 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile C3-symmetric scaffolds and their use for covalent stabilization of the foldon trimer†
Arne
Berthelmann
a,
Johannes
Lach
a,
Melissa A.
Gräwert
bc,
Michael
Groll
b and
Jutta
Eichler
*a
aDepartment of Chemistry and Pharmacy, University of Erlangen-Nurnberg, Schuhstr. 19, 91052 Erlangen, Germany. E-mail: jutta.eichler@fau.de; Fax: +49-9131-852-2587; Tel: +49-9131-852-4117
bCenter for Integrated Protein Science at the Department of Chemistry, Chair of Biochemistry, Technical University of Munich, Lichtenbergstr. 4, 85747 Munich, Germany. E-mail: michael.groll@ch.tum.de; Fax: +49-89-289-13363; Tel: +49-89-289-13361
cEuropean Molecular Biology Laboratory, Hamburg Unit, EMBL c/o DESY, Notkestraße 85, 22603 Hamburg, Germany. E-mail: graewert@embl-hamburg.de; Fax: +49-40-89902-149; Tel: +49-40-89902-115
First published on 18th February 2014
Abstract
C 3-Symmetric trimesic acid scaffolds, functionalized with bromoacetyl, aminooxyacetyl and azidoacetyl moieties, respectively, were synthesized and compared regarding their utility for the trivalent presentation of peptides using three different chemoselective ligation reactions, i.e. thioether and oxime formation, as well as the “click” reaction. The latter ligation method was then used to covalently stabilize the trimer of foldon, a 27 amino acid trimerization domain of bacteriophage T4 fibritin, by linking the three foldon monomers to the triazido-functionalized trimesic acid scaffold. This reaction dramatically enhanced the thermal stability of the trimer, while maintaining the correct fold, as demonstrated by CD spectroscopy and X-ray crystal structure analysis, respectively, of the foldon–scaffold conjugates.
Introduction
Multivalent interactions play an important role in biological systems,1 as exemplified by receptor oligomers.2 Trimeric ligands, such as the tumor necrosis factor (TNF) superfamily,3 are often presented in the shape of C3-symmetric molecules. C3-Symmetric trimeric proteins are also involved in the complex interplay between pathogens, such as the human immunodeficiency virus (HIV-1), and their host cells.4 Interactions of the HIV-1 envelope protein (Env), which is presented as a C3-symmetric trimer on the virus surface,5 with cellular receptors are involved in a cascade of binding events that result in virus entry into the cell.6 Synthetic peptides have proven excellent molecular tools to explore the chemical and structural determinants of protein–protein interactions.7 Accordingly, peptides to be used for studying molecular interactions involving trimeric proteins should also be presented as trimers, which requires ready and fast synthetic access to such trivalent peptides. Addressing this challenge, we have synthesized three differently functionalized trivalent C3-symmetric scaffolds to which peptides can be covalently linked via different chemoselective ligation strategies. For this, we explored three different ligation reactions: thioether formation from an α-haloalkyl and a thiol group,8 the generation of 1,2,3-triazoles through 1,3-dipolar cycloaddition of an azide to an alkyne,9 as well as oxime formation from an aldehyde and a hydroxylamine.10Typically, the purpose of multivalent presentation is to bring into spatial proximity molecules that normally would not self-associate into an oligomer. An alternative strategy is covalent linkage of monomers of a pre-organized oligomer. Such covalently stabilized oligomers could be useful in experiments involving low, i.e. nano- and picomolar concentrations, where the native, non-covalent oligomer may no longer be sufficiently stable. Such a beneficial effect of covalent stabilization has been shown for peptides that present parts of the N-peptide region of HIV-1 gp41 fused to a trimeric coiled coil.11 Covalent stabilization of this trimer by introducing multiple inter-chain disulfide bridges dramatically enhanced the HIV-1 inhibitory activity of these trimeric peptides, most likely due to increased thermodynamic stability of the trimer.12
The C-terminal domain of fibritin, a structural protein of bacteriophage T4, has been shown to be essential for fibritin trimerization and folding.13 This domain, termed foldon, assembles into a β-propeller-like trimeric structure in which each subunit consists of a single β-hairpin.14 Using a peptide presenting only the 27 amino acid foldon sequence, this characteristic fold has been shown to be independent of the structural context of the fibritin protein.15 Furthermore, the trimeric structure of the foldon is largely unaffected by proteins attached to it, rendering this domain an ideal auxilliary to induce or stabilize trimeric structures of peptides and proteins. Examples of such foldon fusion proteins include HIV-1 gp41,16 a tumor necrosis factor ligand,17 as well as a recombinant influenza H5N1 vaccine.18
Here, we asked whether the thermodynamic stability of the foldon trimer could be enhanced, while maintaining the correct fold, by covalently linking the three monomers to a C3-symmetric scaffold (Fig. 1).
