 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ethynylbenzenoid metabolites of Antrodia camphorata: synthesis and inhibition of TNF expression†
Marco
Buccini
a,
Kathryn A.
Punch
a,
Belinda
Kaskow
a,
Gavin R.
Flematti
a,
Brian W.
Skelton
b,
Lawrence J.
Abraham
a and
Matthew J.
Piggott
*a
aSchool of Chemistry and Biochemistry, The University of Western Australia, Perth, WA, Australia. E-mail: matthew.piggott@uwa.edu.au; Fax: +61 8 6488 1005; Tel: +61 8 6488 3170
bCentre for Microscopy, Characterisation and Analysis, University of Western Australia, Perth, WA, Australia
First published on 19th December 2013
Abstract
An improved synthesis of the anti-inflammatory natural product antrocamphin A (2), involving a key Castro–Stephens reaction, is presented, along with the first total synthesis of its congener antrocamphin B (3). Approaches towards the more complex co-metabolite antrodioxolanone (4) were unsuccessful, but a samarium diiodide-mediated pinacol coupling of antrocamphin B did provide the chiral epimers (51). Antrocamphin A (2) inhibits Tumour Necrosis Factor (TNF) reporter gene expression, but its development as an anti-inflammatory agent may be limited by cytotoxicity.
Introduction
We recently questioned the identity of a natural product isolated from the fungus Antrodia camphorata (also called Taiwanofungus camphoratus, niu-chang-chih or jang-jy), a commercially important traditional Chinese medicine, which is becoming increasingly rare in its native Taiwan. The proposed structure 11 (Fig. 1) made this the first reported example of a naturally occurring acid chloride, which seemed incompatible with a physiological existence, or the extraction and isolation procedure. Indeed, synthetic 1 is highly unstable and rapidly hydrolyses on exposure to atmospheric moisture, and trace water in CDCl3, making it difficult to characterise.2 Moreover, the spectroscopic and mass spectrometric properties of 1 do not match those of the natural product.2 These incongruities led us to consider the novel co-metabolites 2–4 reported in the same paper (Fig. 1).1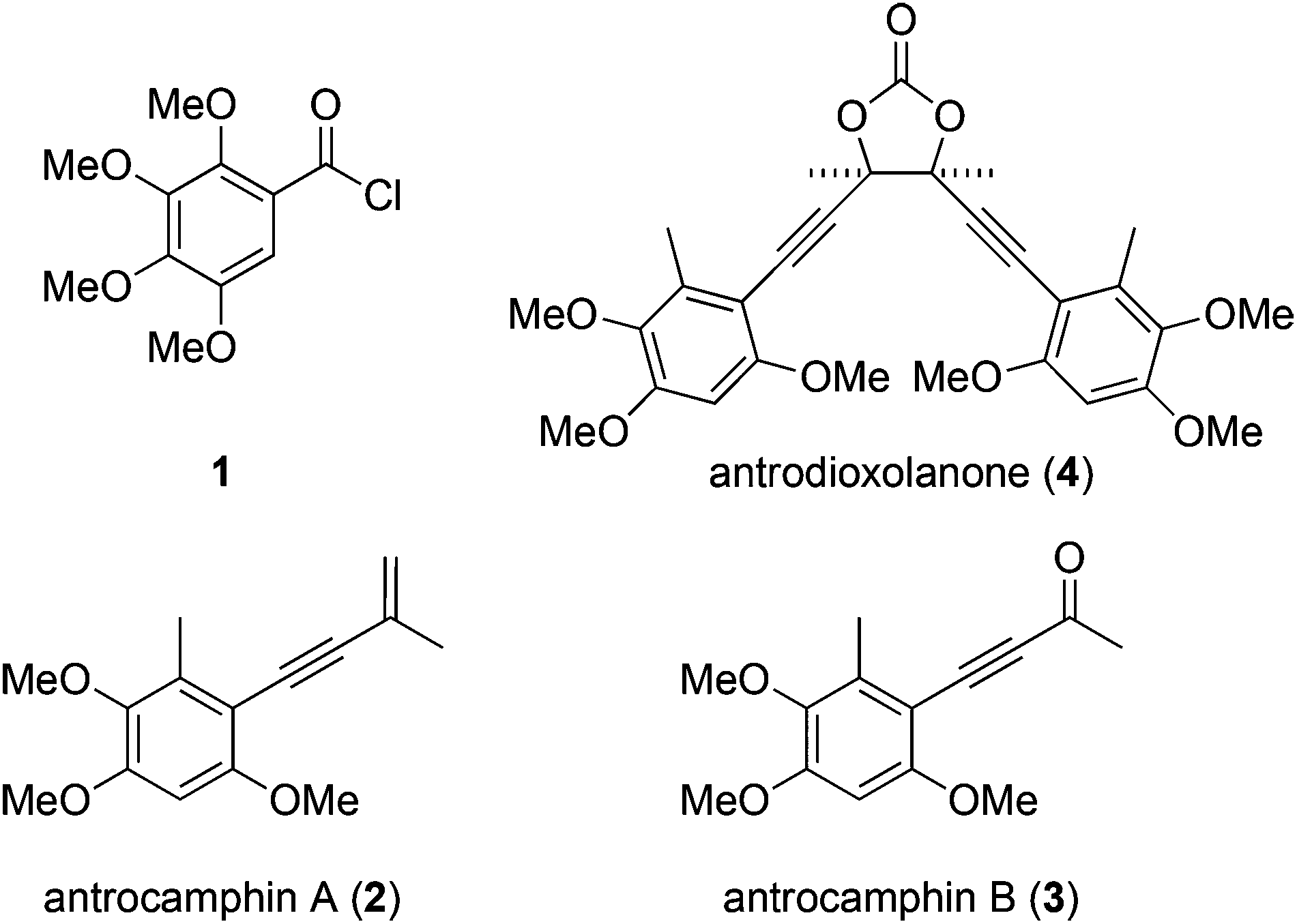 | ||
| Fig. 1 Benzenoid metabolites reportedly isolated from Antrodia camphorata.1 | ||
Antrocamphin A (2) has attracted significant attention due to its anti-inflammatory activity,1,3,4 which is comparable to that of ibuprofen in some assays. Indeed, during the course of our work, Chang, Wu and colleagues reported the first synthesis of 2, along with a series of analogues that were evaluated for anti-inflammatory activities.5 Syntheses of the congener antrocamphin B (3), and the symmetrical, dimeric antrodioxolanone (4) have not previously been reported.
Herein we describe the first synthesis of antrocamphin B (3), an improved synthesis of antrocamphin A (2), and approaches towards antrodioxolanone (4), culminating in the synthesis of its chiral epimers. Studies aimed at elucidating the mode of anti-inflammatory action of antrocamphin A are also reported.
Results and discussion
The antrocamphins (2–3) and antrodioxolanone (4) possess a common benzenoid moiety, which we felt could be exploited in our synthetic endeavours. Thus, a simple retrosynthetic analysis led back to Sonogashira reactions of the iodide 5 (Scheme 1). Chang, Wu and co-workers used a similar approach in their synthesis of antrocamphin A, in which they prepared 5 by the silver trifluoroacetate-mediated iodination of 2,3,5-trimethoxytoluene (10) (see Scheme 2 for structure), in turn derived from o-vanillin in four steps and 46% overall yield.5 Our approach began with Vilsmeier–Haack formylation of 3,5-dimethoxytoluene (6),6 followed by Baeyer–Villiger oxidation7 of the resultant benzaldehyde 7 (Scheme 2). Initially the latter reaction provided the desired phenol 8 contaminated by the corresponding quinone 9, resulting from over oxidation. Although easily separable, the formation of the quinone could be avoided by keeping reaction times short. Methylation of 8 then provided 10. Treatment with N-iodosuccinimide and catalytic trifluoroacetic acid8 furnished a quantitative yield of the desired iodide 5. Alternatively, NaI/Oxone9 provided the iodide more cheaply, although in lower yield. The identity of the iodide was confirmed by a 1D NOESY experiment. Irradiation of the aryl proton at 6.41 ppm led to enhancement of two methoxy signals, which cannot occur in the regioisomer 11.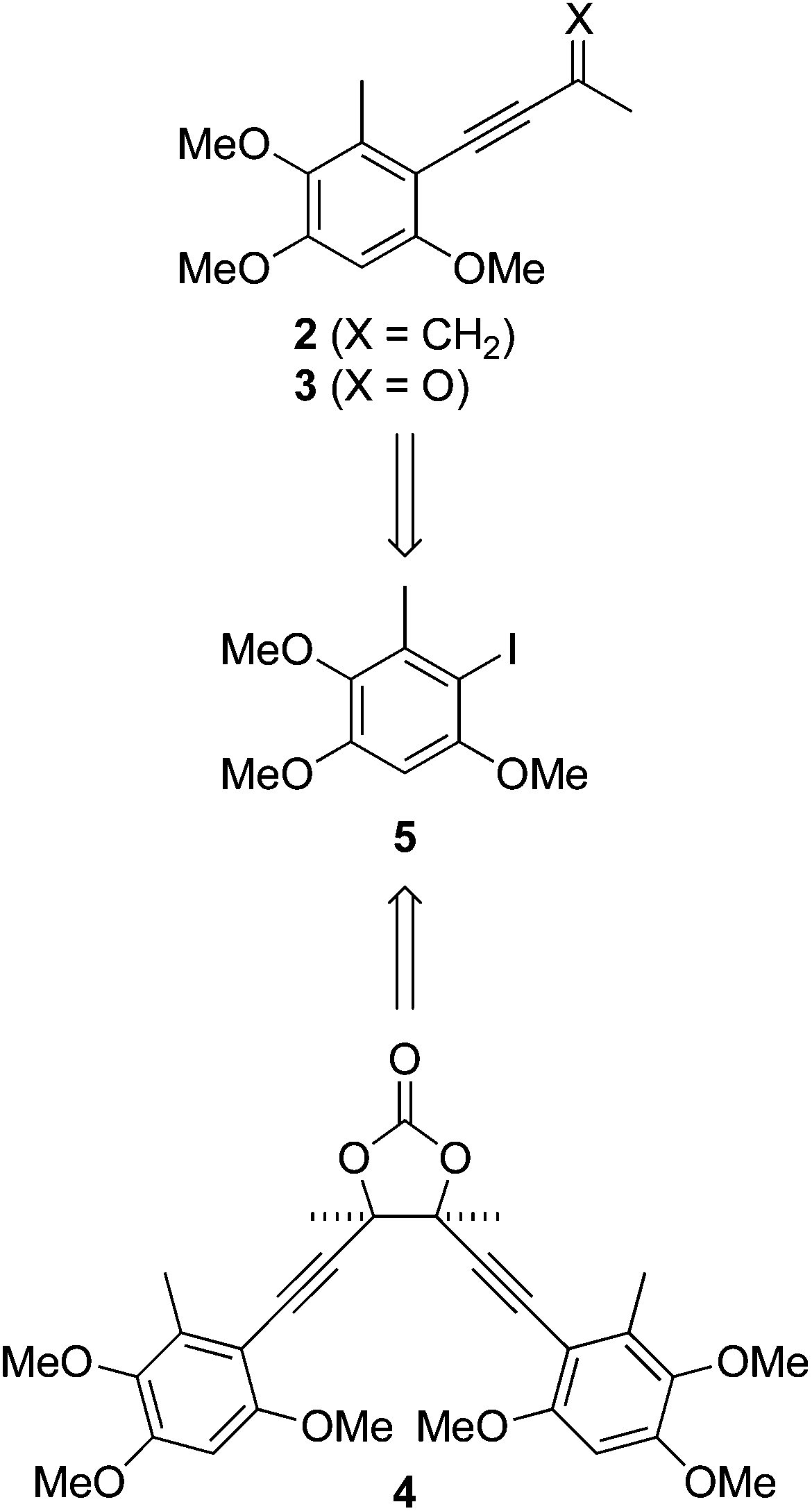 | ||
| Scheme 1 Retrosynthetic analysis. | ||
