 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin†
Mu
Cheng
,
Zyta M.
Ziora
,
Karl A.
Hansford
,
Mark A.
Blaskovich
,
Mark S.
Butler
and
Matthew A.
Cooper
*
Division of Chemistry and Structural Biology, Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: m.cooper@uq.edu.au; Tel: +61-7-3346-2044
First published on 10th March 2014
Abstract
Dalbavancin, a semi-synthetic glycopeptide with enhanced antibiotic activity compared to vancomycin and teicoplanin, binds to the C-terminal lysyl-D-alanyl-D-alanine subunit of Lipid II, inhibiting peptidoglycan biosynthesis. In this study, micro-calorimetry and electrospray ionization (ESI)-MS have been used to investigate the relationship between oligomerisation of dalbavancin and binding of a Lipid II peptide mimic, diacetyl-Lys-D-Ala-D-Ala (Ac2-Kaa). Dalbavancin dimerised strongly in an anti-cooperative manner with ligand-binding, as was the case for ristocetin A, but not for vancomycin and teicoplanin. Dalbavancin and ristocetin A both adopt an ‘closed’ conformation upon ligand binding, suggesting anti-cooperative dimerisation with ligand-binding may be a general feature of dalbavancin/ristocetin A-like glycopeptides. Understanding these effects may provide insight into design of novel dalbavancin derivatives with cooperative ligand-binding and dimerisation characteristics that could enhance antibiotic activity.
Introduction
Acute bacterial skin and skin-structure infections (ABSSSIs), which are commonly caused by the Gram-positive cocci Staphylococcus aureus, Streptococcus pyogenes and to a lesser extent Enterococcus faecalis and E. faecium,1,2 lead to hospitalization and substantial health care costs.3 The emergence of antibiotic resistant strains, such as methicillin-resistant S. aureus (MRSA), has further complicated ABSSSI treatment.4–8 The glycopeptide/lipoglycopeptide antibiotics vancomycin (Fig. 1a) and teicoplanin (Fig. 1b) are normally used for MRSA treatment9,10 but MRSA strains with reduced susceptibility to glycopeptides have emerged and rapidly spread.11,12 In response, second-generation lipoglycopeptides have been developed that include telavancin (Fig. S1a, see ESI†), which was approved by the FDA in 2009, and dalbavancin (Fig. 1c) and oritavancin (Fig. S1b, see ESI†), which are both in late stage clinical development.13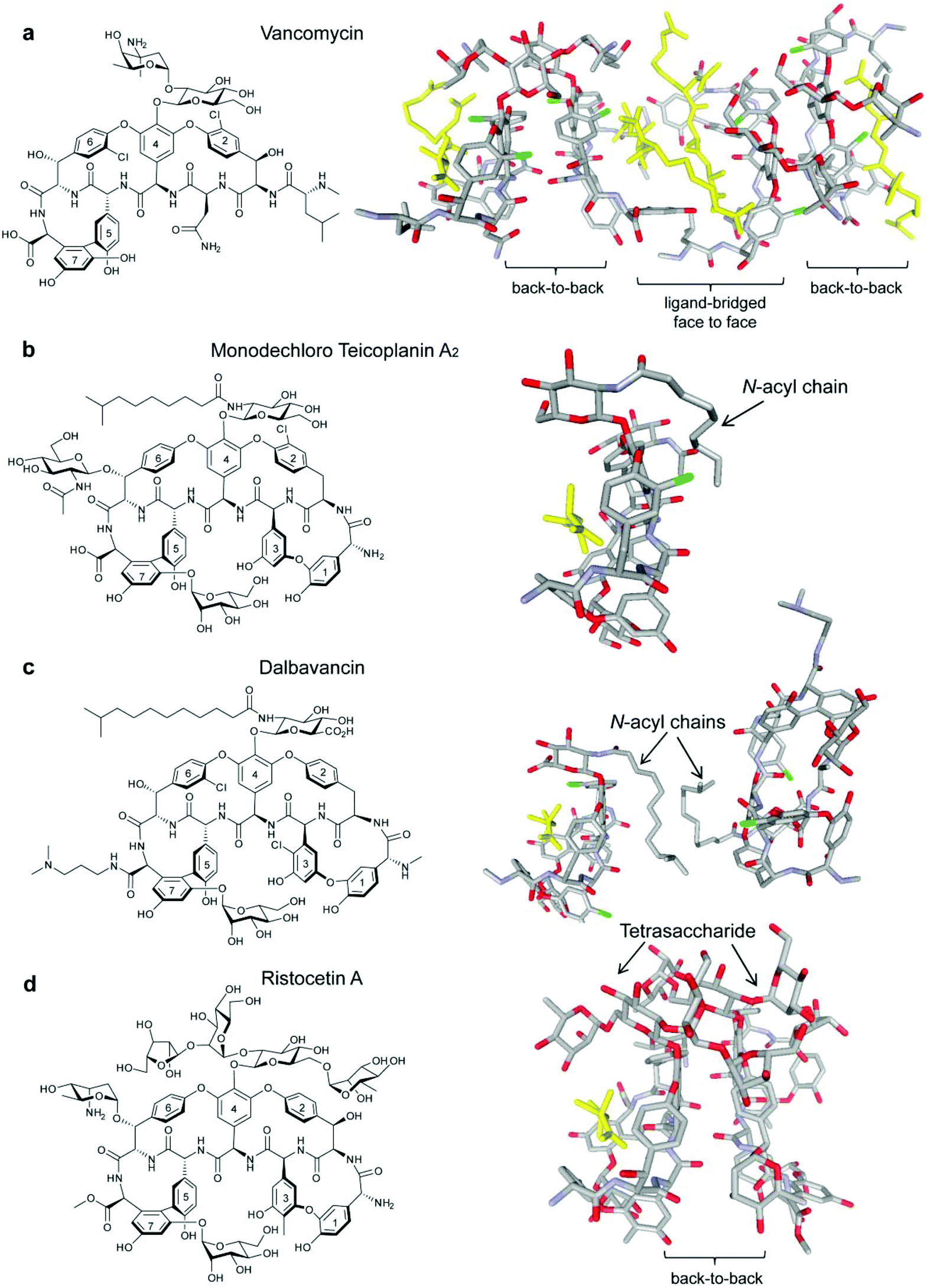 | ||
| Fig. 1 Glycopeptide antibiotics vancomycin (a),19 monodechloro teicoplanin A2 (b),33 dalbavancin (c)19 and ristocetin A (d)19 with published crystal structures highlighted to show bound ligand Lys-D-Ala-D-Ala (yellow) in the Lipid II binding site. Carrier proteins used for crystallisation omitted from (b), (c) and (d). | ||
All three second-generation lipoglycopeptides contain a common heptapeptide backbone that binds to the C-terminal L-lysyl-D-alanyl-D-alanine subunit of peptidoglycan precursors, resulting in inhibition of cell wall biosynthesis and cell death.14 The lipophilic side chains have been proposed to bind to serum proteins, as well as the bacterial membrane, thereby prolonging serum half-life and increasing activity against resistant strains.15,16 Recent NMR studies have suggested that the alkyl side chain of oritavancin interacts with pentaglycyl bridge segments of the cell wall peptidoglycan in S. aureus rather than the membrane.17 Telavancin and oritavancin are classified as vancomycin-type glycopeptides, while dalbavancin belongs to the teicoplanin-type class,18 with an additional macrocyclic ring formed between aryl residues 1 and 3.19 The vancomycin-type glycopeptides, including vancomycin,20 eremomycin,21 balhimycin22 and oritavancin,23 are able to dimerise in aqueous solution with dimerisation being cooperative with ligand-binding.20,21 Teicoplanin does not dimerise,24 but ristocetin A, another teicoplanin-type antibiotic, is the only glycopeptide previously reported to display dimerisation that is anti-cooperative with ligand-binding (Fig. 1d).18,21,24