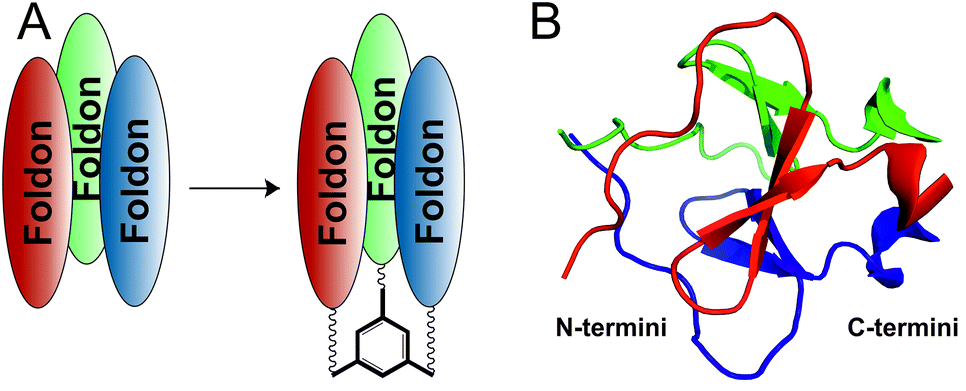 | ||
| Fig. 1 (A) Covalent stabilization of the foldon trimer by N- or C-terminally linking the three monomers to a C3-symmetric scaffold. (B) Location of N-and C-termini, respectively, of the three monomers within the foldon NMR structure (PDB ID: 1RFO).15 | ||
Results and discussion
Synthesis of scaffolds
Benzene-1,3,5-tricarboxylic acid (trimesic acid) 1, containing a 3-fold axis, served as a starting point for the generation of the different scaffolds. The utility of this simple aromatic compound for the synthesis of complex trimeric structures, such as monodisperse dendrimers,19 symmetric LewisX antigens20 or trimeric integrin ligands,21 has been demonstrated before.In order to provide a trivalent amino-functionalized scaffold, N1-Boc-3,6-dioxaoctane-1,8-diamine (Boc-DOOA) 2 was coupled as a spacer to the three carboxylic groups of 1 using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) as a coupling reagent, yielding the Boc-protected scaffold 3 (Scheme 1, Fig. S1 and S2†). EDCI was chosen because it facilitates work-up of the product through simple aqueous extraction of the resulting water-soluble urea derivative.22 Removal of the Boc groups yielded the C3-symmetric scaffold 4 (Scheme 1, Fig. S3 and S4†). The ethylene glycol nature of the diamine spacer 2 was chosen based on its favorable chemical and physical characteristics, which include solubility in organic solvents, such as dichloromethane or chloroform, in which scaffold synthesis was carried out, as well as in aqueous media for the subsequent ligation reaction with peptides. Furthermore, PEG based spacers are flexible, non-toxic and non-immunogenic.23
 | ||
| Scheme 1 Synthesis of the amino-functionalized trimesic acid scaffold 4. | ||
Scaffold 4 provided the basis for the generation of three differently functionalized trimeric scaffolds, through acylation with different acetic acid derivatives (Scheme 2). The bromoacetylated scaffold 5, which served as a precursor for subsequent thioether ligation, was synthesized by acylating the amino groups of 4 with bromoacetic acid. For this reaction, EDCI turned out to be a poor coupling reagent, since it was used as a hydrochloride, resulting in a halide exchange of the bromo alkyl group, yielding the less reactive chloroacetyl scaffold derivative. This reaction could be avoided by using N,N′-diisopropylcarbodiimide (DIC) as the coupling reagent, which yielded the desired bromoacetyl scaffold derivative 5 (Fig. S5 and S6†).
 | ||
| Scheme 2 Functionalization of the amino scaffold 4 for different ligation reactions. (i) Bromoacetic acid/DIC; (ii) azidoacetic acid/EDCI; (iii) (a) AOA/EDCI, (b) TFA/DCM; X: –(CH2)2–O–(CH2)2–O–(CH2)2–. | ||
The triazido scaffold 6 was generated by two different approaches. The first strategy involved coupling of pre-formed azidoacetic acid 824 (Fig. S11 and S12†) in conjunction with EDCI to yield 6 (20% isolated). The second method was based on a pseudo halide exchange of the haloacetylated scaffold 5 with sodium azide in the presence of 18-crown-6-ether.25 Both methods worked equally well, yielding 6 as the desired product (Fig. S7 and S8†).