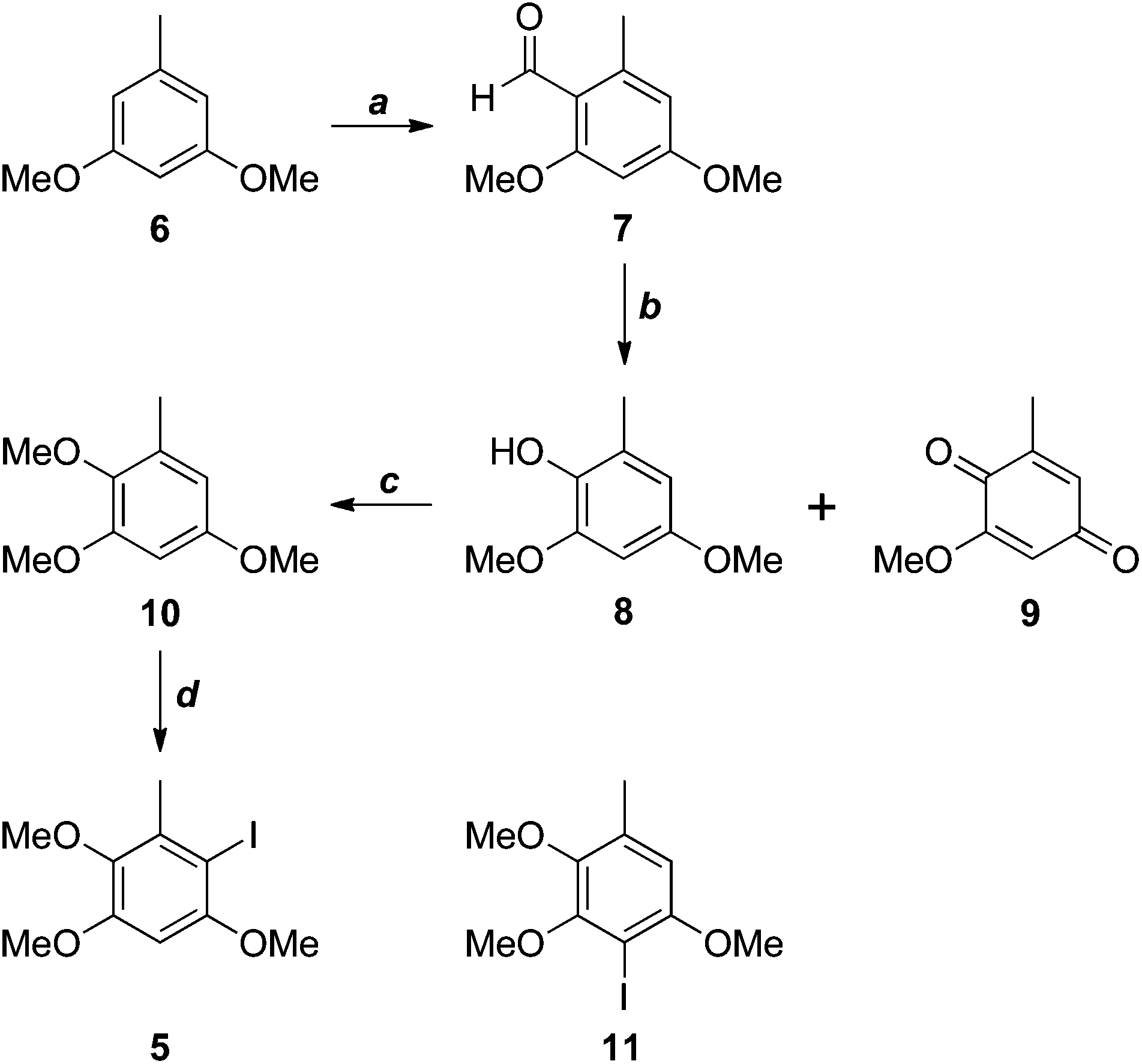 | ||
| Scheme 2 Reagents, conditions and yields: (a) POCl3, DMF, 97%; (b) H2O2, H2SO4, MeOH, 97% (optimised for 8); (c) MeI, K2CO3, DMF, 96%; (d) NIS, TFA, MeCN, quant.; or I2, Oxone, H2O, 77%. | ||
Synthesis of the antrocamphins
Wu and colleagues completed their total synthesis of antrocamphin A with a low-yielding (10%) Sonogashira coupling of iodide 5 with enyne 12 (X = H, Scheme 3).5 We also encountered problems with this reaction. Complete conversion to antrocamphin A (2) was not achieved despite varying the base, increasing the excess of the terminal alkyne, and carrying out the reaction in a sealed vessel. At best, a conversion of 44% (based on the 1H NMR spectrum of the crude product) was obtained, with the desired product accompanied by the homocoupled diyne 13 and unreacted iodide 5. While the volatility of 13 facilitated its simple removal from the crude product, the very similar chromatographic mobility of 5 and antrocamphin A (2) made purification virtually impossible. Accordingly we investigated the Castro–Stephens reaction10 of the copper acetylide 12 (X = “Cu”)11 (Scheme 3). Pleasingly, coupling of this species proceeded smoothly in refluxing pyridine, providing antrocamphin A as a yellow solid in 74% yield. The spectroscopic features of synthetic 2 were consistent with the natural product1 and previously synthesised material.5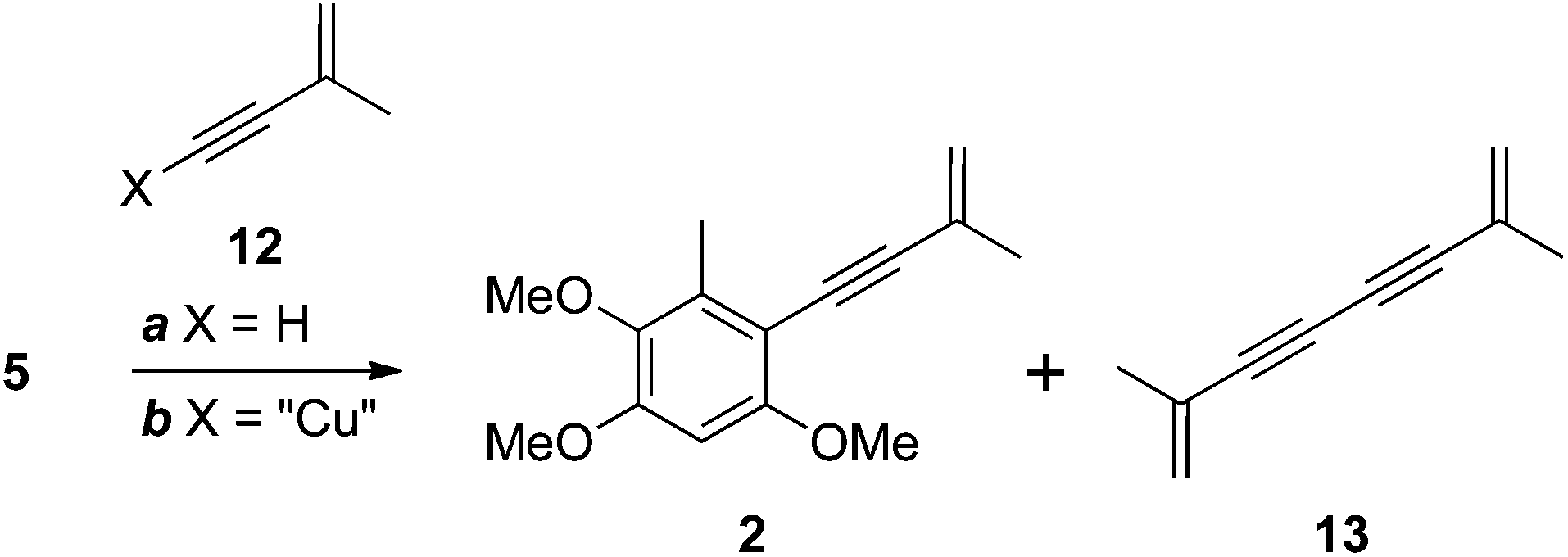 | ||
| Scheme 3 Reagents, conditions and yields: (a) Pd(PPh3)2Cl2, CuI, NEt3, MeCN, 80 °C, sealed tube, 44% (as a mixture with 5); (b) pyridine, reflux, 74% (2). | ||
Wu and co-workers have recently applied an efficient, two-step strategy for the installation of the 3-methylbut-3-en-1-ynyl substituent in a very electron-rich substrate 14 in their synthesis of benzocamphorin F 17 (Scheme 4), a co-metabolite of antrocamphin A from Antrodia camphorata.12
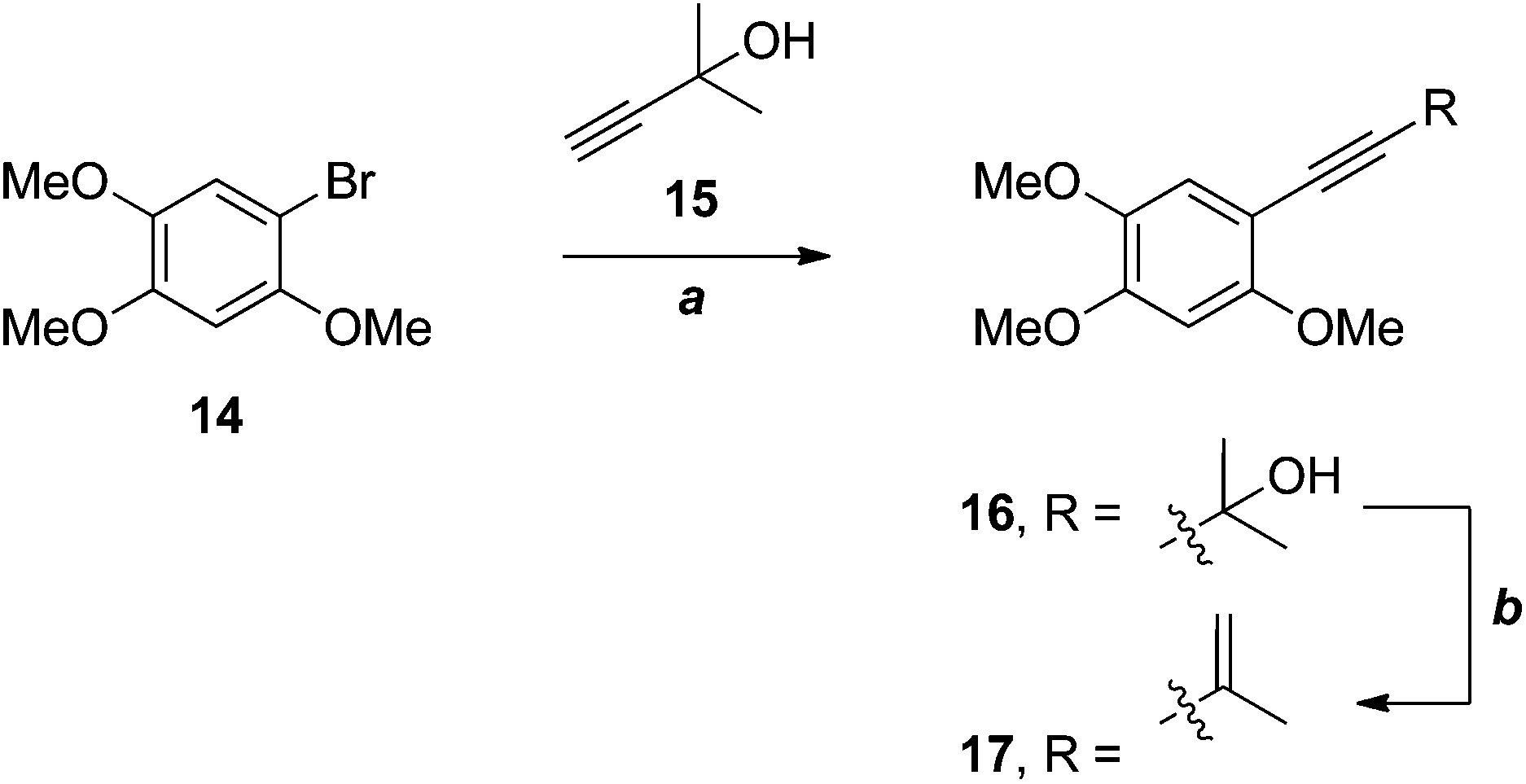 | ||
| Scheme 4 Wu's synthesis of benzocamphorin F.12 (a) Pd(PPh3)4, CuI, DMF, 85%; (b) MsCl, PhMe, microwave, 92%. | ||
Our attention now turned to antrocamphin B (3), which could in principle be derived from the Sonogashira coupling of the iodide 5 with 3-butyne-2-one. However, electron deficient alkynes are poor substrates for the Sonogashira reaction, so we opted for two-step coupling of the propargyl alcohol 18, followed by oxidation (Scheme 5). After some experimentation, the Sonogashira coupling to give 19 was achieved in moderate yield following chromatography and evaporation of the homocoupled diyne 20, which had similar chromatographic mobility to 19. Somewhat surprisingly, the Castro–Stephens reaction of the copper acetylide derived from 18 failed in this case. Oxidation of 19 with MnO213 or, more reliably, under modified Swern conditions14,15 provided antrocamphin B (3), as a bright yellow solid, in excellent yield. The spectroscopic data derived from 3 were consistent with those reported for the natural product.1
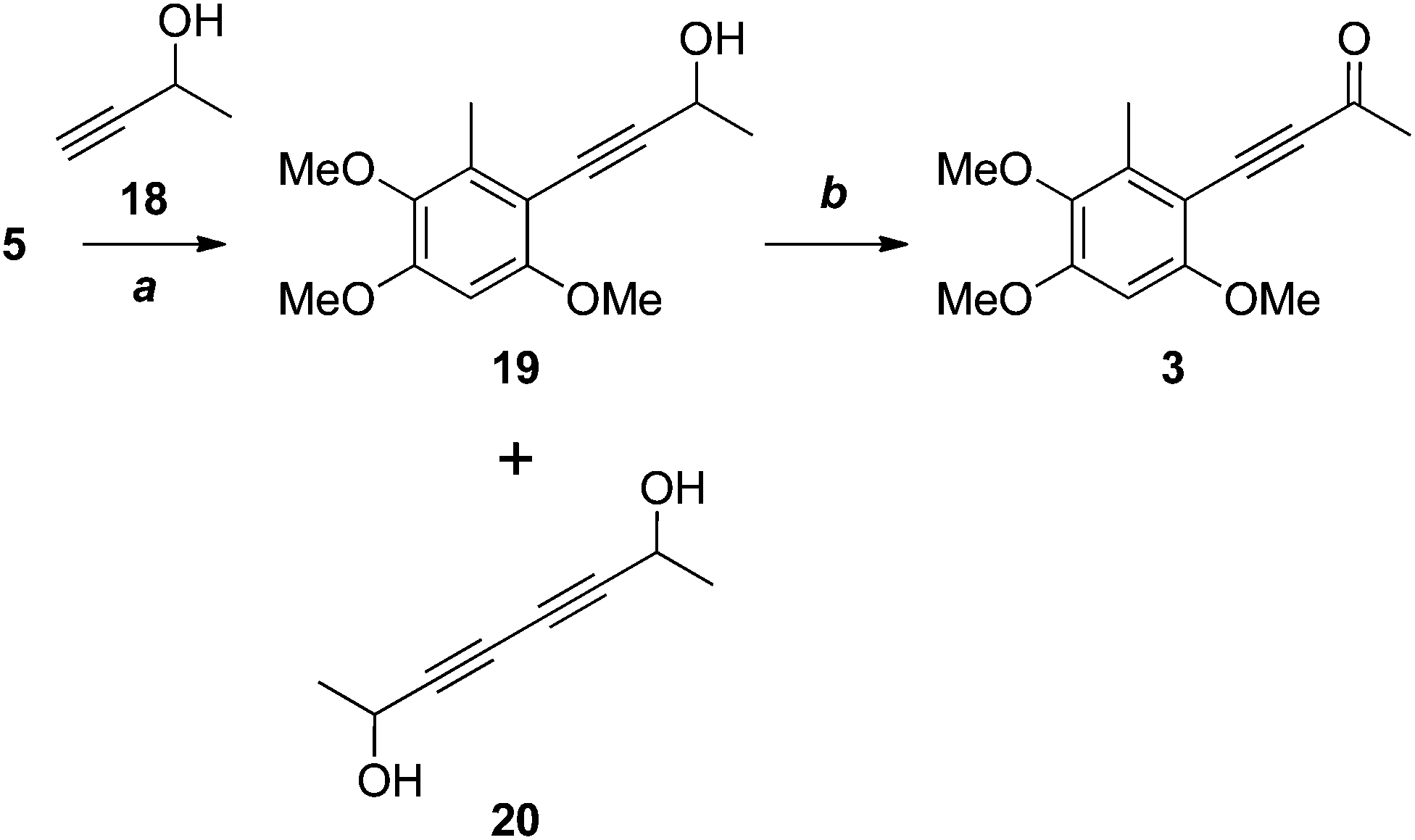 | ||
| Scheme 5 Reagents, conditions and yields: (a) Pd(PPh3)2Cl2, CuI, HNEt2, DMSO, 75 °C, 58%; (b) MnO2, DCM, 80%; or, DMSO, Ac2O, quant. | ||
Approaches towards antrodioxolanone
Application of a key double Sonogashira reaction to the synthesis of the more complex antrodioxolanone (4) required the meso-diol 25, which was prepared in two steps from diacetyl (21) and lithium TMS-acetylide (22), as described previously (Scheme 6).16 Cyclocondensation with triphosgene then gave the novel dioxolanone 26. Unfortunately all attempts to effect the Sonogashira reaction resulted only in the consumption of the diyne 26, with the iodide 5 recovered essentially quantitatively. In an attempt to emulate the Castro–Stephens reaction that was successful in the synthesis of antrocamphin A, 26 was subjected to the conditions that gave copper acetylide 12 (X = “Cu”). Although the identity of the bright yellow precipitate that formed could not be conclusively assigned, on the assumption that the diacetylide had formed, it was heated with iodide 5; however, once again, only 5 was recovered.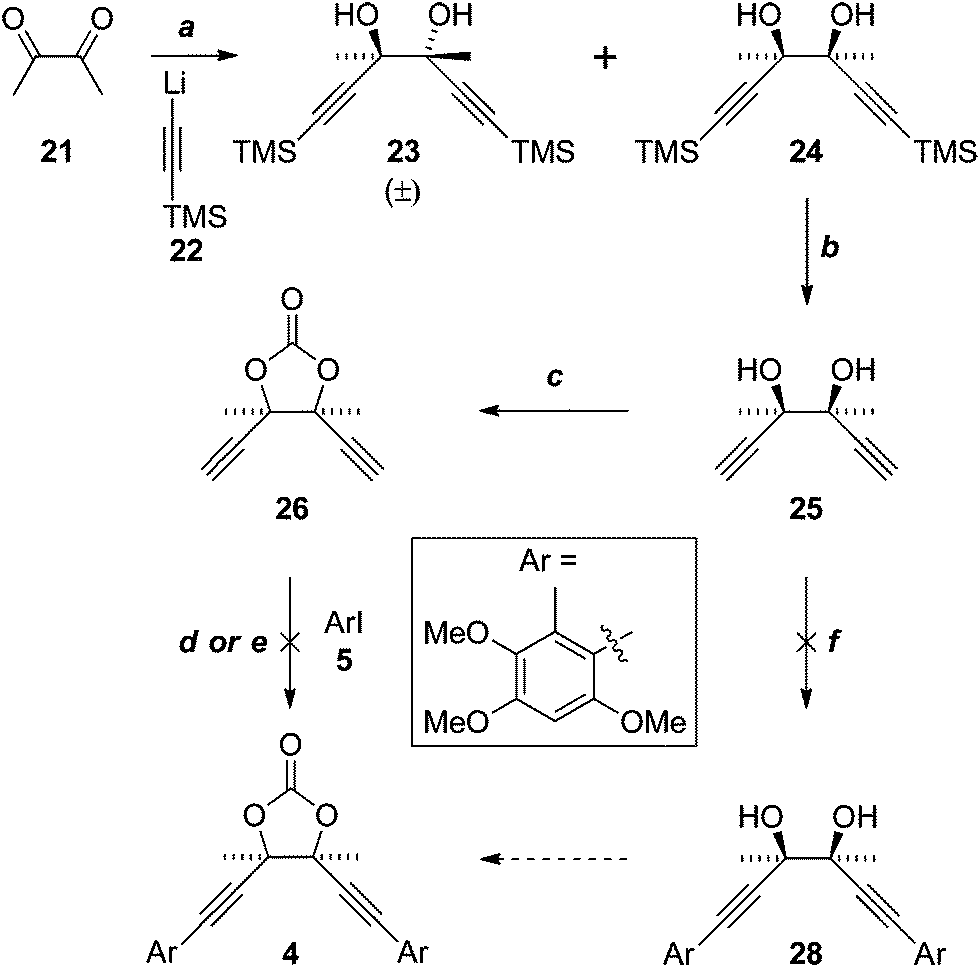 | ||
| Scheme 6 Reagent, conditions and yields: (a) see ref. 16; (b) K2CO3, DCM–MeOH, 96%; (c) CO(OCCl3)2, pyridine, THF, 83%; (d) Pd(PPh3)2Cl2; NEt3 thin film, or with MeCN, or HNEt2–MeCN, all 0%; (e) 1. HONH3Cl, CuSO4·5H2O, NH3, H2O, EtOH (52% based on diacetylide); 2. 5, pyridine, reflux (0%); (f) Pd(PPh3)2Cl2, NEt3 or HNEt2, MeCN, both 0%. | ||