Dalbavancin is a semi-synthetic N,N-dimethyl-1,3-diaminopropane derivative of the teicoplanin-like A40926 Factor B0.25,26 It displays enhanced in vitro activity compared to vancomycin and teicoplanin against methicillin-susceptible S. aureus (MSSA), MRSA, coagulase-negative staphylococci (CoNS) and non-VanA enterococci.27,28 To date, there have been only two studies published on the mode of action of dalbavancin: a recent patent has described oligomerisation detected by electrospray (ESI)-MS, protein-binding measured using MALDI-TOF and binding to diacetyl-Lys-D-Ala-D-Ala in the presence of serum protein using isothermal titration calorimetry (ITC),28 while an X-ray crystal structure of dalbavancin bound to a Lys-D-Ala-D-Ala (Kaa) binding epitope attached to a carrier protein was published in 2012.19 In this X-ray structure, two dalbavancin molecules were loosely associated in a back-to-back dimer via their fatty acyl chains (Fig. 1c).19
In this study, ITC and ESI-MS were used to investigate the relationship between dimerisation/oligomerisation of dalbavancin and binding of its target ligand diacetyl-Lys-D-Ala-D-Ala (Ac2-Kaa) in aqueous solutions and the results were compared to vancomycin, teicoplanin and ristocetin A. Serum components were not used in these experiments to reduce non-specific effects caused by protein binding.29,30 The ITC experiments showed that dalbavancin dimerised in an anti-cooperative manner with ligand-binding, which was also observed for ristocetin A. ESI-MS demonstrated similar oligomerisation behaviours between dalbavancin and ristocetin A in solution. Vancomycin also oligomerised, but weakly in the absence of ligand, whereas teicoplanin did not oligomerise. These data support the hypothesis that the anti-cooperativity between dimerisation and ligand-binding might be a feature of most teicoplanin-type glycopeptides, potentially due to a general ligand-induced ‘closed’ conformation observed in the crystal structures of dalbavancin and ristocetin A.19 Antibiotics with high dimerisation constants are generally potent against bacteria (i.e. eremomycin31 and oritavancin15), and the cooperativity between dimerisation and ligand-binding has previously been proposed to correlate with enhanced antibacterial activity.32 This study might provide some insights for further design and synthesis of novel glycopeptide/lipoglycopeptide derivatives with enhanced activity against resistant strains, in particular with VanA-type enterococci.
Results and discussion
Synthesis of dalbavancin
As dalbavancin was not available commercially, it was synthesised from the glycopeptide A40926-B0 using methods modified from a previous procedure.34 This synthetic dalbavancin gave minimum inhibitory concentration (MIC) values in agreement with previous studies (Table S1, see ESI†).15Dalbavancin dimerises strongly in solution
The presence of a fatty acyl chain contributes to dalbavancin's cLogP value of 1.54 and results in a lack of solubility in aqueous solution at physiological pH. Although serum protein can be used to increase the solubility of dalbavancin at neutral pH,28 protein binding dramatically reduces the free drug concentration of dalbavancin and could cause non-specific effects on dalbavancin–ligand interactions, complicating this analysis. Hence, a protein-free, buffered aqueous solution (0.1 M NaOAc, pH 5.0) was used to dissolve dalbavancin. LC/MS analyses showed that dalbavancin was completely soluble and stable in this buffer (Fig. S2, see ESI†), which was consistent with a previous study of dalbavancin solubility.28Dimerisation of dalbavancin was investigated by ITC dilution experiments, in which highly concentrated solutions of dalbavancin were titrated into a dilution buffer (0.1 M NaOAc, pH 5.0) to detect heat energy changes caused by dissociation of dalbavancin dimers.20 The resulting dissociation was endothermic (Fig. 2a), as was the case for vancomycin (Fig. 2b) and ristocetin A (Fig. 2c), though the heat pulses were broader and took longer to return to the baseline compared to vancomycin and ristocetin A. Teicoplanin showed negligible dose-dependent response beyond that expected for simple heat of dilution (Fig. 2d), which was consistent with a previous report that indicated that it exists exclusively as a monomer.33 Dimerisation constants (Kdim) of vancomycin and ristocetin A in the absence of ligand were in low mM ranges (Table 1), in agreement with previous reports.20,35–37 In contrast, the Kdim value of dalbavancin was approximately 50-fold higher than that for vancomycin or ristocetin A, suggesting that dalbavancin dimerises strongly under these experimental conditions.
