107, 241–246 CrossRef CAS.
- J. R. D. Brown, PhD thesis, University of Manchester, 2013.
- T. A. Houston, ChemBioChem, 2010, 11, 954–957 CrossRef CAS PubMed.
- B. C. Roy and S. Mallik, J. Org. Chem., 1999, 64, 2969–2974 CrossRef CAS.
- K. M. K. Swamy, Y. J. Jang, M. S. Park, H. S. Koh, S. K. Lee, Y. J. Yoon and J. Yoon, Tetrahedron Lett., 2005, 46, 3453–3456 CrossRef CAS PubMed.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260–273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 3. K+ transport by 3. U-tube picrate transport data. Effect of fructose on Na+ transport by 3 or 4. See DOI: 10.1039/c4ob00165f |
| This journal is © The Royal Society of Chemistry 2014 |