The failure of the Sonogashira and Castro–Stephens reactions of 26, and its instability under the reaction conditions, was puzzling. Such reactions of 1,5-diynes are numerous; however, the vast majority of examples involve o-ethynylbenzenes. To the best of our knowledge there are no examples with a bridging 5-membered ring. This suggested that the rigid 5-membered-ring, and the meso configuration of 26, may predispose the dialkyne to π-chelate palladium (as in 29), leading to some unknown mode of degradation, perhaps via oxidative cyclisation to the palladacycle 30 (Scheme 7). Although 3-palladabicyclo[3.2.0]hepta-1,4-dienes such as 30 appear to be unprecedented, the [3.3.0]-palladabicycle 32, synthesised by oxidative addition to the diene 31, has been isolated and characterised spectroscopically.17
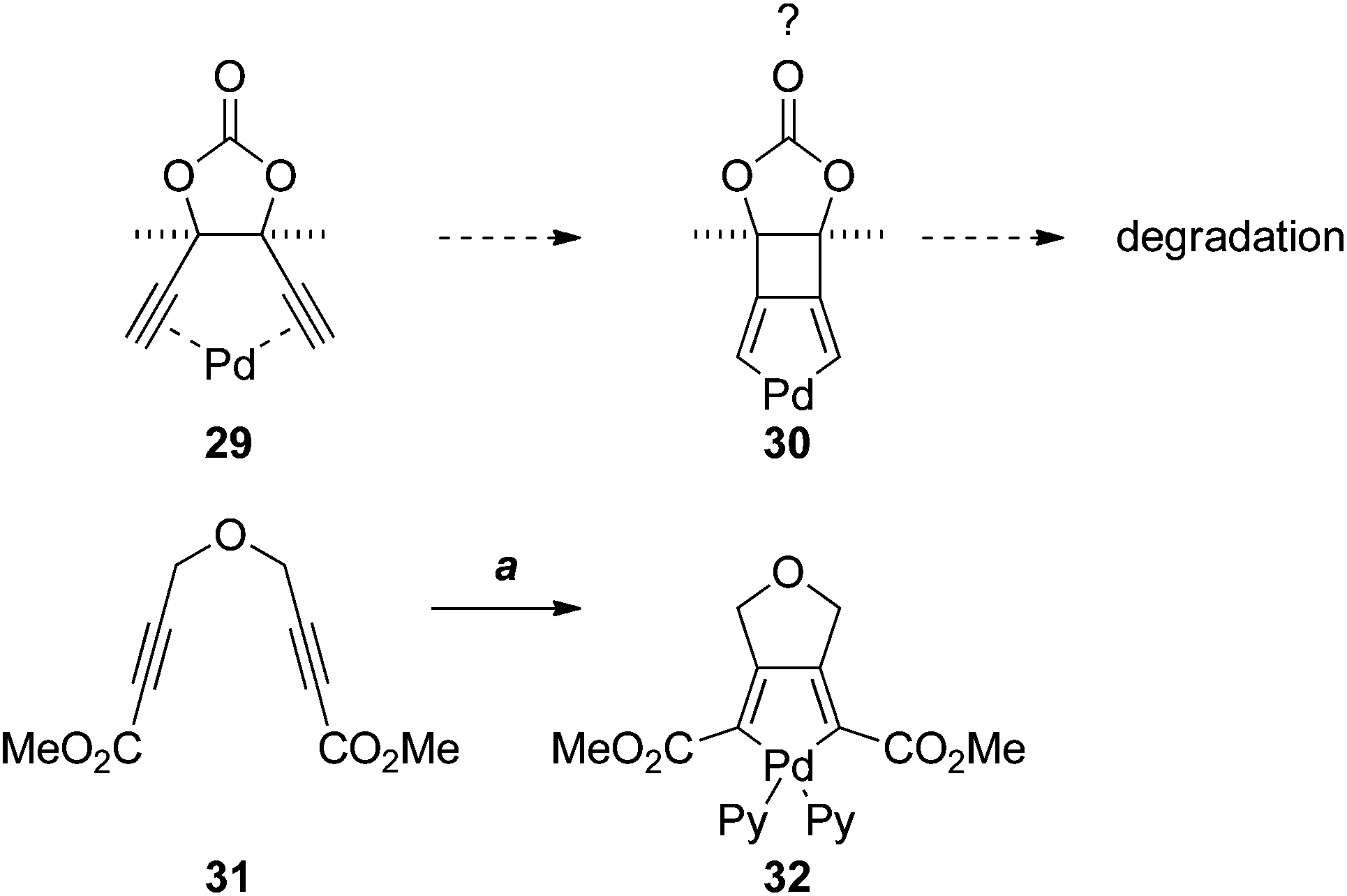 | ||
| Scheme 7 Reagents, conditions (a) 1. Pd2(dba)3·CHCl3, acetone; 2. pyridine (Py), DCM.17 | ||
There are just two reported examples of the double Sonogashira reaction of a 1,5-diyne with bridging sp3-hybridised carbon atoms (Scheme 8).18,19 These precedents suggested that, for our purposes, it may be possible to effect the sp–sp2 coupling prior to formation of the dioxolane, that is, with diol 25 (Scheme 6), in which greater conformational freedom might disfavour oxidative cyclisation. However, in practice, none of the desired coupling product 28 was isolated.
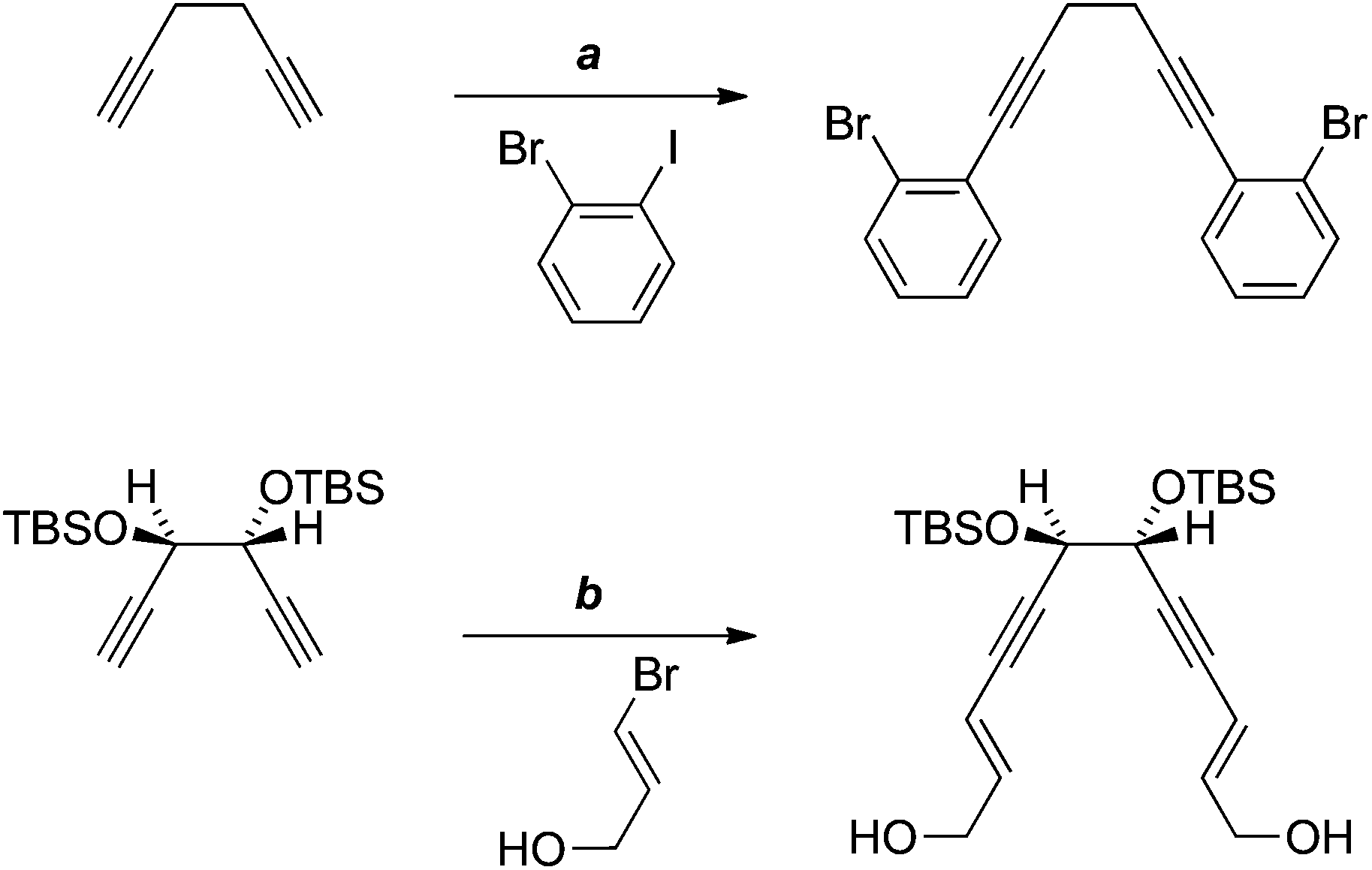 | ||
| Scheme 8 Reagents, conditions and yields: (a) PdCl2(PPh3)2, CuI, NEt3, piperidine, 46%;18 (b) PdCl2(PPh3)2, CuI, HNEt2, 79%.19 | ||
Given the apparent incompatibility of the diynes 25 and 26 with Sonogashira coupling conditions, out attention turned to strategies in which the quaternary stereocentres required for antrodioxolanone are constructed late in the synthesis. The first of these is outlined in Scheme 9. The Sonogashira coupling of TMS-acetylene 29 with iodide 5 suffered from all of the problems associated with the analogous reaction of 12 (X = H, Scheme 3), and as a result the yield of 30 was low. The coupling with the significantly cheaper masked acetylene 31 was much more efficient, and deprotection20 of 32 proceeded smoothly to provide the terminal acetylene 33 in excellent yield.
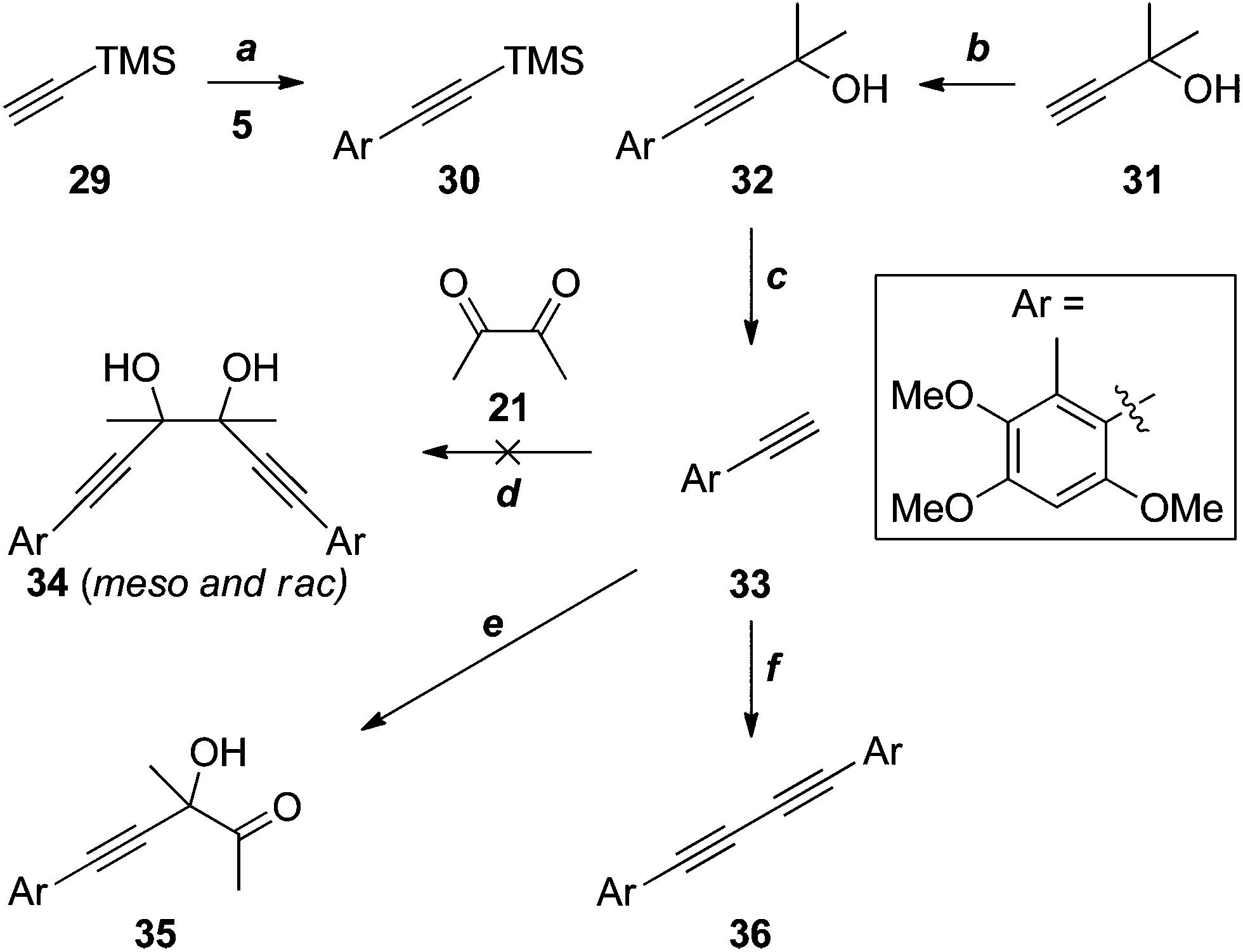 | ||
| Scheme 9 Reagents, conditions and yields: (a) PdCl2(PPh3)2, CuI, NEt3, 21%;(b) PdCl2(PPh3)2, CuI, HNEt2, DMSO, 71%; (c) NaOH, PhMe, reflux, 94%; (d) Na, THF, then 21; or RMgBr, Et2O and/or THF, then 21 (R = Et or i-Pr. All gave complex mixtures); (e) EtMgBr, Et2O, then 21, 11%; (f) BuLi, THF, then 21, 17%; or PrN4OH, DMSO, 21, 10%. | ||
Several attempts at addition of the acetylide, generated in situ by deprotonation of 33 with sodium21 or Grignard reagents,22 to diacetyl (21), resulted in complex mixtures of products, with none of the glycol 34 detected. On one occasion, the mono-addition product 35 was isolated in low yield. When BuLi23 or tetrapropylammonium hydroxide24 were the bases used, the only identifiable product was the diyne 36 arising from oxidative coupling, presumably due to trace contamination by transition metal(s). It is possible that competing deprotonation of diacetyl (21) by the acetylide contributes, at least in part, to the failure of these reactions. However, an attempted reaction of the less basic cerium acetylide25 also failed to give any discernible products.
The failure of these reactions, despite the close precedents cited above, including the double addition reaction of lithium TMS-acetylide in our own hands (21 + 22 → 23, Scheme 6), led us to hypothesise that the electron-rich benzene ring of 33 was somehow negatively impacting the outcome. Indeed, application of the most promising conditions to phenylacetylene (37) gave an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the diastereomeric diols 38 and 39 in reasonable yield (Scheme 10), matching the result reported previously.22 We are unable to explain why the analogous reaction of 33 fails. The cyclocondensation of the diols 38 and 39 gave the corresponding cyclic carbonates 40 and 41, in low yield, after chromatographic separation.
:1 ratio of the diastereomeric diols 38 and 39 in reasonable yield (Scheme 10), matching the result reported previously.22 We are unable to explain why the analogous reaction of 33 fails. The cyclocondensation of the diols 38 and 39 gave the corresponding cyclic carbonates 40 and 41, in low yield, after chromatographic separation.