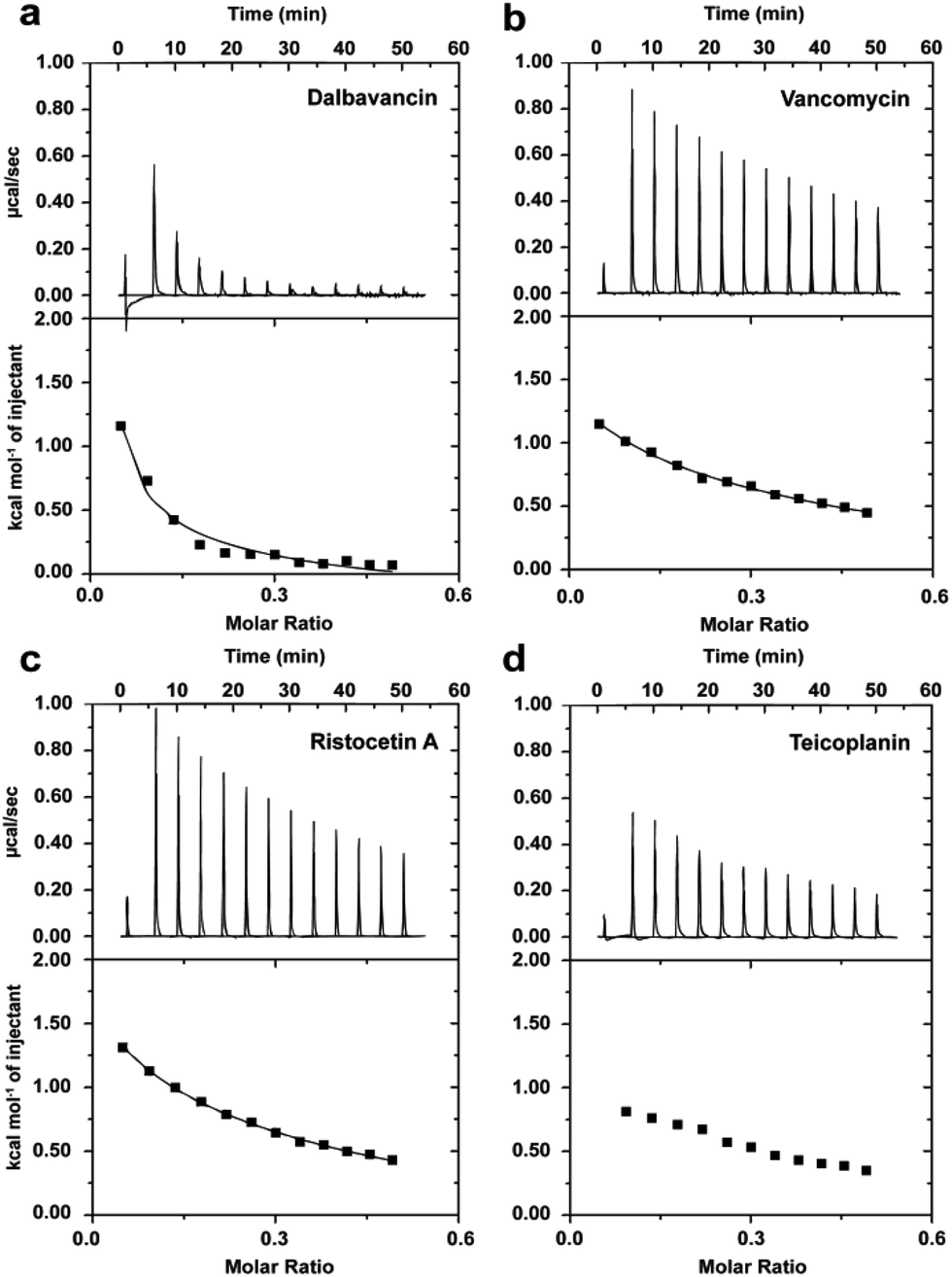 | ||
| Fig. 2 Typical ITC dilution data of dalbavancin (a), vancomycin (b), ristocetin A (c) and teicoplanin (d) in the absence of ligand at 25 °C in 0.1 M NaOAc buffer, pH 5.0. Upper profile: endothermic responses for sequential injections; lower profile: integrated dilution heat effects with theoretical fit to a dimer–monomer dissociation model.20 | ||
| Antibiotic | Ligand | (M−1) | (kJ mol−1) | ||
|---|---|---|---|---|---|
K
dim![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
ΔHdima |
TΔSdima |
ΔGdima |
||
| a Data are means ± SD for n = 3. b The thermodynamics of dalbavancin dimerisation in the presence of Ac2-Kaa could not be determined due to the poor solubility of the complex. | |||||
| Dalbavancin | None | 38400 ± 8260 |
−45.0 ± 2.2 | −18.9 ± 1.7 | −26.1 ± 0.5 |
| Ac2-Kaa | n/ab | n/ab | n/ab | n/ab | |
| Vancomycin | None | 750 ± 80 | −11.5 ± 0.5 | 4.9 ± 0.7 | −16.4 ± 0.3 |
| Ac2-Kaa | 1940 ± 170 | −17.1 ± 0.4 | 1.6 ± 0.2 | −18.7 ± 0.2 | |
| Ristocetin A | None | 920 ± 120 | −14.2 ± 0.5 | 2.7 ± 0.8 | −16.9 ± 0.3 |
| Ac2-Kaa | 690 ± 140 | −20.0 ± 1.3 | −3.9 ± 1.8 | −16.1 ± 0.5 | |
The dimerisation of teicoplanin is sterically impaired by both the N-acetyl-β-D-glucosamine substituent on residue 6 and the fatty acyl chain attached to the glucosamine on residue 4 which lies on the back (convex) interface (Fig. 1b).33 In comparison, the fatty acyl chain in dalbavancin is slightly longer and highly flexible, but it lacks the residue 6 N-acetyl-β-D-glucosamine subunit. In both antibiotics, the lipophilic chains are similarly oriented in the solid state, and there is evidence of self-association in dalbavancin molecules (Fig. 1c) but not in teicoplanin.19 These differentiating features apparently favour dimerisation in dalbavancin but not in teicoplanin.