The hydroxylamine functionality required for subsequent oxime ligation was introduced by coupling bis-Boc protected (aminooxy)acetic acid (AOA) to the triamino scaffold 4, followed by removal of the Boc groups with trifluoroacetic acid (TFA), yielding tris(aminooxy)-functionalized scaffold 7 quantitatively (Fig. S9 and S10†).
Ligation reactions
The utility of the three differently functionalized scaffolds 5, 6 and 7 for the respective ligation reactions was tested using the peptide sequence AKYRP, a short trypsin inhibitor.26 This peptide was synthesized thrice (peptides 9, 10 and 11). The sequence was N-terminally extended by ε-aminohexanoic acid (Ahx) as a spacer, followed by an N-acetylated cysteine (peptide 9, Fig. S13†), N-acetylated propargyl glycine (peptide 10, Fig. S14†), and 4-formylbenzoic acid (peptide 11, Fig. S15†), respectively. For the cleavage of peptide 11 from the resin, the scavenger composition of the TFA cocktail had to be modified in order to avoid side reactions, in particular reduction of the aldehyde to the alcohol. Therefore, thioanisole was omitted from the cleavage cocktail, and formylbenzoic acid was added as an additional scavenger. All three peptides could be synthesized in good yields of 42–58%.The functionalized scaffolds 5, 6 and 7, as well as peptides 9, 10 and 11, subsequently served as mutually reactive precursors for the three ligation reactions to be studied (Scheme 3). To analyze and compare the kinetics of these ligations, the reaction progress was followed by LC-MS over time.
 | ||
| Scheme 3 Ligation reactions. (A) Thioether ligation; (B) 1,3-dipolar cycloaddition; (C) oxime ligation. (i) Carbonate buffer pH 9.6; (ii) 6 M CuSO4, 18 M sodium ascorbate; (iii) citrate buffer pH 2.5. X: –(CH2)2–O–(CH2)2–O–(CH2)2–; peptide: Ahx-AKIYRP. | ||
Formation of a thioether from haloacetylated and thiol containing precursors in aqueous media (Scheme 3A) is well established.27 This reaction, however, is not absolutely chemoselective. Peptide dimerization through disulfide formation is a prominent side reaction, which can be reversed by reducing the disulfide using tris(2-carboxyethyl)phosphine (TCEP).28 The ligation reaction was performed at pH 9.6 to provide a highly nucleophilic thiolate species of 9. As shown in Fig. 2A, the trimeric product 12 (Fig. S16†) was generated instantaneously upon mixing of precursors 5 and 9. The first LC-MS sample was taken after approximately 1 min at which time the conversion rate was already 55%, indicating a very fast reaction. Kinetic analysis by HPLC indicated 85% conversion to the thioether trimer within 30 min. The disulfide dimer of 9 was detected merely as a minor side product, while the remaining side products were identified as mono- and di-substituted scaffolds, respectively. These results demonstrate that thioether ligation is an excellent method for the generation of trivalent peptides. Furthermore, this method can also be used to trimerize proteins using free cysteine residues in their sequences as ligation points.
 | ||
| Fig. 2 Kinetics of ligation reactions: (A) thioether ligation (generation of 12), (B) 1,3-dipolar cycloaddition (generation of 13), (C) oxime ligation (generation of 14). Monomeric peptides were used at 3 mM and scaffolds at 1 mM, resulting in a concentration of 1 mM for the generated trivalent peptides. | ||
Copper(I)-catalyzed 1,3-dipolar cycloaddition of azides to alkynes generates 1,4-substituted (1,2,3)-triazoles (Scheme 3B) and is better known as “click chemistry”.29 This type of chemoselective reaction has been extensively used to generate diverse bioactive molecules, including combinatorial peptidotriazole libraries,30 cyclopeptide analogs,31 or β-turn mimics.32 The click reaction is controlled by several parameters, most importantly the source of Cu(I). A convenient way to provide the catalytic Cu(I) species is in situ reduction of Cu(II)-salts with a suitable reductive agent, such as ascorbic acid. Another critical factor is the solvent. While an aqueous environment is favorable for reactions involving unprotected peptides, protected or resin-bound peptides, as well as other, less polar compounds, require a polar organic solvent. To cover both scenarios, two solvent mixtures were tested, i.e. a water–isopropanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and a water–DMF (1:1) mixture.