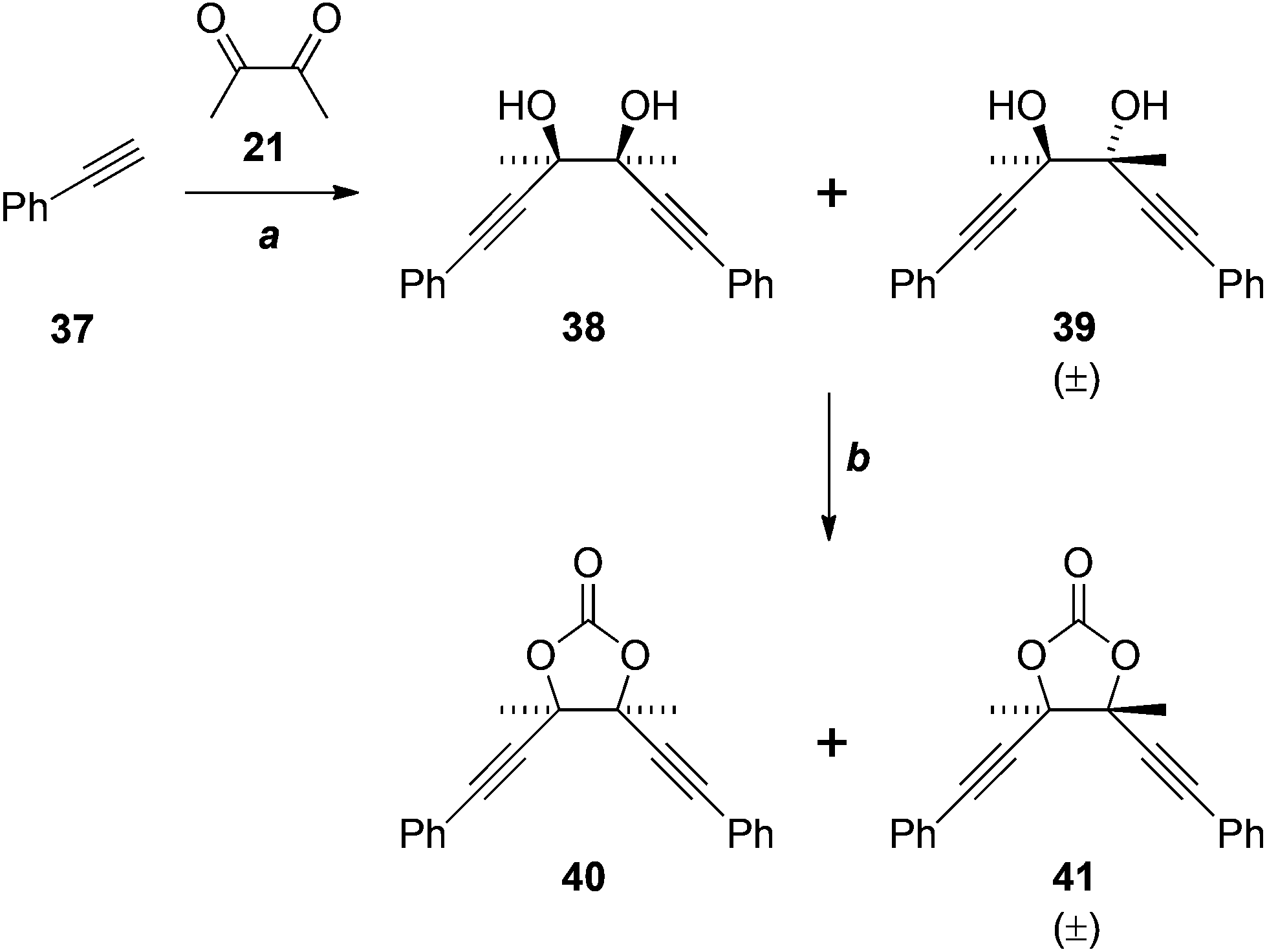 | ||
| Scheme 10 Reagents, conditions and yields: (a) EtMgBr, Et2O, then 21, 68% (1:1 mixture of 38 and 39); (f) CO(OCCl3)2, pyridine, THF, 28% (40), 19% (41). | ||
Given the possible complication of deprotonation of diacetyl by acetylide nucleophiles, we investigated the alternative addition of more reactive methylmetallic nucleophiles to dione 47 (Scheme 11), which lacks appreciably acidic protons. Thus, the Sonogashira reaction of propargyl alcohol (42) with iodide 5, and oxidation of the resultant aryl acetylene 43, gave aldehyde 44, which underwent efficient pinacol coupling to give the glycols 45. It was impossible to determine the diastereomeric ratio from the 1H NMR spectrum of this mixture due to coincident signals; however, this became apparent upon conversion to the cyclic carbonates 46. Although it was not possible to distinguish the cis (meso) from the trans (rac) isomers, the 1H NMR spectrum of the mixture did reveal a ∼13:20 ratio of diastereomers. This was somewhat immaterial, as the dione 47, devoid of stereocentres, was the target. Treatment of 45 with MnO2 led to significant oxidative cleavage, regenerating 44; this was avoided under modified Swern conditions, providing dione 47 in excellent yield. Unfortunately, the attempted addition reaction of methylmagnesium iodide26 failed entirely, with no evidence for the formation of the desired glycol 34, or any other identifiable material. With methyllithium,27 terminal acetylene 33 was the only product identified. Presumably this arises from scission of an addition intermediate such as 48. The instability of such intermediates may partially explain the failure to access the sterically congested glycol moiety required for the synthesis of antrodioxolanone via nucleophilic addition chemistry.
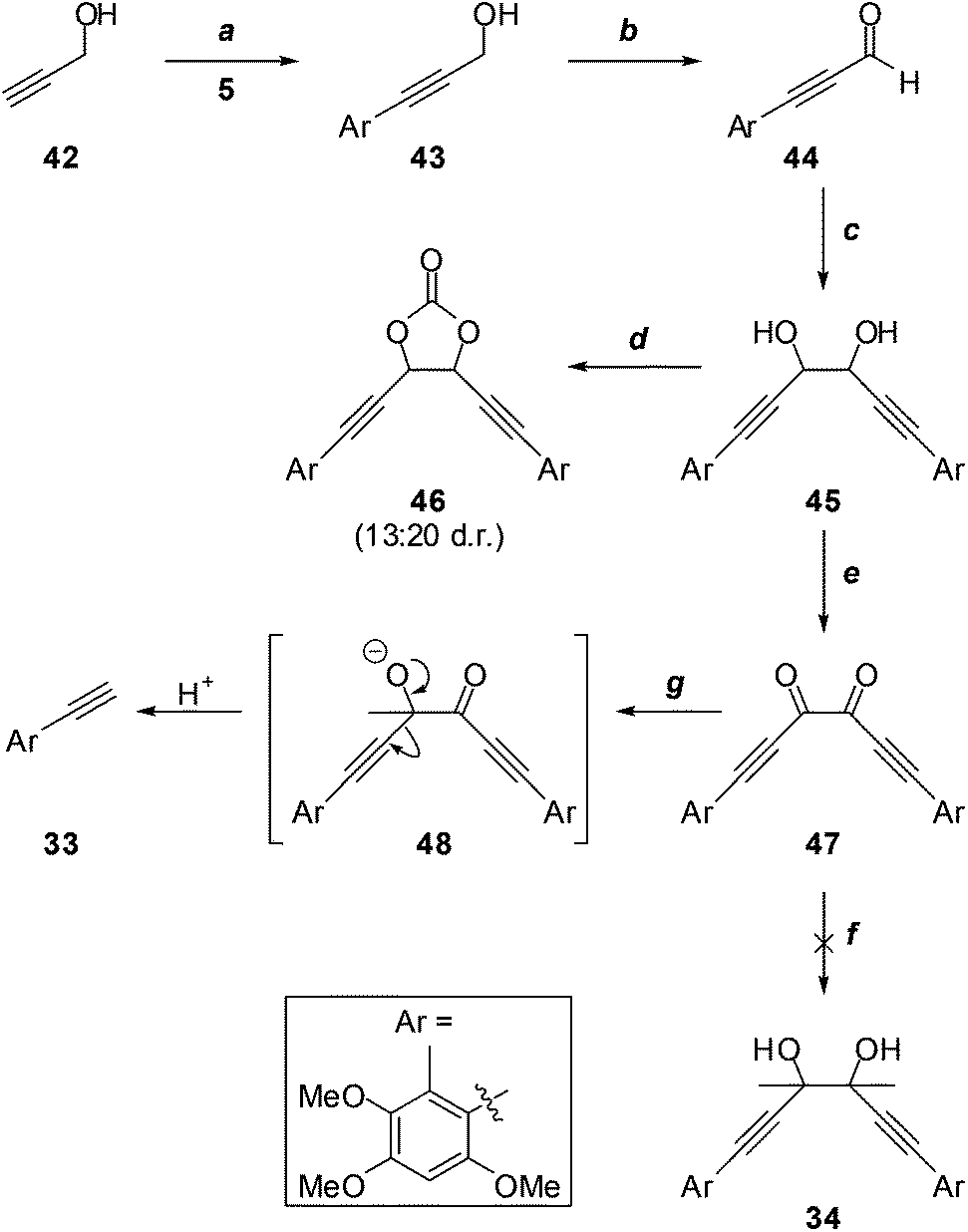 | ||
| Scheme 11 Reagents, conditions and yields: (a) PdCl2(PPh3)2, CuI, HNEt2, DMSO, 67%; (b) MnO2, DCM, 67% or Ac2O, DMSO, 55%; (c) Cu/Zn, AcOH, THF, 82%; (d) CO(OCCl3)2, pyridine, DCM, 45%; (e) MnO2, DCM, 14% (+30% 44) or Ac2O, DMSO, 99%; (f) MeMgBr, Et2O and or THF, 0%; MeLi, Et2O–THF, 50% (33). | ||
In the original report on the isolation of antrodioxolanone (4), it was noted that the natural product may arise biogenetically through an “intermolecular cyclization at the acetyl group” of 3.1 Indeed, given the symmetry of antrodioxolanone (4), a pinacol coupling of antrocamphin B (3) seemed a plausible biosynthetic step, and an appealing means to construct the contiguous quaternary stereocentres in a total synthesis. This realisation led us to explore the pinacol coupling of the ynal 44 described above (Scheme 11). In parallel, we also investigated pinacol couplings of antrocamphin B.
We are unaware of any biosynthetic examples of pinacol couplings. However, photochemically-induced pinacol coupling of an aryl alkynyl ketone has been observed upon irradiation at 300 nm.28 It is conceivable that a non-enzymatic, sunlight-induced pinacol coupling of antrocamphin B (3) might be involved in the biosynthesis of antrodioxolanone (4). This led us to irradiate solutions of 3 (Scheme 12). However, in all cases no reaction was detected by TLC.
 | ||
| Scheme 12 Reagents, conditions and yields: (a) Cu/Zn, AcOH, THF, 22% (49), 8% (50); (b) Na, PhBr, PhMe, 23% (19), 4% (33); (c) NEt3, UV (TLC lamp) or i-PrOH, AcOH, ambient lab light then direct sunlight. | ||
More conventional metal-mediated pinacol couplings were then investigated. The Cu/Zn couple that worked well for ynal 44 (Scheme 11) did indeed give a pinacol coupling product with antrocamphin B (3), but unfortunately accompanied by semi-reduction of the alkynes to give a trans-diene 49. Although this appeared to be a single diastereomer, it was not possible to define the relative configuration with the spectroscopic data available. In addition, the dihydrostilbene 50, arising from reductive coupling at the benzylic position, was isolated in low yield. X-ray crystallography revealed this to be the meso isomer (Fig. 2), although the formation of the chiral diastereomers cannot be ruled out, as not all products of this reaction were able to be purified. The attempted sodium/bromobenzene-promoted pinacol coupling29 of 3 resulted in degradation, whereas no reaction was observed with this reductant system in carbon tetrachloride or cyclohexane solutions. In toluene, the secondary alcohol 19, and the terminal alkyne 33, presumably resulting from scission of the alkoxide precursor to 19, were the only detectable products. Treatment of 3 with TiCl4–TBAI, which is an effective promoter of pinacol coupling for aryl methyl ketones,30 gave a complex mixture of products.
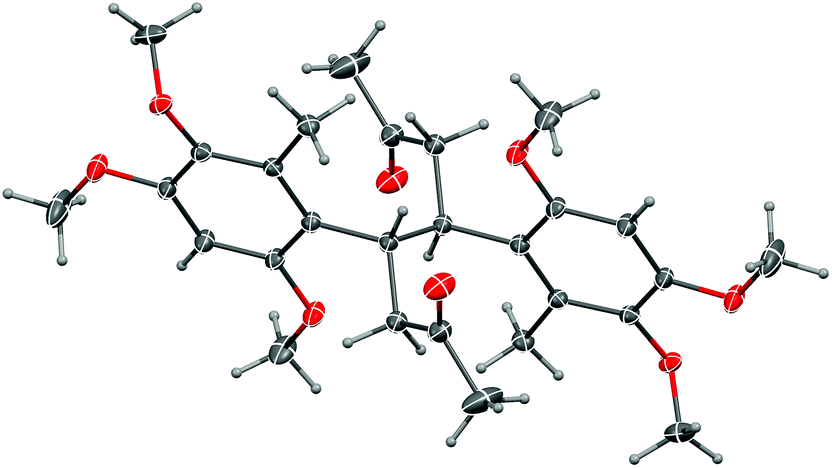 | ||
| Fig. 2 Representation of the crystal structure of 50. Ellipsoids are shown at 50% probability amplitudes with hydrogen atoms assigned arbitrary radii. | ||
Of the reducing agents investigated, only SmI2,14,15 provided the pinacol coupling products 34a and 34b, in 55% yield, but unfortunately favouring the chiral isomers 34b 10:1 (Scheme 13). The diastereomers were separable by HPLC but, disappointingly, and hampered by material availability, attempts to convert the meso isomer into antrodioxolanone were unsuccessful. When the 10:1 mixture of diastereomeric glycols was treated with triphosgene, only the chiral trans-isomer, (±)-epi-antrodioxolanone (51) was isolated in very low yield, as confirmed with an X-ray crystal structure (Fig. 3). The yield of 51 could neither be improved using NEt3 or DMAP as catalysts, nor carbonyldiimidazole as electrophile, and presumably results from steric congestion in the bis-tertiary glycol. A final attempt at the one-pot pinacol coupling/cyclisation using SmI2 and methyl chloroformate31 gave 51 directly, albeit in low yield, with none of the meso natural product 4 detected.
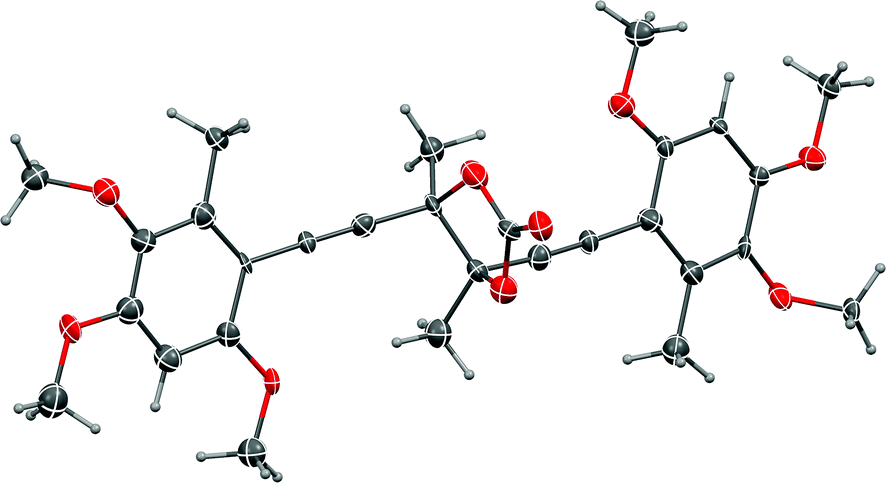 | ||
| Fig. 3 Representation of the crystal structure of (±)-epi-antrodioxolanone (51). R,R-Enantiomer shown. Ellipsoids are shown at 50% probability amplitudes with hydrogen atoms assigned arbitrary radii. | ||
 | ||
| Scheme 13 Reagents, conditions and yields: (a) SmI2, THF, 55% (1:10 mixture of 34a:34b); (b) CO(OCCl3)2, pyridine, DCM, 16% (c) 1. SmI2, THF; 2. ClCO2Me (32%). | ||
Anti-inflammatory activity
Following their initial isolation and structure elucidations, the antrocamphins and antrodioxolanone were assessed for anti-inflammatory effects through their impact on superoxide anion production by neutrophils, induced by the inflammatory cytokine fMLP (N-formyl-Met-Leu-Phe).1 Antrocamphin B and antrodioxolanone showed no activity in this assay, but antrocamphin A suppressed superoxide production with an IC50 of 9 ± 3 μM, more effectively than ibuprofen (IC50 = 28 ± 3 μM).1 Synthetic antrocamphin A was later also shown to inhibit the fMLP-induced excretion of elastase by human neutrophils, and many analogues of the natural product more potently inhibited superoxide generation by these cells.5Additional mode of action studies on antrocamphin A were conducted by Wang and coworkers.3 The natural product dose-dependently suppressed the production of inflammatory cytokines NO and prostaglandin E2 in lipopolysaccharide-challenged macrophages (RAW 264.7 cells). The expression of inflammatory enzymes cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) were also downregulated by antrocamphin A. The authors hypothesised that this could be due to suppression of NFκB, a transcription factor that is a central player in the inflammatory cascade. Cytosolic NFκB is bound by the inhibitor IκB. Phosphorylation of IκB by the kinase IKK causes the NFκB–IκB complex to dissociate, allowing NFκB to enter the nucleus, where it induces transcription of a host of genes involved in the inflammatory response. Nuclear accumulation of NFκB was indeed dose-dependently decreased by antrocamphin A. Concurrently, expression of IκB increased, while that of the phosphorylated form of IKK, decreased.3
We have recently developed a cellular assay to determine the effects of novel thalidomide derivatives on the NFκB activation pathway, as a measure of anti-inflammatory activity.32–34 To measure inhibition of NFκB pathway signalling, a Tumour Necrosis Factor (TNF) transcriptional reporter cell line was constructed by linking the green fluorescent protein (GFP) reporter gene to the NFκB-responsive human TNF promoter. The construct was then inserted into the genome of the human T cell line, Jurkat E6-1, to generate the reporter line, FRT-Jurkat TNF, as previously described.35,36 As a measure of TNF promoter activity, GFP activity can be quantitated by flow cytometry. This method has the added advantage of being able to concurrently assess the cytotoxicity of each compound, by comparing forward- and side-scatter of light during flow cytometry.