The reported MIC values of dalbavancin are lower than that of vancomycin and teicoplanin, with MIC values against MRSA of 0.12 to 0.25 μg mL−1, compared to MIC values of vancomycin and teicoplanin against MRSA ranging from 1 to 2 μg mL−1 and from 2 to 8 μg mL−1, respectively.27 The improved antibacterial activity of dalbavancin may be correlated not only with bacterial membrane anchoring,19 which could serve to enhance the local concentration of antibiotic at the site of peptidoglycan biosynthesis on the membrane, but also with its strong dimerisation behaviour, as is the case for oritavancin.23
The ITC dilution results of this study indicated that dimerisation was driven by favourable enthalpy (ΔHdim) in all antibiotics with the exception of teicoplanin, but there were significant differences in the entropic component (TΔSdim) (Table 1). The thermodynamic parameters of ristocetin A were similar to vancomycin in the absence of ligand, consistent with a previous ITC study.20 While dalbavancin dimerisation was more exothermic than vancomycin or ristocetin A, there was concomitant unfavourable dimerisation entropy. It is conceivable that self-association of the fatty acyl chains in the dalbavancin could bury the hydrophobic surface area from solvent, leading to a considerable entropy of solvation. In contrast, the exothermic dimerisation of dalbavancin may be attributed to the formation of amide-amide hydrogen-bonds between heptapeptide backbones, the favourable van der Waals interactions between non-polar groups and the orthogonal π–σ interactions between aromatic rings of dalbavancin complexes, as was the case for vancomycin and ristocetin A.35,38 Additional ionic interactions may also favourably contribute to the overall dimerisation enthalpy of dalbavancin.28 However, the hydrophobic interactions between the carbohydrate group and aromatic rings are important in stabilizing the dimer species of vancomycin39 and ristocetin A.18
Dalbavancin dimerisation and ligand binding are anti-cooperative
Dalbavancin dimerisation was next studied in the presence of the ligand Ac2-Kaa by ITC dilution measurements using conditions that predominantly favoured the dalbavancin–ligand complex. This avoided complications caused by the change in ligand-bound state during the dilution/dissociation process as previously described.20 Vancomycin and ristocetin A were used for comparative purposes. Our data (Table 1) confirmed the cooperativity and the anti-cooperativity in vancomycin and ristocetin A, respectively, which was consistent with previous studies.20,21 However, it was impossible to determine the dimerisation of dalbavancin in the presence of ligand in solution due to the poor solubility of the dalbavancin–ligand complex. Although dalbavancin and the peptide ligand were both soluble in acetate buffer, when concentrated aqueous solutions of the two were mixed, precipitation was observed.The ligand-binding of dalbavancin in solution was investigated by ITC binding measurements at concentrations that populate monomeric or dimeric forms. The ITC data indicated that both were exothermic processes (Fig. 3a). The association constant (Kass) of the tripeptide ligand Ac2-Kaa toward the dalbavancin monomer was increased approximately 3-fold compared to monomeric vancomycin and 4-fold compared to monomeric ristocetin A (Fig. 3b). This Kass value was reduced around 2-fold when binding to dalbavancin dimer, as was the case with ristocetin A, in which the Kass value of ligand-binding of dimeric ristocetin A was reduced 10-fold. In contrast, the Kass value of ligand-binding toward the vancomycin dimer was increased approximately 1.4-fold compared to its monomer, consistent with a previously reported value.21 The ITC binding data in this study demonstrated that dimerisation reduced ligand-binding affinity in cases of dalbavancin and ristocetin A, whereas dimerisation of vancomycin enhanced ligand-binding. Therefore, dalbavancin dimerises in an anti-cooperative manner with ligand-binding, in a similar fashion to ristocetin A.
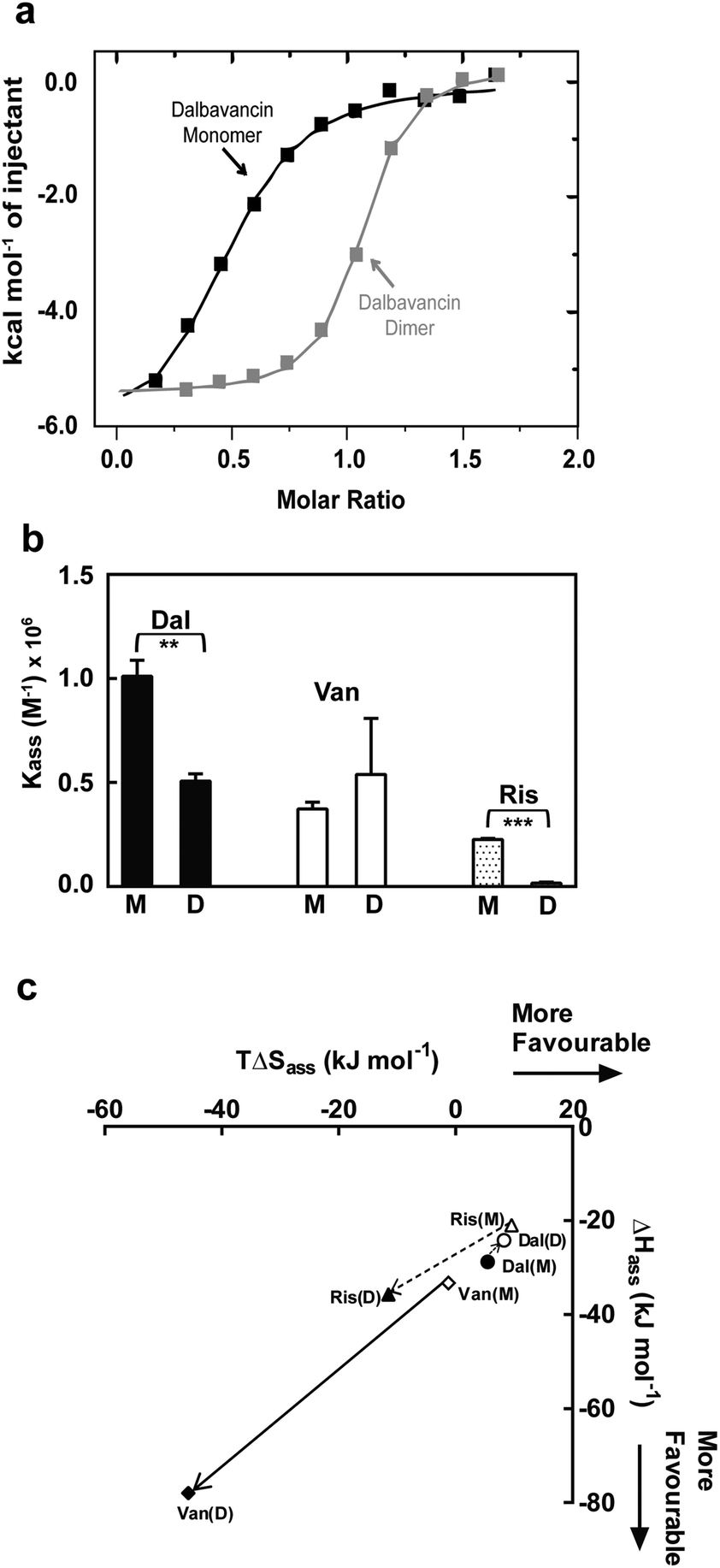 | ||
| Fig. 3 ITC binding data of dalbavancin (Dal), vancomycin (Van) and ristocetin A (Ris) at 25 °C in 0.1 M NaOAc (pH 5.0) showing anti-cooperativity of ligand binding to dimer for dalbavancin and ristocetin A. (a) Integrated titration curves upon complexation of Ac2-Kaa with dalbavancin monomer (black) and dimer (grey), with theoretical fit to a single site binding mode.40 (b) The binding constant (Kass) of antibiotic monomers (M) and dimers (D). Statistical comparison of Kass values was performed by the two-tailed Student's t-test ( **, p < 0.01; ***, p < 0.001). (c) Enthalpy–entropy plots showing thermodynamics of ligand binding for three antibiotics (Dal: ○; Van: ◊; Ris: Δ). Means ± SD for n = 3. | ||