In the current study, antrocamphin A dose-dependently reduced the amount of expression by the TNF-reporter line (Fig. 4). The results suggest an IC50 for NFκB-induced expression inhibition of approximately 100 μM; however, it was not possible to determine this value accurately as, at the higher concentrations, there was a significant effect on cell viability (Fig. 4). Although the issue seems to have been avoided in earlier publications, antrocamphin A does exhibit dose-dependent cytotoxicity towards RAW 264.7 cells, causing approximately 30% cell death at 20 μg mL−1.3 More recently, antrocamphin A was shown to be toxic to four human tumour-derived cell lines – Doay (breast medulloblastoma), Hep2 (laryngeal carcinoma), MCF-7 (breast adenocarcinoma) and HeLa (cervical epithelioid carcinoma) – with ED50 values ≤10 μg mL−1.4
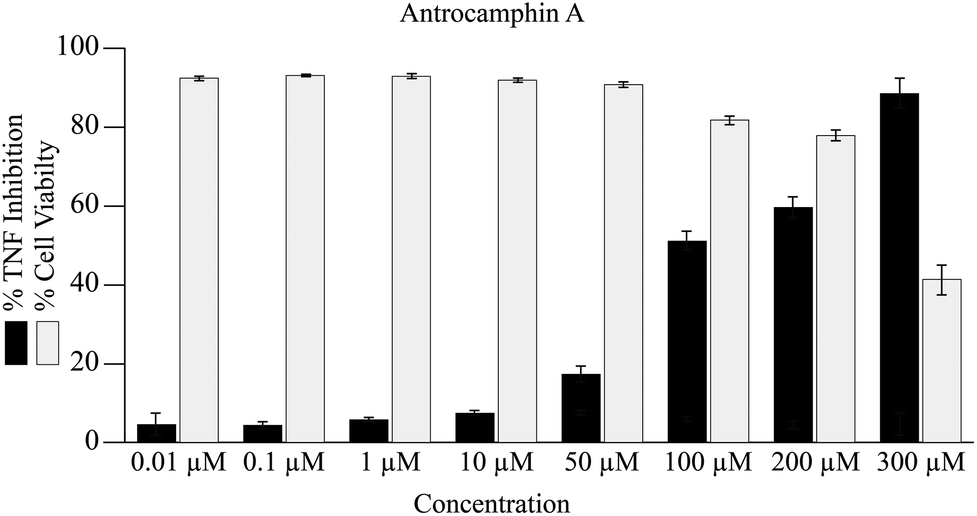 | ||
| Fig. 4 Inhibition of TNF-reporter gene expression and consequence on cell viability following treatment with antrocamphin A (2) for 24 h. Data represent geometric mean expression levels of GFP driven by a TNF promoter, measured by flow cytometry (n = 3, bars represent mean ± SEM). Cell viability was assessed by comparing forward- and side-scatter as a measure of cellular size and granularity. The cell population in each sample that exhibited low granularity were considered dead, as confirmed by propidium iodide staining. | ||
It seems that the biological activity of antrocamphin B (3) has not been considered since its isolation. We also assessed the ability of this compound to suppress NFκB TNF-promoter mediated transcription. Although there is evidence for the inhibition of TNF transcription, there is no clear dose–response relationship and the data are clearly complicated by the cytotoxicity of the compound (Fig. 5, IC50 = 10.7 ± 0.3 μM [std. dev]). This is not surprising given that antrocamphin B is a Michael acceptor.
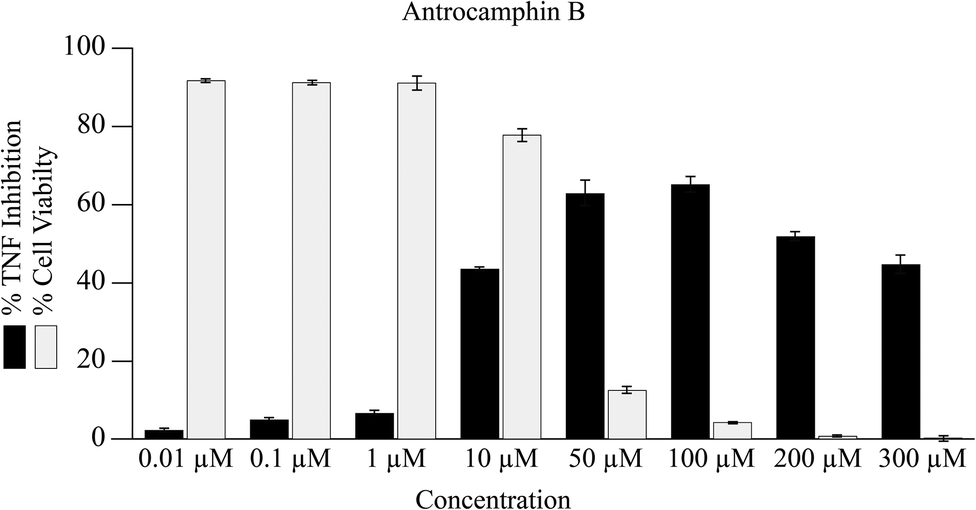 | ||
| Fig. 5 Inhibition of TNF promoter transcriptional activity and consequence on cell viability following treatment with antrocamphin B (3) for 24 h. Data represent geometric mean expression levels of GFP driven by the TNF promoter measured by flow cytometry (n = 3, bars represent mean ± SEM). Cell viability was assessed as described in Fig. 4 caption. | ||
Some synthetic intermediates and analogues of the antrocamphins and antrodioxolanone were also briefly assessed in the TNF inhibition assay (Fig. 6). Interestingly, 40 the analogue of antrodioxolanone (4) possessing phenyl substituents in place of the oxygenated aromatic substituents in the natural product, did inhibit TNF-induced expression at 100 μM with little effect on cell viability. The mixture of its trans-diastereomers 41 on the other hand, was quite cytotoxic, killing approximately 60% of cells at 10 μM. The ynal 44 also inhibited TNF expression, but with accompanying cell death. None of the compounds displayed activity warranting more rigorous examination.
 | ||
| Fig. 6 Inhibition of TNF reporter gene expression and consequence on cell viability following treatment with synthetic intermediates and analogues of the antrocamphins and antrodioxolanone for 24 h. Data represent geometric mean expression levels of GFP driven by a TNF promoter measured by flow cytometry. | ||
Conclusions
An improved synthesis of the anti-inflammatory natural product antrocamphin A (2), involving a key Castro–Stephens reaction, has been devised, along with the first synthesis of its congener antrocamphin B (3). Several approaches to the synthesis of the more complex antrodioxolanone (4) were thwarted, including a route involving a possibly biomimetic pinacol coupling of antrocamphin B. This latter strategy did, however, provide racemic epi-antrodioxolanone (51). The sterically congested 4,5-diethynyldioxolanone core of antrodioxolanone is unique amongst natural products and its stereoselective synthesis, in the presence of electron rich pendant aromatic rings, presents quite a challenge.Antrocamphin A (2) was shown to inhibit TNF expression with modest potency, supporting an earlier hypothesis3 that its anti-inflammatory effects arise, at least in part, by interfering with the nuclear localisation of the transcription factor NFκB. The potency of action determined herein is approximately an order of magnitude less than downstream measures of anti-inflammatory activity reported previously, which is not surprising for a drug acting on a signalling pathway that regulates the expression of genes associated with the inflammatory response.
Despite being a constituent of a Chinese traditional medicine that has presumably been used without severe adverse effects for some time, there is mounting evidence that antrocamphin A is toxic to some cell types. Whether this cytotoxicity is linked with its interference in the inflammatory signalling cascade and/or is selective towards cancerous cell lines, and whether antrocamphin A and related compounds exhibit safe therapeutic indices, remains to be established.
Experimental
General details
General details are as described previously.37Crystallography
Crystallographic data for 50 and 51 were collected at 100(2) K on an Oxford Diffraction Gemini or Xcalibur diffractometer fitted with Mo Kα radiation. Following multi-scan absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the program SHELXL-97.38 All H-atoms were added at calculated positions and refined by use of a riding model with isotropic displacement parameters based on those of the parent atoms. Anisotropic displacement parameters were employed for the non-hydrogen atoms.2,4-Dimethoxy-6-methylbenzaldehyde (7)6
POCl3 (14.5 mL, 0.156 mol) was added dropwise to a solution of 3,5-dimethoxytoluene (19 g, 0.12 mol) in DMF (100 mL) at 0 °C. The reaction was allowed to warm to room temperature over 24 h before being poured slowly into cold H2O (100 mL). After 15 min the suspension was further diluted with H2O (300 mL) and saturated NaHCO3 (100 mL), stirred overnight, then extracted with EtOAc (4 × 50 mL). The extract was dried and evaporated to give 7 as a white powder (21.9 g, 97%) sufficiently pure for the next step, m.p. = 64–65 °C [lit.6 64–65 °C]. 1H NMR (400 MHz) δ 10.48 (s, 1H, CHO), 6.32 (s, 2H, 2 × ArH), 3.87 (s, 3H, OMe), 3.85 (s, 3H, OMe), 2.58 (s, 3H, Me). The 1H NMR data are similar to those acquired at 300 MHz previously reported.392,4-Dimethoxy-6-methylphenol (8)
30% Aqueous H2O2 (15.2 mL, 149 mmol) was added dropwise to a stirred solution of 7 (21.9 g, 122 mmol) and concentrated H2SO4 (0.25 mL, 4.7 mmol) in MeOH (100 mL) at 0 °C. After 20 min the precipitate that had formed was filtered, washed with H2O (3 × 20 mL) and dried under vacuum to give 8a white solid (19.8 g, 97%), m.p. = 103–104 °C [lit.40 103–104 °C]. 1H NMR (400 MHz) δ 6.35 (d, J = 2.8 Hz, 1H, ArH), 6.29 (d, J = 2.8 Hz, 1H, ArH), 5.26 (s, 1H, OH), 3.85 (s, 3H, OMe), 3.75 (s, 3H, OMe), 2.24 (s, 3H, Me). The 1H NMR data are similar to those acquired at 60 MHz previously reported.402,3,5-Trimethoxytoluene (10)41
MeI (1.36 mL, 22.8 mmol) was added to a stirred suspension of 8 (2.90 g, 17.2 mmol) and K2CO3 (4.7 g, 34 mmol) in dry DMF (30 mL) under argon. The reaction mixture was stirred in the dark for 24 h then quenched with ice-cold 1 MHCl (200 mL). The aqueous phase was extracted with ether (6 × 80 mL). The extract was washed with saturated NaHCO3 (50 mL), saturated NH4Cl (50 mL) and water (50 mL), then dried and evaporated to give 10 as a colourless oil (2.96 g, 96%). 1H NMR (500 MHz) δ 6.35 (1H, d, J = 3.0 Hz, H6), 6.28 (1H, d, J = 3.0 Hz, H4), 3.83 (3H, s, MeO), 3.76 (3H, s, MeO), 3.74 (3H, s, MeO), 2.25 (3H, s, Me). The 1H NMR data are similar to those acquired at 80 MHz previously reported.412,3,5-Trimethoxy-6-iodotoluene (5)
Method 1: N-iodosuccinimide (3.70 g, 16.6 mmol) was added to a stirred solution of 10 (2.70 g, 14.8 mmol) and trifluoroacetic acid (350 μL, 4.5 mmol) in dry MeCN (60 mL) under argon. The reaction mixture was stirred in the dark for 30 min then poured into ice-water (300 mL) and extracted with DCM (4 × 60 mL). The extract was washed with H2O (50 mL), dried and evaporated to give 5 as a yellow solid (4.60 g, quant.), which crystallised from MeOH as white needles, m.p. = 90–93 °C. Rf (10% EtOAc–hexanes) 0.4. 1H NMR (500 MHz): δ 6.41 (1H, s, H6), 3.88 (3H, s, 5-MeO), 3.86 (3H, s, 1-MeO), 3.72 (3H, s, 4-MeO), 2.43 (3H, s, Me). 13C NMR (125 MHz): δ 154.9 (ArO), 153.4 (ArO), 141.6 (ArO), 136.4 (Ar-Me), 95.0 (Ar-H), 82.3 (CI), 60.8 (MeO), 57.0 (MeO), 56.1 (MeO), 21.8 (Me). MS (EI) m/z 308 (M, 100%), 293 (79), 265 (30), 250 (13); HRMS observed: 307.9910 C10H13IO3˙+ requires: 307.9909. Microanalysis found: C 39.1, H 4.1%; calculated for C10H13IO3: C 39.0, H 4.3%. The 1H NMR data are identical to those acquired at 200 MHz and reported previously.5Method 2: A mixture of Oxone (0.28 g, 0.46 mmol), 10 (0.16 g, 0.89 mmol) and NaI (0.14 g, 0.91 mmol) in H2O (7 mL) was heated under reflux for 4 h, then cooled, diluted with H2O (30 mL) and extracted with EtOAc (3 × 10 mL). The extract was washed with 10% Na2S2O4 (2 × 10 mL), dried and evaporated to give a yellow solid, which crystallised from MeOH to give 10 as pale-yellow needles (0.21 g, 77%), spectroscopically identical with the material described above.
1,2,5-Trimethoxy-3-methyl-4-(3-methylbut-3-en-1-yn-1-yl)benzene, antrocamphin A (2)
Copper isopropenylacetylide (12b)42 (77 mg, 0.60 mmol) was added to a stirred solution of 5 (62 mg, 0.20 mmol) in anhydrous pyridine (1.5 mL) under argon, and the reaction mixture was heated under reflux for 24 h. After cooling, the reaction mixture was filtered, and the filtrate was diluted with H2O (50 mL) and extracted with ether (4 × 30 mL). The ether extract was evaporated to give a yellow oil, which was subjected to RSF. Elution with EtOAc–hexanes 1:19 gave 2 as a yellow solid (36 mg, 74%), which crystallised from hexanes as a yellow powder, m.p. = 39–41 °C [lit.1,5 oil]. Rf (20% EtOAc–hexanes) 0.5; IR νmax cm−1: 2197 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C). 1H NMR (500 MHz, CDCl3) δ 6.33 (s, 1H, H6′), 5.37 (m, 1H, H4), 5.25 (m, 1H, H4), 3.88 (s, 3H, MeO), 3.87 (s, 3H, MeO), 3.72 (s, 3H, MeO), 2.36 (s, 3H, 3′-Me), 2.01 (t, J3-Me,4 = 1 Hz, 3H, 3-Me); 13C NMR (125.8 MHz; CDCl3) δ 157.4 (ArO), 153.6 (ArO), 141.3 (ArO), 135.5 (3′-Me), 127.5 (C3), 120.9 (C4), 105.1 (C4′), 97.7 (C2), 94.6 (C6′), 83.7 (C1), 60.6 (MeO), 56.5 (MeO), 56.0 (MeO), 23.9 (3-Me), 14.2 (3′-Me); MS (EI) m/z 248 (63%), 246 (M, 100), 233 (53), 231 (64); HRMS observed: 246.1257 C15H18O3˙+ requires: 246.1256. The spectroscopic data match those reported previously.1,5
C). 1H NMR (500 MHz, CDCl3) δ 6.33 (s, 1H, H6′), 5.37 (m, 1H, H4), 5.25 (m, 1H, H4), 3.88 (s, 3H, MeO), 3.87 (s, 3H, MeO), 3.72 (s, 3H, MeO), 2.36 (s, 3H, 3′-Me), 2.01 (t, J3-Me,4 = 1 Hz, 3H, 3-Me); 13C NMR (125.8 MHz; CDCl3) δ 157.4 (ArO), 153.6 (ArO), 141.3 (ArO), 135.5 (3′-Me), 127.5 (C3), 120.9 (C4), 105.1 (C4′), 97.7 (C2), 94.6 (C6′), 83.7 (C1), 60.6 (MeO), 56.5 (MeO), 56.0 (MeO), 23.9 (3-Me), 14.2 (3′-Me); MS (EI) m/z 248 (63%), 246 (M, 100), 233 (53), 231 (64); HRMS observed: 246.1257 C15H18O3˙+ requires: 246.1256. The spectroscopic data match those reported previously.1,5
4-(3,4,6-Trimethoxy-2-methylphenyl)but-3-yn-2-ol (19)
A Young's flask was charged with 5 (2.62 g, 8.51 mmol), CuI (71 mg, 5 mol%), Pd(PPh3)2Cl2 (77 mg, 1.5 mol%), DMSO (20 mL) and Et2NH (4 mL, 0.04 mol), then briefly evacuated and back filled with argon. But-3-yn-2-ol (18) (1.0 mL, 13 mmol) was added and the flask was sealed [CAUTION: safety shield]. The mixture was stirred at 65 °C for 16 h, then cooled to room temperature, and diluted with H2O (100 mL) and 1 M HCl (20 mL). The aqueous solution was extracted with EtOAc (3 × 30 mL), dried and evaporated to give a brown oil, which was subjected to flash chromatography. Elution with 40% EtOAc–hexanes gave 19 (1.24 g, 58%) as a pale yellow solid, which crystallised from MeOH as pale yellow needles, m.p. = 119–124 °C. Rf (20% EtOAc–hexanes) 0.1; IR νmax cm−1: 3600–3100 (OH), 2219 (CC); 1H NMR (500 MHz, CDCl3) δ 6.32 (s, 1H, H5′), 4.83 (m, 1H, H2), 3.88 (s, 3H, MeO), 3.86 (s, 3H, MeO), 3.71 (s, 3H, MeO), 2.34 (s, 3H, 2′-Me), 2.06 (br d, JOH,2 = 5 Hz, 1H, OH), 1.57 (d, J1,2 = 7 Hz, 3H, 1-Me); 13C NMR (125.8 MHz; CDCl3) δ 157.5 (ArO), 153.7 (ArO), 141.3 (ArO), 135.7 (C2′), 104.2 (C1′), 98.1 (C3 or 4), 94.4 (C5′), 79.1 (C3 or 4), 60.6 (MeO), 59.3 (C2), 56.4 (MeO), 56.0 (MeO), 24.8 (2′-Me), 14.2 (C1); MS (EI) m/z 250 (M, 36%) 232 (85), 86 (63), 84 (100); HRMS observed: 250.1202 C14H18O4˙+ requires: 250.1205; Microanalysis found: C 66.4, H 7.1%; calculated for C14H18O4 C 67.2, H 7.2%.
4-(3,4,6-Trimethoxy-2-methylphenyl)but-3-yn-2-one, antrocamphin B (3)
Method 1: activated MnO2 (420 mg, 4.8 mmol) was added to a stirred solution of 19 (60 mg. 0.24 mmol) in anhydrous DCM (2 mL) under argon. The reaction mixture was stirred for 24 h then vacuum filtered through a Celite plug and washed through with DCM. Evaporation of the filtrate gave 5 as a yellow solid (47 mg, 80%), which crystallised from MeOH as bright yellow needles, m.p. = 101–108 °C. Rf (20% EtOAc–hexanes) 0.15; IR νmax cm−1: 2180 (CC), 1646 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (500 MHz, CDCl3) δ 6.32 (s, 1H, H5′), 3.91 (s, 3H, MeO), 3.89 (s, 3H, MeO), 3.73 (s, 3H, MeO), 2.46 (s, 3H, 1-Me), 2.39 (s, 3H, 2′-Me); 13C NMR (125.8 MHz; CDCl3) δ 184.7 (CO), 159.9 (ArO), 156.3 (ArO), 141.3 (ArO), 137.4 (C2′), 101.4 (C1′), 96.4 (C3), 94.0 (C5′), 88.0 (C4), 60.6 (MeO), 56.3 (MeO), 56.0 (MeO), 32.9 (1-Me), 14.2 (2′-Me); MS (EI) m/z 248 (M, 100%) 233 (85), 205 (13); HRMS observed: 248.1044, C14H16O4 requires: 248.1049. The spectroscopic data matched those reported.1
O). 1H NMR (500 MHz, CDCl3) δ 6.32 (s, 1H, H5′), 3.91 (s, 3H, MeO), 3.89 (s, 3H, MeO), 3.73 (s, 3H, MeO), 2.46 (s, 3H, 1-Me), 2.39 (s, 3H, 2′-Me); 13C NMR (125.8 MHz; CDCl3) δ 184.7 (CO), 159.9 (ArO), 156.3 (ArO), 141.3 (ArO), 137.4 (C2′), 101.4 (C1′), 96.4 (C3), 94.0 (C5′), 88.0 (C4), 60.6 (MeO), 56.3 (MeO), 56.0 (MeO), 32.9 (1-Me), 14.2 (2′-Me); MS (EI) m/z 248 (M, 100%) 233 (85), 205 (13); HRMS observed: 248.1044, C14H16O4 requires: 248.1049. The spectroscopic data matched those reported.1
Method 2: Ac2O (10 mL, 0.11 mol) was added to a stirred solution of 19 (1.24 g, 4.95 mmol) in DMSO (40 mL). After 24 h the reaction mixture was diluted with H2O (100 mL) and the resultant precipitate was collected by vacuum filtration. The filtrate was extracted with EtOAc (3 × 30 mL). The extract was dried, combined with the precipitate, and evaporated to give 3 as a yellow solid (1.21 g, quant.), identical with the material described above.
meso-3,4-Dimethyl-hexa-1,5-diyne-3,4-diol (25)
K2CO3 (155 mg, 1.13 mmol) was added to a stirred solution of meso-3,4-dimethyl-1,6-bis(trimethylsilyl)hexa-1,5-diyne-3,4-diol (24)43 (210 mg, 0.75 mmol) in MeOH–DCM (1:1, 3 mL) under argon. The resulting slurry was stirred for 3 h then vacuum filtered through Celite and rinsed through with DCM. The filtrate was evaporated to give a colourless oil, which was subjected to RSF. Elution with 20% EtOAc–hexanes gave 25 as a white solid (97 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 2.80 (s, 2H, OH), 2.54 (s, 2H, CH), 1.55 (s, 6H, Me). The 1H NMR spectrum matched the data reported.43
meso-4,5-Diethynyl-4,5-dimethyl-1,3-dioxolan-2-one (26)
A solution of triphosgene (60 mg, 0.2 mmol) in DCM (0.5 mL) was added dropwise to a stirred solution of 25 (35 mg, 0.25 mmol) and pyridine (99 mg, 1.25 mmol) in anhydrous DCM (0.5 mL) at −78 °C under argon. The reaction mixture was warmed to 0 °C slowly (over 3 h) and stirred at 0 °C for 1 h, then quenched with saturated NH4Cl (20 mL) and extracted with DCM (4 × 20 mL). The extract was washed with 1 M HCl (20 mL), brine (20 mL), saturated NaHCO3 (20 mL) and brine (20 mL), dried and evaporated to give 26 as a white solid (35 mg, 83%), which crystallised from hexanes–EtOAc as a white powder, m.p. = 59–62 °C. Rf (20% EtOAc–hexanes) 0.28; IR νmax cm−1: 3287 (CH), 2133 (CC), 1798 (CO); 1H NMR (500 MHz, CDCl3) δ 2.83 (s, 2H, 2 × CH), 1.71 (s, 6H, 2 × Me); 13C NMR (125.8 MHz; CDCl3) δ 151.8 (CO), 81.7 (C4/5-C alkyne), 78.7 (CH), 78.6 (C4/5), 22.3 (Me); MS (CI) m/z 165 [M]˙+ (100), 103 (10); HRMS observed: 165.0551, C9H9O3˙+ requires: 165.0552.
1,2,5-Trimethoxy-3-methyl-4-(trimethylsilylethynyl)benzene (30)
A stirred solution of 5 (308 mg, 1.11 mmol) in triethylamine (1.25 mL) was evacuated and back filled with argon (×3) then treated with trimethylsilylacetylene (150 μL, 1.1 mmol), Pd(PPh3)2Cl2 (7 mg, 1 mol%) and CuI (9 mg, 5 mol%). The reaction vessel was sealed [CAUTION: safety shield] then stirred at 60 °C for 72 h. 1H NMR analysis of an aliquot after this time showed the starting material was only 16% consumed. Additional equivalents of catalysts and trimethylsilylacetylene (amounts as above) were added and the reaction mixture heated at 60 °C in a sealed tube for another 72 h. The process was repeated with fresh equivalents of catalyst and alkyne (amounts as above) for another 96 h then the reaction mixture was diluted with H2O (50 mL) and extracted with ether (4 × 40 mL). The extract was dried and evaporated to give an orange oil, which was subjected to RSF. Elution with 10% EtOAc–hexanes gave 30 as a white solid (59 mg, 21%), which crystallised from hexanes–EtOAc as a white powder, m.p. = 56–59 °C. Rf (10% EtOAc–hexanes) 0.3; IR νmax cm−1: 2147 (CC); 1H NMR (500 MHz, CDCl3) δ 6.30 (s, 1H, H6′), 3.87 (s, 3H, MeO), 3.86 (s, 3H, MeO), 3.71 (s, 3H, MeO), 2.35 (s, 3H, Me), 0.26 (s, 9H, Si(Me)3); 13C NMR (125.8 MHz; CDCl3) δ 157.9 (ArO), 153.8 (ArO), 141.3 (ArO), 136.1 (3′-Me), 105.0 (C4′ or 1 or 2), 101.3 (C4′ or 1 or 2), 100.3 (C4′ or 1 or 2), 94.5 (C6′), 60.6 (MeO), 56.5 (MeO), 55.9 (MeO), 14.2 (3′-Me), 0.41 (Si(Me)3); MS (EI) m/z 278 [M]˙+, (100%), 263 (83), 248 (23), 233 (24); HRMS observed: 278.1345, C15H22O3Si requires: 278.1338.
2-Methyl-4-(3,4,6-trimethoxy-2-methylphenyl)but-3-yne-2-ol (32)
A stirred mixture of 5 (3.23 g, 10.5 mmol), CuI (64 mg, 3.4 mol%), Pd(PPh3)2Cl2 (89 mg, 1.3 mol%) and DMSO (30 mL) in a Young's flask was evacuated and backfilled with argon (×3). Et2NH (5 mL, 0.05 mol) and 2-methylbut-3-yn-2-ol (31) (3 mL, 0.03 mole) were added and the vessel was sealed and stirred at 70 °C for 24 h. The reaction mixture was cooled to room temperature, diluted with H2O (150 mL) and 1 M HCl (30 mL), and extracted with EtOAc (4 × 30 mL). The extract was washed with brine (30 mL), dried and evaporated to give a brown oil, which was subjected to flash chromatography. Elution with 40% EtOAc–hexanes yielded 32 (1.98 g, 71%) as a white solid, m.p. = 94–96 °C. Rf (40% EtOAc–hexanes): 0.2; IR (KBr) νmax cm−1: 3600–3000 (OH), 2219 (CC). 1H NMR (400 MHz): δ 6.42 (1H, s, H5′), 3.87 (3H, s, OMe), 3.85 (3H, s, OMe), 3.71 (3H, s, OMe), 2.33 (3H, s, C2′-Me), 2.13 (1H, s, OH), 1.64 (6H, s, C2-Me). 13C NMR (100 MHz) δ 157.6 (C4′ or C6′), 153.7 (C4′ or C6′), 141.4 (C3′), 135.7 (C2′), 104.6 (C1′), 101.1 (C3 & C4), 94.8 (C5′); 66.2 (C2), 60.7 (OMe), 56.6 (OMe), 56.1 (OMe), 32.0 (C1 & C2-Me), 14.3 (C2′-Me). MS (EI) m/z: 264.1 [M]˙+ (18), 249.1 [M − Me]+ (15), 246.1 [M − H2O]˙+ (100). HRMS (EI): observed, 264.1363. C15H20O4˙+ requires 264.1362.
2-Ethynyl-1,4,5-trimethoxy-3-methylbenzene (33)
Crushed, dry NaOH (0.18 g, 4.4 mmol) was added to a stirred solution of 32 (0.69 g, 2.6 mmol) in toluene (12 mL) and the mixture was heated under reflux. After 6 h the mixture was cooled to room temperature, diluted with H2O (50 mL) and 1 M HCl (10 mL) and extracted with EtOAc (3 × 10 mL). The extract was washed with brine (30 mL), dried and evaporated to give a brown solid, which was subjected to flash chromatography. Elution with 10% EtOAc–hexanes yielded 32 as a white solid (0.51 g, 94%), m.p. = 86–90 °C. Rf (20% EtOAc–hexanes): 0.2; IR (KBr) νmax cm−1: 3284 (C–H), 2150 (CC). 1H NMR (400 MHz): δ 6.34 (s, 1H, H5), 3.88 (s, 6H, 2 × OMe), 3.72 (s, 3H, OMe), 3.45 (s, 1H, CH), 2.37 (3H, s, Me). 13C NMR (100 MHz) δ 158.1 (C4 or C6), 159.7 (C4 or C6), 141.0 (C3), 136.0 (C2), 103.5 (C1), 94.1 (C5), 83.6 (CH); 78.8 (ArC), 60.4 (OMe), 56.2 (OMe), 55.8 (OMe), 14.0 (Me). MS (EI) m/z: 206 [M]˙+ (100%), 191 [M − Me]+ (99). HRMS (EI) observed: 206.0948, C10H9O3˙+ requires: 206.0943.
3-Hydroxy-3-methyl-5-(3,4,6-trimethoxy-2-methylphenyl)pent-4-yn-2-one (35)
A 0.20 M solution of EtMgBr (5.0 mL, 1.0 mmol) in Et2O was added dropwise to a stirred solution of 33 (0.21 g, 1.0 mmol) in Et2O (5 mL) under argon. The reaction mixture was heated under reflux for 2.5 h, then cooled to room temperature and treated dropwise with a solution of 2,3-butanedione (0.18 mL, 2.1 mmol) in Et2O (2 mL). The reaction mixture was heated for 24 h under reflux, then cooled, diluted with H2O (50 mL) and extracted with EtOAc (3 × 20 mL). The extract was washed with brine (30 mL), dried and evaporated to give a yellow oil, which was subjected to flash chromatography. Elution with 10% EtOAc–hexanes yielded 35 as a white solid (32 mg, 11%), m.p. = 86–88 °C. Rf (40% EtOAc–hexanes): 0.25; IR (thin film) νmax cm−1: 3100–3700 (OH), 2218 (CC), 1720 (CO). 1H NMR (400 MHz): δ 6.30 (s, 1H, H5′), 4.12 (s, 1H, OH), 3.87 (s, 3H, OMe), 3.82 (s, 3H, OMe), 3.70 (s, 3H, OMe), 2.49 (s, 3H, C2′-Me), 2.32 (s, 3H, H1), 1.72 (s, 3H, C3-Me). 13C NMR (100 MHz) δ 206.2 (C2), 157.9 (C4′ or C6′), 154.0 (C4′ or C6′), 141.1 (C3′), 135.1 (C2′), 102.6 (C1′), 94.8 (C5), 94.3 (C5′), 81.1 (C4), 73.2 (C3), 60.5 (OMe), 56.2 (OMe), 55.8 (OMe), 27.3 (C1), 23.4 (C3-Me), 14.1 (C2′-Me). MS (EI) m/z: 292 [M]˙+ (20), 276 [M − OH]+ (77), 233 (100). HRMS (EI): observed, 292.1312. C16H20O5˙+ requires 292.1311.