Recent crystal structures of dalbavancin and ristocetin A with bound ligand, when compared to similar glycopeptide ligand-free structures, show that ligand binding induces a conformational change in which the two ends of the heptapeptide are drawn closer together, with the mannose attached to residue 7 reaching across to the biaryl ether linkage of residues 1 and 3.18,19 This ligand-bound monomer ‘closed’ conformation may interfere with dimerisation of these antibiotics, possibly disrupting formation of the ‘back-to-back’ network of hydrogen bonds. In contrast, the lack of crosslinking between residues 1 and 3 in vancomycin-type glycopeptides16 (i.e. vancomycin, eremomycin, balhimycin and oritavancin) presumably imparts greater flexibility and thus allows ligand-induced dimerisation.21,23 Thus, variations in conformational flexibility appears to dictate the cooperativity observed in vancomycin-type antibiotics and the anti-cooperativity in ristocetin A and dalbavancin.
Enthalpy (ΔHass) versus entropy (TΔSass) plots of ligand-binding for dalbavancin, vancomycin and ristocetin A are shown in Fig. 3c. ΔHass against TΔSass for vancomycin–ligand binding was close to a linear correlation, in which the TΔSass reduction was similar to the ΔHass increment going from a monomer to a dimer. However, this was not the case for ristocetin–ligand binding due to a slightly reduced entropic contribution going from monomeric to dimeric forms. Ligand-binding of the dalbavancin dimer was more favoured entropically, but less favoured enthalpically than with its monomer. While dimerisation of both ristocetin A and dalbavancin was anti-cooperative with ligand-binding, the thermodynamic contributions to this behaviour differed. The two halves of the ristocetin A dimer are known to bind ligand with different affinities due to the asymmetric tetrasaccharide orientation in the ristocetin A dimer,41,42 which contributes to the anti-cooperative ligand-binding of ristocetin A.18 Removing the tetrasaccharide moiety and the residue 7 mannose in ristocetin A (known as ristocetin ψ) has been reported to change the anti-cooperative behaviour to cooperative.21 Dalbavancin lacks this tetrasaccharide group and thus the ligand-induced ‘closed’ conformation might provide the major contribution to the anti-cooperativity in dalbavancin.
In the absence of ligands the dimerisation constant of ristocetin A is similar to vancomycin (Table 1), but it is less active than vancomycin in vitro,43 most likely due to its anti-cooperativity. Additionally, a previous study reported that linking the vancosamine groups of two vancomycin molecules reduced the MIC value against vancomycin-resistant E. faecium from >16 μg mL−1 to 1 μg mL−1.32 It is notable that dalbavancin has poor activity against VanA-type enterococci with a MIC value of 32 μg mL−1,44,45 whereas oritavancin, which shares a similar dimerisation constant with dalbavancin, is highly active with a MIC of 0.25 μg mL−1.23 Hence, it could be hypothesised that the anti-cooperativity between dimerisation and ligand-binding might contribute to the poor activity of dalbavancin against VanA-type enterococci.
Binding stoichiometry of dalbavancin monomers and dimers are different
The stoichiometry (N) obtained from ITC binding experiments reflects the number of moles of ligand Ac2-Kaa required to saturate all the available binding sites of the antibiotic.28 The binding stoichiometry values of vancomycin and ristocetin A ranged from 0.9 to 1.2 for both monomeric and dimeric antibiotic species (Fig. S3a–S3d, see ESI†), which corresponded to a 1:1 binding complex for the monomer and 2:2 for the dimer.46 Our data is in agreement with a previous study.40 For teicoplanin and dalbavancin, the calorimeter cell was pre-rinsed with the experimental concentration of glycopeptides to prevent non-specific binding of these lipophilic antibiotics to the metal surface of the calorimeter cell. The binding stoichiometry of the teicoplanin monomer was 0.8, which fitted to a 1:1 binding.46 Interestingly, the ligand-binding of the dalbavancin monomer fitted to a 2:1 dalbavancin:ligand complex (N closer to 0.5), while its dimer bound to ligand in a 1:1 ratio (N closer to 1, Fig. S3e–S3f, see ESI†). In the presence of serum protein, dalbavancin monomers have been reported to bind in a 1:1 ratio to the same tripeptide ligand Ac2-Kaa.28 The fatty acyl chain in the dalbavancin monomer is highly flexible,19,33 and can interfere with ligand binding. Immobilization of fatty acyl chains either by dimerisation or by protein binding is likely to prevent lipophilic groups from blocking the binding pocket of dalbavancin, thereby allowing for complete occupancy of the binding sites of dalbavancin.
Dalbavancin and ristocetin A oligomerise in the absence of ligands
The potential formation of dalbavancin multimers (dimers or oligomers) in solution of varying concentrations was investigated using ESI-MS and compared to vancomycin, ristocetin A and teicoplanin. Potential multimers would be observed as the [nM + (n + 1)H](n+1)+ mass ion species, where n is a positive integer indicating the multiplicity of the multimer (e.g. n = 2 when the multimer is a dimer and n = 4 when the multimer is a tetramer), M is the mass of the monomer and (n + 1)+ indicates the charge.28 For example, the dimer and tetramer species were assigned respectively as the [2M + 3H]3+ and [4M + 5H]5+ mass ion peaks. The MS data (Fig. S4–S7, see ESI†) showed the presence of multimers for dalbavancin, ristocetin A and vancomycin, but not for teicoplanin. Although the primary multimeric form was a dimer, the formation of various oligomers including trimers and tetramers were also observed. Dalbavancin and ristocetin A were able to form higher order oligomers in solution, such as pentamers and hexamers, at concentrations above 100 μM (Fig. 4), which were absent for vancomycin. The weak oligomerisation of vancomycin observed in this ESI-MS was consistent with a previous study, which demonstrated that the formation of vancomycin oligomeric species in solution was ligand mediated.47 In this ligand-free ESI-MS study, dalbavancin was found to oligomerise in a dose-dependent manner, as was the case with ristocetin A (Fig. 4). An increase in antibiotic concentration was found to correspond to the increased multimer mass traces and hence an increase in the population ratio of antibiotic multimer to monomer.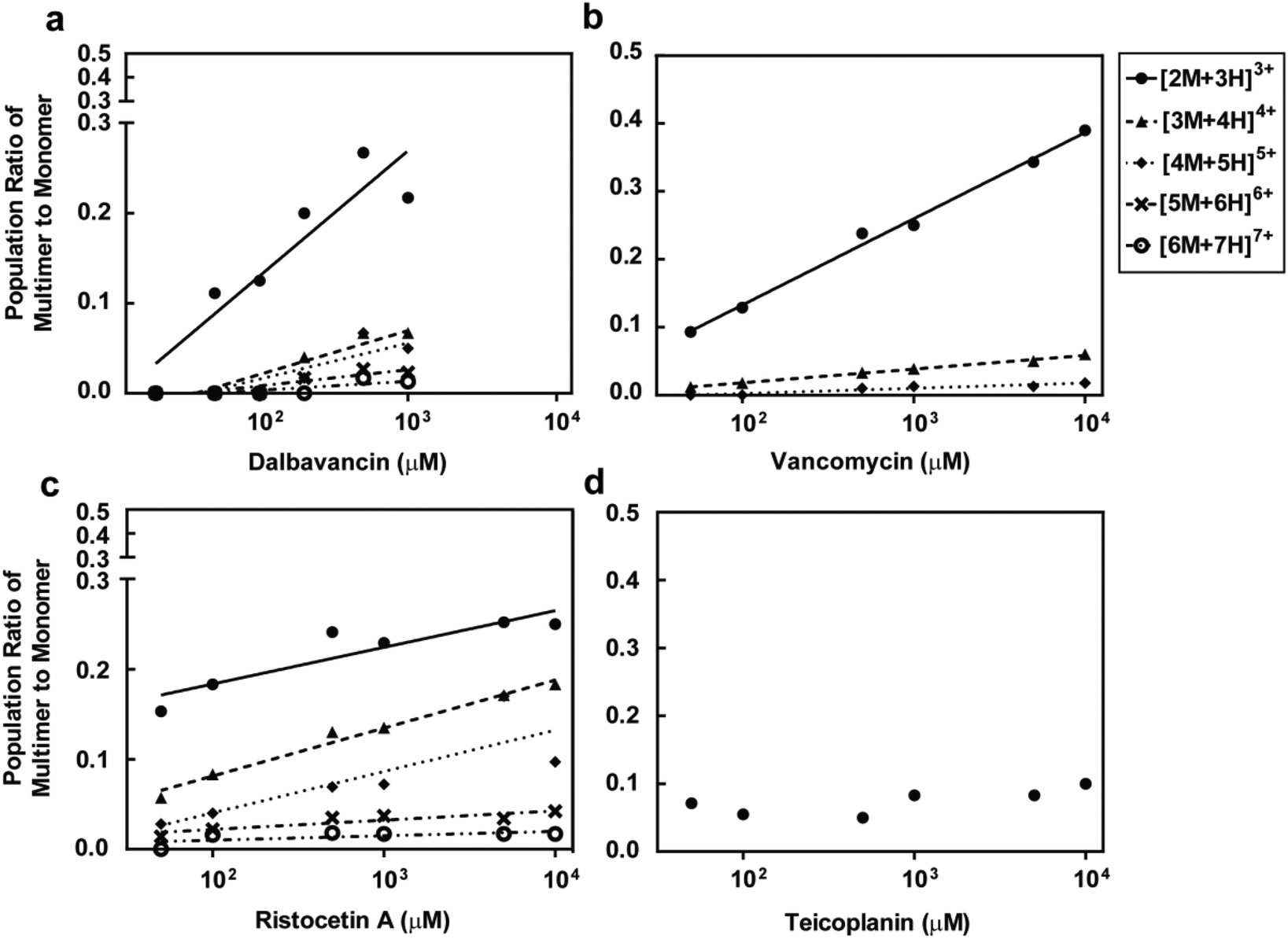 | ||
| Fig. 4 Concentration of dalbavancin (a), vancomycin (b), ristocetin A (c) and teicoplanin (d) versus the ratio of antibiotic multimer to monomer population. There are concentration-dependent increases in the ratios of multimer to monomer in all antibiotics except teicoplanin. | ||
Experimental section
Antibiotics, ligand and bacteria
Vancomycin hydrochloride hydrate salt, teicoplanin containing >80% teicoplanin A2, ristomycin monosulfate containing >90% ristocetin A and A40926 ≥ 80% purity (HPLC) were purchased from Sigma-Aldrich and used without further purification. Dalbavancin (TFA salt) with 97% purity (HPLC) was synthesized based on a published procedure34 as described in the ESI.† The peptide ligand diacetyl-L-Lys-D-Ala-D-Ala (Ac2-Kaa) was purchased from Chiron-Mimotopes (Melbourne, Australia). MRSA ATCC 43300 and Streptococcus pneumoniae (multi-drug-resistant) ATCC 700677 were purchased from American Type Culture Collection (Manassas, VA, USA). E. faecium (VanA) was a clinical isolate supplied by Prof. D. Paterson from the University of Queensland Centre for Clinical Research (UQCCR, Brisbane, Australia).ITC
Calorimetry experiments were performed using a MicroCal Omega Auto-Itc200 (GE Healthcare, Australia) at 25 °C in triplicate. Each experiment consisted of sequential injections from the titration syringe into the calorimeter cell (cell volume ∼0.2 mL) with stirring at 1000× rpm. The interval time between each injection was 240 s. Computer simulations (curve fitting) were performed using the MicroCal Origin version 7.0 software package adapted for ITC data analysis to yield stoichiometry (N), enthalpy (ΔHass), entropy (ΔSass) and association constant (Kass) for the single site binding mode or dissociation constant (Kdiss) and enthalpy (ΔHdiss) for the dimer–monomer dissociation mode. The Gibbs free energy (ΔG) was calculated using the Gibbs–Helmholtz thermodynamic and van't Hoff equations as previously described.46ITC: dimer–monomer dissociation
Calorimetric data for the dilution of dalbavancin and control antibiotics (vancomycin, ristocetin A and teicoplanin) solutions in the absence and presence of ligand Ac2-Kaa were determined using a modified calorimetric dilution method.20 Briefly, a 3 mM antibiotic solution in Buffer A (0.1 M NaOAc, pH 5.0) was consecutively injected (13 × 3 μL per injection, first injection of 0.5 μL) into the calorimeter cell initially containing Buffer A alone. Dilution experiments involving ligand employed consecutive injections of the antibiotic–ligand mixture (3 mM of antibiotic and 9 mM of free Ac2-Kaa) dissolved in Buffer A into the stirred calorimeter cell initially containing the same concentration of ligand Ac2-Kaa. Data were corrected for small injection/mixing effects from buffer controls that were performed separately and analyzed under identical conditions by omitting the first injection.ITC: single site binding