1,4-Bis(3,4,6-trimethoxy-2-methylphenyl)buta-1,3-diyne (36)
A 1.29 M solution of BuLi in hexanes (0.80 mL, 1.0 mmol) was added dropwise to a stirred solution of 32 (0.21 g, 1.0 mmol) and anhydrous THF (10 mL) under argon at −78 °C. The solution was allowed to warm to room temperature over 1 h, then cooled to −78 °C. A solution of 2,3-butanedione (0.05 mL, 0.6 mmol) in dry THF (1 mL) was added dropwise and the reaction mixture was allowed to warm to room temperature. After 24 h the solution was diluted with saturated NH4Cl (20 mL) and H2O (50 mL), then extracted with EtOAc (3 × 15 mL). The extract was washed with brine (30 mL), dried and evaporated to give a brown solid, which was subjected to flash chromatography. Elution with 40% EtOAc–hexanes yielded 36 as a white solid (36 mg, 17%), m.p. = 210–214 °C. Rf (40% EtOAc–hexanes): 0.25; IR (KBr) νmax cm−1: 2342 & 2140 (CC). 1H NMR (600 MHz): δ 6.32 (s, 2H, H5′), 3.89 (s, 6H, OMe), 3.88 (s, 6H, OMe), 3.72 (s, 6H, OMe), 2.40 (s, 6H, C2-Me). 13C NMR (100 MHz) δ 159.0 (C4′ or C6′), 154.1 (C4′ or C6′), 141.0 (C3′), 136.6 (C2′), 104.0 (C1′), 94.1 (C5′), 80.7 (C1 or C2); 77.6 (C1 or C2), 60.4 (OMe), 56.2 (OMe), 55.8 (OMe), 14.2 (C2-Me). MS (EI) m/z: 410 [M]˙+ (100), 395 [M − Me]+ (22). HRMS (EI): observed, 410.1730. C24H26O6˙+ requires 410.1729.
4,5-Dimethyl-4,5-bis(phenylethynyl)-1,3-dioxolan-2-one (cis/meso40 and (±)-trans41)
A solution of triphosgene (0.18 g, 0.62 mmol) in DCM (1 mL) was added dropwise to a stirred solution of 38/39 (∼1:1 mixture of diastereomers)44 (0.20 g, 0.70 mmol) and pyridine (0.17 mL, 2.1 mmol) in DCM (2 mL) under argon at 0 °C. The reaction allowed to warm to room temperature slowly. After 2 h the solution was diluted with H2O (20 mL) and extracted with DCM (3 × 20 mL). The extract was washed with brine (20 mL), dried and evaporated to give a yellow oil, which was subjected to flash chromatography. Elution with 20% EtOAc–hexanes yielded 41 (42 mg, 19%) as a pale yellow solid, m.p. = 122–127 °C. Rf (20% EtOAc–hexanes): 0.35; IR (thin film) νmax cm−1: 2252 (CC), 1803 (CO). 1H NMR (400 MHz): δ 7.48 (m, 4H, ArH), 7.38–7.26 (m, 6H, ArH), 2.02 (s, 6H, Me). 13C NMR (100 MHz) δ 152.2 (C2), 131.9 (CH), 129.6 (CH), 128.5 (CH), 120.8 (ArC), 90.4 (ArC = C2′), 83.0 (C1′ or C4/5), 82.6 (C1′ or C4/5), 24.9 (Me). MS (EI) m/z: 272 [M − CO2]˙+ (1), 256 [M − CO3]˙+ (18), 128.0 (100). HRMS (ES): observed, 358.1440. [C21H17O3 + MeCN]+ requires 358.1438.
Further elution gave 40 (62 mg, 28%) as a pale yellow oil. Rf (20% EtOAc–hexanes): 0.25; IR (thin film) νmax cm−1: 2230 (CC), 1815 (CO). 1H NMR (400 MHz): δ 7.30 (4H, m, ArH), 7.32–7.38 (2H, m, ArH), 7.29–7.31 (4H, m, ArH), 1.84 (6H, s, Me). 13C NMR (100 MHz) δ 152.3 (C2), 132.0 (PhC), 129.4 (PhC), 128.4 (PhC), 121.9 (PhC), 89.6 (C4 & C5), 84.4 (C1′ or C2′), 81.8 (C1′ or C2′), 22.5 (Me). MS (EI) m/z: 272 [M − CO2]˙+ (8), 256 [M − CO3]˙+ (28), 128 (100). HRMS (ES): observed, 358.1441. [C21H17O3 + MeCN]+ requires 358.1438.
3-(3,4,6-Trimethoxy-2-methylphenyl)prop-2-yn-2-ol (43)
A stirred mixture of iodo-1,4,5-trimethoxy-3-methylbenzene (5) (4.07 g, 13.2 mmol), CuI (92 mg, 4.0 mol%), Pd(PPh3)2Cl2 (0.12 g, 1.5 mol%) and DMSO (24 mL) in a Young's flask at 50 °C was evacuated and backfilled with argon (×3). Et2NH (5 mL, 50 mmol) and prop-2-yn-1-ol (42) (2.4 mL, 42 mmol) were added and the vessel sealed. After stirring for 3 d at 50 °C fresh additions of CuI (89 mg, 3.9 mol%), Pd(PPh3)2Cl2 (99 mg, 1.3 mol%) and 42 (1.2 mL, 21 mmol) were made. After a further 24 h at 50 °C, the reaction was cooled to room temperature diluted with H2O (100 mL) and 1 M HCl (30 mL). The aqueous solution was extracted with EtOAc (4 × 30 mL), washed with brine (30 mL), dried and the solvent evaporated to give a brown oil, which was subjected to flash chromatography. Elution with 40% EtOAc–hexanes yielded 43 (2.08 g, 67%) as a white solid, m.p. = 108–114 °C. Rf (40% EtOAc–hexanes): 0.17; IR (thin film) νmax cm−1: 3000–3600 (OH), 2218 (CC). 1H NMR (400 MHz): δ 6.33 (s, 1H, H5′), 4.56 (d, J = 5.6 Hz, 2H, H1), 3.88 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.72 (s, 3H, OMe), 2.35 (s, 3H, C2′-Me), 1.79 (t, J = 5.6 Hz, 1H, OH). 13C NMR (100 MHz) δ 157.5 (C4′ or C6′), 153.7 (C4′ or C6′), 141.1 (C3′), 135.7 (C2′), 104.0 (C1′), 94.2 (C5′), 94.0 (C3); 80.6 (C2), 60.4 (OMe), 56.2 (OMe), 55.8 (OMe), 51.9 (C1), 14.1 (C2′-Me). MS (EI) m/z: 236.0 [M]˙+ (100), 221.0 [M − Me]+ (84), 205 [M − MeOH]˙+ (38). HRMS (EI): observed, 236.1044. C13H16O4˙+ requires 236.1049.
3-(3,4,6-Trimethoxy-2-methylphenyl)propionaldehyde (44)
Method 1: a suspension of activated MnO2 (1.63 g, 18.7 mmol) in a solution of 43 (0.24 g, 1.1 mmol) in DCM (5 mL) was stirred for 16 h. The reaction mixture was vacuum filtered through Celite and washed through with DCM (4 × 5 mL). The filtrate was evaporated to yield 44 as a pale yellow solid (0.16 g, 67%) identical with the material described below.
Method 2: Ac2O (10 mL, 0.11 mol) was added to a stirred solution of 43 (1.24 g, 4.95 mmol) in dry DMSO (40 mL). After 24 h the solution was diluted with H2O (60 mL) and NEt3 (10 mL), then extracted with EtOAc (4 × 20 mL). The extract was washed with brine (20 mL), dried and evaporated to yield 44 as a pale yellow solid (1.21 g, quant.), m.p. = 126–129 °C. Rf (40% EtOAc–hexanes): 0.3; IR (thin film) νmax cm−1: 2166 (CC), 1645 (CO). 1H NMR (600 MHz): δ 9.46 (s, 1H, CHO), 6.33 (s, 1H, H5′), 3.92 (s, 3H, OMe), 3.90 (s, 3H, OMe), 3.73 (s, 3H, OMe), 2.39 (s, 3H, C2′-Me). 13C NMR (150 MHz) δ 176.8 (CO), 160.4 (C6′ or C4′), 156.9 (C6′ or C4′), 141.4 (C3′), 137.9 (C2′), 101.0 (C1′), 96.9 (C2 or C3), 93.9 (C5′), 92.9 (C2 or C3), 60.7 (OMe); 56.3 (OMe), 56.0 (OMe), 14.3 (C2′-Me). MS (EI) m/z: 234 [M]˙+ (100), 219.0 [M − Me]+ (42). HRMS (EI): observed, 234.0898. C13H14O4˙+ requires 234.0892.
1,6-Bis(3,4,6-trimethoxy-2-methylphenyl)hexa-1,5-diyn-3,4-diol (45)(∼13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :20 mixture of diastereomers based on 46)
:20 mixture of diastereomers based on 46)
Freshly prepared Cu/Zn couple45 (3.1 g, 1:1 –CuSO4·5H2O:Zn w/w) and AcOH (1 mL, 20 mmol) were added to a stirred solution of 44 (1.01 g, 4.28 mmol) in THF (30 mL). After 3 d the reaction mixture was diluted with H2O (100 mL) and saturated NaHCO3 (10 mL) then vacuum filtered through Celite and washed through with DCM (3 × 10 mL). The filtrate was concentrated under reduced pressure to ∼100 mL and the resulting precipitate was collected by vacuum filtration and air-dried, giving 45 as a pale yellow solid (0.82 g, 82%), m.p. = 192–195 °C. Rf (70% EtOAc–hexanes): 0.25; IR (thin film) νmax cm−1: 3000–3500 (OH), 2222 (CC). 1H NMR (400 MHz): δ 6.31 (s, 2H, H5′), 4.81 (d, J = 6.4 Hz, 2H, H3/H4), 3.87 (s, 6H, OMe), 3.82 (s, 6H, OMe), 3.69 (s, 6H, OMe), 3.01 (d, J = 6.4 Hz, 2H, OH), 2.31 (s, 6H, C2′-Me). 13C NMR (100 MHz) δ 158.1 (C4′ or C6′), 154.2 (C4′ or C6′), 141.3 (C3′), 135.9 (C2′), 104.0 (C1′), 94.4 (C5′), 93.3 (C1 or C2); 82.5 (C1 or C2), 68.2 (C3 & C4), 60.7 (OMe), 56.5 (OMe), 56.1 (OMe), 14.4 (C2′-Me). MS (EI) m/z: 455 [M − Me]+ (25), 453 [M − OH]+ (71). HRMS (EI): observed, 470.1937. C26H30O8˙+ requires 470.1941.
4,5-Bis((3,4,6-trimethoxy-2-methylphenyl)ethynyl)-1,3-dioxolan-2-one (46)(∼13:20 mixture of diastereomers)
A solution of triphosgene (0.19 g, 0.67 mmol) in DCM (2 mL) was added dropwise to a stirred solution of 45 (mixture of diastereomers) (32 mg, 68 μmol) and pyridine (0.10 mL, 1.3 mmol) in DCM (6 mL) under argon at 0 °C. The reaction mixture was allowed to warm to room temperature slowly. After 45 min the solution was diluted with H2O (20 mL) and extracted with DCM (3 × 10 mL). The extract was washed with brine (20 mL), dried and evaporated, and the residue was subjected to flash chromatography. Elution with 60% EtOAc–hexanes gave 46 as a pale yellow solid (15 mg, 45%), m.p. = 165–170 °C. IR (thin film) νmax cm−1: 2226 (CC), 1810 (CO). MS (ES) m/z: 515 [M + H + H2O]+ (50), 497 [M + H]+ (100), 471 [M + H − CO]+ (18), 453 [M + H − CO2]+ (89). HRMS (ES): observed 497.1821. C27H29O9+ requires 497.1806.
Isomer 1 (major): Rf (60% EtOAc–hexanes): 0.35. 1H NMR CDCl3 (400 MHz) δ 6.33 (s, 2H, H5′), 5.59 (2H, s, H4/5), 3.89 (s, 6H, OMe), 3.86 (s, 6H, OMe), 3.72 (s, 6H, C3′–OMe), 2.34 (s, 6H, C2′-Me). 13C NMR CDCl3 (150 MHz) δ 158.2 (C4′ or C6′), 155.0 (C4′ or C6′), 153.3 (C2), 141.2 (C3′), 136.5 (C2′), 102.2 (C1′), 94.21 (C5′), 87.7 (C1′′ or C2′′), 86.32 (C1′′ or C2′′), 73.4 (C4/5), 60.6 (C3′–OMe), 56.3 (OMe), 56.0 (OMe), 14.2 (C2′-Me).
Isomer 2 (minor): Rf (60% EtOAc–hexanes): 0.3; 1H NMR CDCl3 (400 MHz) δ 6.28 (s, 2H, H5′), 5.77 (s, 2H, H4/5), 3.87 (s, 6H, OMe), 3.73 (s, 6H, OMe), 3.66 (s, 6H, C3′–OMe), 2.24 (s, 6H, C2′-Me). 13C NMR CDCl3 (100 MHz) δ 158.2 (C4′ or C6′), 154.4 (C4′ or C6′), 153.1 (C2), 140.8 (C3′), 136.2 (C2′), 102.4 (C1′), 93.7 (C5′), 87.5 (C1′′ or C2′′), 85.9 (C1′′ or C2′′), 71.3 (C4/5), 60.2 (C3′–OMe), 55.8 (OMe), 55.6 (OMe), 13.8 (C2′-Me).
1,6-Bis(3,4,6-trimethoxy-2-methylphenyl)hexa-1,5-diyn-3,4-dione (47)
Ac2O (5 mL, 0.05 mol) was added to a stirred solution of 45 (0.69 g, 1.5 mmol) and DMSO (20 mL). After 5 h the reaction mixture was cooled to 0 °C and diluted with H2O (50 mL). The resulting precipitate was vacuum filtered to give an orange solid. The filtrate was diluted with 28% aqueous NH3 (8 mL), then extracted with EtOAc (3 × 20 mL). The extract was washed with brine (20 mL), dried and evaporated and combined with the precipitate to give 47 as an orange solid (0.66 g, 90%), m.p. = 231–235 °C. Rf (60% EtOAc–hexanes): 0.35; IR (thin film) νmax cm−1: 2177 (CC), 1662 (CO). 1H NMR (600 MHz): δ 6.31 (s, 2H, H5′), 3.93 (s, 6H, OMe), 3.88 (s, 6H, OMe), 3.73 (s, 6H, OMe), 2.47 (s, 6H, C2′-Me). 13C NMR (150 MHz) δ 173.3 (CO), 161.1 (C4′ or C6′), 157.3 (C4′ or C6′), 141.3 (C3′), 138.6 (C2′), 100.8 (C1′), 98.0 (C1/6), 94.8 (C2/5); 93.7 (C5′), 60.5 (OMe), 56.3 (OMe), 55.9 (OMe), 14.2 (C2′-Me). HRMS (ES): observed, 467.1702. C26H27O8+ requires 467.1706.
Attempted pinacol coupling of antrocamphin B (3) with Zn/Cu couple
Freshly prepared Cu/Zn couple45 (3.01 g, 1:1 –CuSO4·5H2O:Zn w/w) and AcOH (1.5 mL, 26 mmol) were added to a stirred solution of antrocamphin B (3) (1.21 g, 4.88 mmol) in THF (40 mL). After 30 h the reaction suspension was vacuum filtered through Celite and washed through with DCM (3 × 15 mL). The filtrate was evaporated residue was subjected to flash chromatography. Elution with 40% EtOAc–hexanes gave meso-4,5-bis(3,4,6-trimethoxy-2-methylphenyl)octane-2,7-dione (50) as a pale yellow solid (92 mg, 8%), m.p. = 203–206 °C. Rf (60% EtOAc–hexanes): 0.35; IR (thin film) νmax cm−1: 1710 (CO). 1H NMR (400 MHz): δ 6.35 (s, 2H, H5′), 4.20–4.22 (m, 2H, H3/6 or H4/5), 3.88 (s, 6H, OMe), 3.84 (s, 6H, OMe), 3.71 (s, 6H, OMe), 2.97–3.03 (m, 2H, C3/6 or C4/5), 2.43 (s, 6H, C2′-Me), 2.22–2.27 (m, 2H, H3/6 or H4/5). 13C NMR (100 MHz) δ 209.2 (C2/7′), 154.8 (C4′ or C6′), 151.6 (C4′ or C6′), 141.1 (C3′), 133.1 (C2′), 122.3 (C1′), 95.3 (C5′), 60.6 (OMe), 55.8 (OMe), 55.7 (OMe), 45.9 (C3/6 or C4/5), 37.3 (C3/6 or C4/5), 30.5 (C1/8), 14.2 (C2′-Me). MS (ES) m/z: 541 [M + K]+, 525 [M + Na]+. HRMS (ES): observed 503.2626. C28H39O8+ requires 503.2639.
Further elution with 60% EtOAc–hexanes gave 3,4-dimethyl-1,6-bis(3,4,6-trimethoxy-2-methylphenyl)hexa-1,5-diene-3,4-diol (49) as a white solid (0.28 g, 22%), m.p. = 138–141 °C. Rf (60% EtOAc–hexanes): 0.30; IR (thin film) νmax cm−1: 3200–3600 (OH. 1H NMR (600 MHz): δ 6.58 (d, J = 16.2 Hz, 2H, H6), 6.36 (s, 2H, H5′), 6.26 (d, J = 16.2 Hz, 2H, H5), 3.86 (s, 6H, OMe), 3.71 (s, 12H, 2 × OMe), 2.48 (s, 6H, OH), 2.25 (s, 6H, C2′-Me), 1.45 (s, 6H, H3/4-Me). 13C NMR (100 MHz) δ 154.0 (C4′ or C6′), 151.9 (C4′ or C6′), 141.5 (C3′), 137.9 (C6), 131.3 (C1′ or C2′), 122.9 (C5), 118.7 (C1′ or C2′), 95.1 (C5′), 78.2 (C4), 60.6 (OMe), 56.0 (OMe), 22.4 (C4-Me), 13.4 (C2′-Me). MS (ES) m/z: 541 [M + K]+ (100), 539 [M + K − H2]+ (20), 525 [M + Na]+ (50), 523 [M + Na − H2]+ (35), 507 [M + Na − H2O] (10). HRMS (ES): observed 525.2468. C28H38O8Na+ requires 525.2459.
3,4-Dimethyl-1,6-bis(3,4,6-trimethoxy-2-methylphenyl)hexa-1,5-diyne-3,4-diol (34)
A flame dried Schlenk flask under argon was charged with samarium metal (75 mg, 0.49 mmol), 1,2-diiodoethane (65 mg, 0.23 mmol) and dry THF (4 mL), purging and back-filling with argon after each addition. After 30 min of stirring the reaction mixture containing SmI2 was treated with a solution of antrocamphin B (3) (63 mg, 0.25 mmol) in dry THF (1 mL). After 2.5 h the reaction was quenched with H2O (20 mL) and the aqueous solution was extracted with EtOAc (3 × 10 mL). The extract was washed with brine (10 mL), dried and evaporated, and the residue was subjected to flash chromatography. Elution with 60% EtOAc–hexanes gave 34 (10:1 mixture of rac:meso isomers) (35 mg, 55%) as a pale yellow solid. IR (thin film) νmax cm−1: 3100–3700 (OH), 2222 (CC).
The isomers were separated by semi-preparative HPLC using a Hewlett Packard 1050 system equipped with a multiple wavelength detector (MWD). Separation was achieved using a 250 × 10 mm i.d., 5 μm, Apollo C18 reversed phase column (Grace-Davison) with a 33 mm × 7 mm guard column of the same material. The column was eluted at 4 mL min−1 with 30% (v/v) acetonitrile–water and 1 mL was injected. The chiral isomers eluted first giving 34b as a white solid: m.p. = 126–130 °C. Rf (60% EtOAc–hexanes): 0.2. 1H NMR CDCl3 (600 MHz) δ 6.30 (s, 2H, H5′), 3.86 (s, 6H, OMe), 3.81 (s, 6H, OMe), 3.69 (s, 6H, OMe), 3.37 (s, 2H, OH), 2.33 (s, 6H, C2′-Me), 1.72 (6H, s, C3/4-Me). 13C NMR CDCl3 (150 MHz) δ 157.9 (C4′ or C6′), 153.8 (C4′ or C6′), 141.2 (C3′), 135.2 (C2′), 104.3 (C1′), 97.3 (C1/6 or C2/5), 94.5 (C5′), 80.6 (C1/6 or C2/5), 75.3 (C3/4), 60.6 (OMe), 56.4 (OMe), 56.0 (OMe), 23.9 (C3/4-Me), 14.3 (C2′-Me).
Further elution gave the mesoisomer 34a as a white solid: m.p. = 138–142 °C. Rf (60% EtOAc–hexanes): 0.2. 1H NMR CDCl3 (600 MHz) δ 6.29 (s, 2H, H5′), 3.86 (s, 6H, OMe), 3.79 (s, 6H, OMe), 3.69 (s, 6H, OMe), 3.23 (s, 2H, OH), 2.29 (s, 6H, C2′-Me), 1.72 (s, 6H, C3/4-Me). 13C NMR CDCl3 (150 MHz) δ 157.9 (C4′ or C6′), 153.7 (C4′ or C6′), 141.2 (C3′), 135.4 (C2′), 104.4 (C1′), 97.9 (C1/6 or C2/5), 94.5 (C5′), 80.4 (C1/6 or C2/5), 74.9 (C3/4), 60.5 (OMe), 56.4 (OMe), 55.9 (OMe), 22.9 (C3/4-Me), 14.2 (C2′-Me). MS (ES) m/z: 537 [M + K]+ (18), 521 [M + Na]+ (80), 481 [M − OH]+ (40). HRMS (ES): observed 499.2338. C28H35O8+ requires 499.2326.
(±)-trans-4,5-Dimethyl-4,5-bis((3,4,6-trimethoxy-2-methylphenyl)ethynyl)-1,3-dioxolan-2-one (51)
Method 1: a solution of triphosgene (0.13 g, 0.42 mmol) in DCM (2 mL) was added dropwise to a stirred solution of 34 (10:1 rac:meso mixture) (0.18 g, 0.36 mmol) and pyridine (0.10 mL, 1.3 mmol) in DCM (8 mL) under argon at 0 °C, then the reaction was allowed to warm to room temperature slowly. After 4 h the solution was diluted with H2O (30 mL) and extracted with DCM (3 × 20 mL). The extract was washed with brine (20 mL), dried and evaporated, and the residue was subjected to flash chromatography. Elution with 60% EtOAc–hexanes gave 51 (30 mg, 16%) as a pale yellow/orange solid, m.p. = 85–90 °C. Rf (40% EtOAc–hexanes): 0.35; IR (thin film) νmax 2207 (CC), 1787 (CO). 1H NMR CDCl3 (600 MHz) δ 6.33 (s, 2H, H5′), 3.87 (s, 6H, OMe), 3.85 (s, 6H, OMe), 3.72 (s, 6H, OMe), 2.36 (s, 6H, C2′-Me), 1.70 (s, 6H, C4/5-Me). 13C NMR CDCl3 (150 MHz) δ 158.5 (C4′ or C6′), 154.7 (C4′ or C6′), 153.0 (CO), 141.2 (C3′), 135.8 (C2′), 102.9 (C1′), 94.4 (C5′), 90.1 (C4/5 or C1′′), 86.0 (C2′′), 83.5 (C4/5 or C1′′), 60.6 (OMe), 56.4 (OMe), 56.0 (OMe), 25.5 (C4/5-Me), 14.27 (C2′-Me). HRMS (ES): observed 525.2132. C29H33O9+ requires 525.2125.
Method 2: A flame dried Schlenk flask under argon was charged sequentially with samarium metal (76 mg, 0.51 mmol), 1,2-diiodoethane (0.13 g, 0.47 mmol) and dry THF (4 mL), purging and back-filling with argon after each addition. After 30 min of stirring a solution of antrocamphin B (3) (69 mg, 0.28 mmol) in dry THF (1 mL) was added via cannula to the preformed solution of SmI2. After 1.5 h the reaction mixture was quenched with methyl chloroformate (30 μL, 0.39 mmol) and the reaction mixture was stirred for a further 30 min before being diluted with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The extract was washed with brine (10 mL), dried and evaporated, and the residue was subjected to flash chromatography. Elution with 40% EtOAc–hexanes gave 51 as a pale yellow solid (23 mg, 32%) identical with the material described above.
TNF expression inhibition assays
These were conducted as described previously.32Acknowledgements
We acknowledge the facilities and scientific and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy, Characterisation, and Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. MRB is, and KAP was, the recipient of an Australian Postgraduate Award. We thank Professors Mark Ogden and George Koutsantonis for valuable discussions regarding alkyne-Pd complexation.Notes and references
- J.-J. Chen, W.-J. Lin, C.-H. Liao and P.-C. Shieh, J. Nat. Prod., 2007, 70, 989–992 CrossRef CAS PubMed.
- K. A. Punch, E. L. Ghisalberti and M. J. Piggott, J. Nat. Prod., 2011, 74, 1348–1350 CrossRef CAS PubMed.
- Y.-H. Hsieh, F.-H. Chu, Y.-S. Wang, S.-C. Chien, S.-T. Chang, J.-F. Shaw, C.-Y. Chen, W.-W. Hsiao, Y.-H. Kuo and S.-Y. Wang, J. Agric. Food Chem., 2010, 58, 3153–3158 CrossRef CAS PubMed.
- L.-S. Shi, C.-H. Chao, D.-Y. Shen, H.-H. Chan, C.-H. Chen, Y.-R. Liao, S.-J. Wu, Y.-L. Leu, Y.-C. Shen, Y.-H. Kuo, E. J. Lee, K. Qian, T.-S. Wu and K.-H. Lee, Bioorg. Med. Chem., 2011, 19, 677–683 CrossRef CAS PubMed.
- C.-L. Lee, C.-H. Huang, H.-C. Wang, D.-W. Chuang, M.-J. Wu, S.-Y. Wang, T.-L. Hwang, C.-C. Wu, Y.-L. Chen, F.-R. Chang and Y.-C. Wu, Org. Biomol. Chem., 2011, 9, 70–73 CAS.
- M. V. Sargent and P. O. Stransky, J. Chem. Soc., Perkin Trans. 1, 1982, 2373–2377 RSC.
- M. Matsumoto, K. Kobayashi and Y. Hotta, J. Org. Chem., 1984, 49, 4740–4741 CrossRef CAS.
- A.-S. Castanet, F. Colobert and P.-E. Broutin, Tetrahedron Lett., 2002, 43, 5047–5048 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and S. Kazemi, Can. J. Chem., 2009, 87, 1675 CrossRef CAS.
- R. D. Stephens and C. E. Castro, J. Org. Chem., 1963, 28, 3313–3315 CrossRef CAS.
- F. G. Schreiber and R. Stephenson, J. Chem. Soc., Perkin Trans. 1 (1972–1999), 1977, 90–92 CAS.
- Y.-R. Liao, P.-C. Kuo, J.-W. Liang, Y.-C. Shen and T.-S. Wu, Int. J. Mol. Sci., 2012, 13, 10432–10440 CrossRef CAS PubMed.
- A. Padwa and G. S. K. Wong, J. Org. Chem., 1986, 51, 3125 CrossRef CAS.
- E. D. Goddard-Borger, E. L. Ghisalberti and R. V. Stick, Eur. J. Org. Chem., 2007, 3925–3934 CrossRef CAS.
- S. Kobayashi and T. Kobayashi, Molecules, 2000, 5, 1062 CrossRef.
- I. V. Alabugin, V. I. Timokhin, J. N. Abrams, M. Manoharan, R. Abrams and I. Ghiviriga, J. Am. Chem. Soc., 2008, 130, 10984 CrossRef CAS PubMed.
- Y. Yamamoto, A. Nagata, Y. Arikawa, K. Tatsumi and K. Itoh, Organometallics, 2000, 19, 2403–2405 CrossRef CAS.
- H. S. Blanchette, S. C. Brand, H. Naruse, T. J. R. Weakley and M. M. Haley, Tetrahedron, 2000, 56, 9581–9588 CrossRef CAS.
- Y.-T. He, S. Xue, T.-S. Hu and Z.-J. Yao, Tetrahedron Lett., 2005, 46, 5393–5397 CrossRef CAS PubMed.
- J. G. Rodriguez and T. Laparra, Tetrahedron, 2009, 65, 2551 CrossRef CAS PubMed.
- R. B. Davis and P. Hurd, J. Am. Chem. Soc., 1954, 77, 3284 CrossRef.
- F. J. Wilson and W. M. Hyslop, J. Chem. Soc., 1923, 123, 2612 RSC.
- A. Boussonniere, R. Beneteau, N. Zimmerman, J. Lebreton and F. Denes, Chem.–Eur. J., 2011, 17, 5613 CrossRef CAS PubMed.
- T. Ishikawa, T. Mizuta, K. Hagiwara, T. Aikawa, T. Kudo and S. Saito, J. Org. Chem., 2002, 68, 3702 CrossRef PubMed.
- D. Crich, A. B. Pavlovic and D. J. Wink, Synth. Commun., 1999, 29, 359 CrossRef CAS.
- R. Brettle and S. M. Shibib, J. Chem. Soc., Perkin Trans. 1, 1981, 2912 RSC.
- C. Clausen, R. Wartchow and H. Butenschon, Eur. J. Org. Chem., 2001, 93 CrossRef CAS.
- B. Guerin and L. J. Johnston, J. Org. Chem., 1989, 54, 3176–3181 CrossRef CAS.
- H. Zhao, D. J. Li, L. Liu and Q. X. Guo, Chem. Commun., 2003, 506 RSC.
- T. Tsuritani, S. Ito, H. Shinokubo and K. Oshima, J. Org. Chem., 2000, 65, 5066 CrossRef CAS.
- L. Lu, J. M. Fang, G. H. Lee and W. Yu, J. Chin. Chem. Soc., 1997, 44, 279 CAS.
- S. G. Stewart, C. J. Braun, S.-L. Ng, M. E. Polomska, M. Karimi and L. J. Abraham, Bioorg. Med. Chem., 2010, 18, 650–662 CrossRef CAS PubMed.
- S. G. Stewart, D. Spagnolo, M. E. Polomska, M. Sin, M. Karimi and L. J. Abraham, Bioorg. Med. Chem. Lett., 2007, 17, 5819–5824 CrossRef CAS PubMed.
- S. Y. Yeung, S. Kampmann, K. A. Stubbs, B. W. Skelton, B. J. Kaskow, L. J. Abraham and S. G. Stewart, MedChemComm, 2011, 2, 1073–1078 RSC.
- M. Karimi, L. C. Goldie, M. N. Cruickshank, E. K. Moses and L. J. Abraham, Eur. J. Hum. Genet., 2009, 17, 1454–1462 CAS.
- M. Karimi, L. C. Goldie, D. Ulgiati and L. J. Abraham, BioTechniques, 2007, 42, 217–224 CrossRef CAS PubMed.
- M. Buccini, S. Y. Jeow, L. Byrne, B. W. Skelton, T. M. Nguyen, C. L. L. Chai and M. J. Piggott, Eur J. Org. Chem., 2013, 2013, 3232–3240 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Wang, Z. Zhang and B. Yu, J. Org. Chem., 2005, 70, 8884–8889 CrossRef CAS PubMed.
- I. M. Godfrey, M. V. Sargent and J. A. Elix, J. Chem. Soc., Perkin Trans. 1, 1974, 11, 1353–1354 RSC.
- J. A. Elix and V. J. Portelli, Aust. J. Chem., 1990, 43, 1773 CrossRef CAS.
- F. G. Schreiber and R. Stephenson, J. Chem. Soc., Perkin Trans. 1, 1977, 90–92 RSC.
- I. V. Alabugin, V. I. Timokhin, J. N. Abrams, M. Manoharan, R. Abrams and I. Ghiviriga, J. Am. Chem. Soc., 2008, 130, 10984–10995 CrossRef CAS PubMed.
- F. J. Wilson and W. M. Hyslop, J. Chem. Soc., 1923, 123, 2612–2618 RSC.
- R. S. Shank and H. Shechter, J. Org. Chem., 1959, 24, 1825–1826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds and crystallographic data. CCDC 970520 for 50 and 970521 for 51. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob42333f |
| This journal is © The Royal Society of Chemistry 2014 |