Ligand-binding experiments of monomeric and dimeric antibiotics were performed using a modified calorimetric titration method.40 Briefly, the concentrated ligand Ac2-Kaa solution (14- to 20-fold higher than the antibiotic solution) dissolved in Buffer A was sequentially injected (12 × 2 μL per injection, first injection of 0.5 μL) into the calorimeter cell charged either with antibiotic monomer solutions (0.01 mM for dalbavancin and 0.025 mM for vancomycin, ristocetin A and teicoplanin) or with antibiotic dimer solutions (0.2 mM for dalbavancin and 2 mM for vancomycin and ristocetin A) dissolved in Buffer A. Pre-rinsing the calorimeter cell with dalbavancin and teicoplanin solutions at the experimental concentration was required to avoid non-specific binding of antibiotics to the metal surface of the calorimeter cell (conditions were determined in separate experiments). Heat of reaction was corrected by the heat of dilution of ligand solution determined in separate experiments.ESI-MS
Experiments were performed using an Applied Biosystem API QStar Pulsar Mass Spectrometer equipped with a TurboIonSpray source, a Triple Quadrupole analyzer, operating in positive ion mode. The MS conditions were performed as previously described28 with some modifications. Dalbavancin was dissolved in H2O–isopropanol (v/v = 8/2) and serially diluted to give concentrations from 1 mM to 0.02 mM. Vancomycin, ristocetin A and teicoplanin were used as controls using concentrations from 10 mM to 0.05 mM in H2O–isopropanol (v/v = 8/2). The antibiotic solutions were injected (10 μL) into a flow of H2O–isopropanol (v/v = 8/2) (15 μL mL−1) and the MS data acquired from 500 to 2000 Da. Data were analyzed with the software version Analyst QS 1.1.Conclusions
In summary, our ITC and ESI-MS data show that dalbavancin dimerises in an anti-cooperative manner with ligand binding, which had only been previously reported for ristocetin A.18,21,24 Analysis of published crystallographic structures suggested that dalbavancin and ristocetin A share a similar constrained ligand-bound induced ‘closed’ conformation, which is absent in vancomycin-type glycopeptide antibiotics that dimerise and bind ligand cooperatively. Hence, it is conceivable that antibiotics with a similar ligand-induced ‘closed’ conformation might display dalbavancin/ristocetin A-like anti-cooperativity in dimerisation and ligand-binding. Given the reported correlation between vancomycin-type cooperativity and improved in vitro antibacterial activity, our findings suggest that further modifications to dalbavancin derivatives, such as removing the cross-linkage between aromatic rings 1 and 3 in dalbavancin, may help to overcome resistant bacteria, in particular VanA-type enterococci.Acknowledgements
This research was supported financially by the NHMRC grants APP631632, APP1026922 and AF511105. MC was supported by an Australian Postgraduate Award (APA) PhD scholarship. ZMZ, KAH, MSB and MAB were supported by a Wellcome Trust Seeding Drug Discovery Award (094977/Z/10/Z). We would like to thank Ms S. Ramu and Mrs A. Kavanagh for MIC determinations, and also thank Ms R. Pelingon and Mr D.J. Edwards for dalbavancin purification.Notes and references
- S. Y. Lee, J. L. Kuti and D. P. Nicolau, Surg. Infect., 2005, 6, 283–295 CrossRef PubMed.
- G. J. Moran, F. M. Abrahamian, F. Lovecchio and D. A. Talan, J. Emerg. Med., 2012, 44, e397–e412 CrossRef PubMed.
- M. J. Zervos, K. Freeman, L. Vo, N. Haque, H. Pokharna, M. Raut and M. Kim, J. Clin. Microbiol., 2012, 50, 238–245 CrossRef PubMed.
- D. M. Livermore, Clin. Microbiol. Infect., 2004, 10, 1–9 CrossRef PubMed.
- P. Nordmann, T. Naas, N. Fortineau and L. Poirel, Curr. Opin. Microbiol., 2007, 10, 436–440 CrossRef CAS PubMed.
- F. R. Deleo and H. F. Chambers, J. Clin. Invest., 2009, 119, 2464–2474 CrossRef CAS PubMed.
- S. Y. C. Tong, E. J. Bishop, R. A. Lilliebridge, A. C. Cheng, Z. Spasova-Penkova, D. C. Holt, P. M. Giffard, M. I. McDonald, B. J. Currie and C. S. Boutlis, J. Infect. Dis., 2009, 199, 1461–1470 CrossRef PubMed.
- G. L. French, Int. J. Antimicrob. Agents, 2010, 36, 3–7 CrossRef.
- W. M. M. Kirby, Rev. Infect. Dis., 1981, 3, S236–S239 CrossRef PubMed.
- G. R. Matzke, G. G. Zhanel and D. R. P. Guay, Clin. Pharmacokinet., 1986, 11, 257–282 CrossRef CAS PubMed.
- S. Sujatha and I. Praharaj, Interdiscip. Perspect. Infect. Dis., 2012, 781679, DOI:10.1155/2012/781679.
- A. Liapikou and A. Torres, Expert Opin. Emerg. Drugs, 2013, 18, 291–305 CrossRef CAS PubMed.
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2013, 66, 571–591 CrossRef CAS PubMed.
- D. Kahne, C. Leimkuhler, L. Wei and C. Walsh, Chem. Rev., 2005, 105, 425–448 CrossRef CAS PubMed.
- G. G. Zhanel, D. Calic and F. Schweizer, Drugs, 2010, 71, 526–526 Search PubMed.
- P.-A. Ashford and S. P. Bew, Chem. Soc. Rev., 2012, 41, 957–978 RSC.
- S. J. Kim, K. S. E. Tanaka, E. Dietrich, A. R. Far and J. Schaefer, Biochemistry, 2013, 52, 3405–3414 CrossRef CAS PubMed.
- V. Nahoum, S. Spector and P. Loll, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 67, 592–592 Search PubMed.
- N. J. Economou, V. Nahoum, S. D. Weeks, K. C. Grasty, I. J. Zentner, T. M. Townsend, M. W. Bhuiya, S. Cocklin and P. J. Loll, J. Am. Chem. Soc., 2012, 134, 4637–4645 CrossRef CAS PubMed.
- D. McPhail and A. Cooper, J. Chem. Soc., Faraday Trans., 1997, 93, 2283–2289 RSC.
- J. P. Mackay, U. Gerhard, D. A. Beauregard, M. S. Westwell, M. S. Searle and D. H. Williams, J. Am. Chem. Soc., 1994, 116, 4581–4590 CrossRef CAS.
- H. Shiozawa, R. Zerella, B. Bardsley, K. L. Tuck and D. H. Williams, Helv. Chim. Acta, 2003, 86, 1359–1370 CrossRef CAS.
- N. E. Allen and T. I. Nicas, FEMS Microbiol. Rev., 2003, 26, 511–532 CrossRef CAS.
- J. P. Mackay, U. Gerhard, D. A. Beauregard, R. A. Maplestone and D. H. Williams, J. Am. Chem. Soc., 1994, 116, 4573–4580 CrossRef CAS.
- G. Candiani, M. Abbondi, M. Borgonovi, G. Romano and F. Parenti, J. Antimicrob. Chemother., 1999, 44, 179–192 CrossRef CAS PubMed.
- E. J. C. Goldstein, D. M. Citron, C. V. Merriam, Y. Warren, K. Tyrrell and H. T. Fernandez, Antimicrob. Agents Chemother., 2003, 47, 1968–1971 CrossRef CAS PubMed.
- S. Lopez, C. Hackbarth, G. Romano, J. Trias, D. Jabes and B. P. Goldstein, J. Antimicrob. Chemother., 2005, 55, 21–24 CrossRef PubMed.
- M. Stogniew, L. Colombo and R. Ciabatti, US Pat., 8 143 212, 2012 Search PubMed.
- A. Leighton, A. B. Gottlieb, M. B. Dorr, D. Jabes, G. Mosconi, C. VanSaders, E. J. Mroszczak, K. C. M. Campbell and E. Kelly, Antimicrob. Agents Chemother., 2004, 48, 940–945 CrossRef CAS.
- M. B. Dorr, D. Jabes, M. Cavaleri, J. Dowell, G. Mosconi, A. Malabarba, R. J. White and T. J. Henkel, J. Antimicrob. Chemother., 2005, 55(Suppl 2), ii25–ii30 CAS.
- K. R. Maples, C. Wheeler, E. Ip, J. J. Plattner, D. Chu, Y.-K. Zhang, M. N. Preobrazhenskaya, S. S. Printsevskaya, S. E. Solovieva, E. N. Olsufyeva, H. Heine, J. Lovchik and C. R. Lyons, J. Med. Chem., 2007, 50, 3681–3685 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Hughes, S. Y. Cho, N. Winssinger, H. Labischinski and R. Endermann, Chemistry, 2001, 7, 3824–3843 CrossRef CAS.
- N. J. Economou, I. J. Zentner, E. Lazo, J. Jakoncic, V. Stojanoff, S. D. Weeks, K. C. Grasty, S. Cocklin and P. J. Loll, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 520–533 CAS.
- A. Malabarba, R. Ciabatti, R. Scotti, B. P. Goldstein, P. Ferrari, M. Kurz, B. P. Andreini and M. Denaro, J. Antibiot., 1995, 48, 869–883 CrossRef CAS.
- U. Gerhard, J. P. Mackay, R. A. Maplestone and D. H. Williams, J. Am. Chem. Soc., 1993, 115, 232–237 CrossRef CAS.
- D. A. Beauregard, D. H. Williams, M. N. Gwynn and D. J. C. Knowles, Antimicrob. Agents Chemother., 1995, 39, 781–785 CrossRef CAS.
- H. Linsdell, C. Toiron, M. Bruix, G. Rivas and M. Menendez, J. Antibiot., 1996, 49, 181–193 CrossRef CAS.
- M. Batchelor, D. Zhou, M. A. Cooper, C. Abell and T. Rayment, Org. Biomol. Chem., 2010, 8, 1142–1148 CAS.
- T. Kikuchi, S. Karki, I. Fujisawa, Y. Matsushima, Y. Nitanai and K. Aoki, Bull. Chem. Soc. Jpn., 2010, 83, 391–400 CrossRef CAS.
- M. Rekharsky, D. Hesek, M. Lee, S. O. Meroueh, Y. Inoue and S. Mobashery, J. Am. Chem. Soc., 2006, 128, 7736–7737 CrossRef CAS PubMed.
- Y. R. Cho, A. J. Maguire, A. C. Try, M. S. Westwell, P. Groves and D. H. Williams, Chem. Biol., 1996, 3, 207–215 CrossRef CAS.
- B. Bardsley and D. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 1925–1929 RSC.
- A. Malabarba, T. I. Nicas and R. C. Thompson, Med. Res. Rev., 1997, 17, 69–137 CrossRef CAS.
- A. Malabarba and B. P. Goldstein, J. Antimicrob. Chemother., 2005, 55, 15–20 CrossRef PubMed.
- S. D. Pope and A. M. Roecker, Pharmacotherapy, 2006, 26, 908–918 CrossRef CAS PubMed.
- L. Damian, Methods Mol. Biol., 2013, 1008, 103–118 CAS.
- P. J. Loll, A. Derhovanessian, M. V. Shapovalov, J. Kaplan, L. Yang and P. H. Axelsen, J. Mol. Biol., 2009, 385, 200–211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C3OB42428F |
| This journal is © The Royal Society of Chemistry 2014 |