:1) and a water–DMF (1:1) mixture.
Similar to the thioether ligation, kinetic analysis (Fig. 2B) demonstrated a fast reaction of peptide 10 with triazido scaffold 6. In isopropanol, the reaction was almost complete within 10 min, with a conversion rate to the trimeric product 13 (Fig. S17†) of 86% (isopropanol) and 90% (DMF), respectively, illustrating the outstanding value of click chemistry for the chemoselective attachment of alkyne peptides to C3-symmetric azido scaffolds. Mono- and disubstituted scaffolds were found as side products (11% and 9%, respectively), whereas only traces of unreacted 10 were detectable after 30 minutes.
Finally, oxime ligation33 was used to generate trivalent peptide 14 by reacting the nucleophilic hydroxylamines of scaffold 7 with peptide 11, which was N-terminally acylated with 4-formylbenzoic acid, providing the aldehyde moiety for oxime formation (Scheme 3C). Analysis of reaction kinetics for the formation of 14 (Fig. S18†) indicated a maximal yield of 80% after 20 minutes (Fig. 2C), which could not be further improved over time. As expected,34 addition of aniline as a catalyst accelerated the reaction, but did not improve the final yield (data not shown). Interestingly, the main side product here was unreacted 11 (18%) rather than the respective mono- and disubstituted scaffolds. It should also be noted that the trimerization yields of parallel experiments involving an analog of 11, in which the aldehyde moiety was replaced by a ketone (pyruvoyl moiety), were much lower and did not exceed 23% after 10 hours.
Comparing the results of the above three ligation strategies, it is apparent that thioether ligation and click reaction yielded the desired trivalent peptides in excellent yields and purities, while the results of the oxime ligation were less satisfactory. Furthermore, all three methods have their specific strengths and limitations. Thioether ligation yields metabolically stable and largely non-reactive molecules that are well compatible with biological systems. In fact, methionine, one of the proteinogenic amino acids, contains a thioether in its side chain. Possible side reactions associated with thioether ligation include an attack of other nucleophilic moieties on the bromoacetylated scaffold, impairing the site-selectivity of the ligation reaction. Similarly, the carbonyl carbon of the aldehyde or ketone moieties involved in oxime ligation may also be attacked by nucleophiles other than the cognate aminooxy group of the scaffold. Furthermore, the limited choice of scavengers to be used during the acidic cleavage of the aldehyde peptide precursor from the resin may impair the purity of longer peptides containing multiple side chain protection groups. In addition, due to the susceptibility of oximes to hydrolysis, this ligation reaction may not be ideal for the generation of molecules to be used in biological systems. Triazoles, on the other hand, which are the product of the click reaction, are thought to be fairly stable in a range of different milieus. It should be noted, however, that the nitrogen atoms of the triazole ring are potentially strong hydrogen bond acceptors in biomolecular interactions, which may mask the native interactions. These specifics should be taken into account when selecting a ligation strategy for a specific application.
Covalent stabilization of the foldon trimer
Based on the excellent results obtained using the click reaction to attach peptide monomers to the trimesic acid scaffold, as well as the robustness of this reaction and ease of synthesis of precursors, we chose the click reaction to covalently stabilize the trimer of the 27 amino acid peptide foldon. The three dioxaoctane spacer units of scaffold 6 were thought to be adequate to cover the distances between the N-(13.7 Å) and C- (5.15 Å) termini of the three foldon monomers within the trimer structure. In addition to the unmodified foldon sequence (15, Fig. S19†), we synthesized two peptides, in which a propargylglycine residue was added either to the N- or C-terminus of the foldon sequence (16 and 17, Fig. S20 and S21,† see Table 1 for peptide sequences). These functionalized peptides were then “clicked” to the triazido scaffold 6, yielding conjugates 18 and 19 (Fig. 3 and S22 through S25†), whose folding and thermal stabilities were subsequently analyzed by CD spectroscopy and X-ray crystal structure analysis, respectively. | ||
| Fig. 3 Covalently stabilized foldon trimers, in which the three monomers are linked N- (18, left) and C- (19, right) terminally respectively, via triazole linkers, to a trimesic acid scaffold. | ||
Comparison of the CD spectra of 15, 18 and 19 indicated that covalent attachment of the three foldon monomers to the scaffold does not affect folding of the trimer, as all three CD spectra are very similar (Fig. 4).
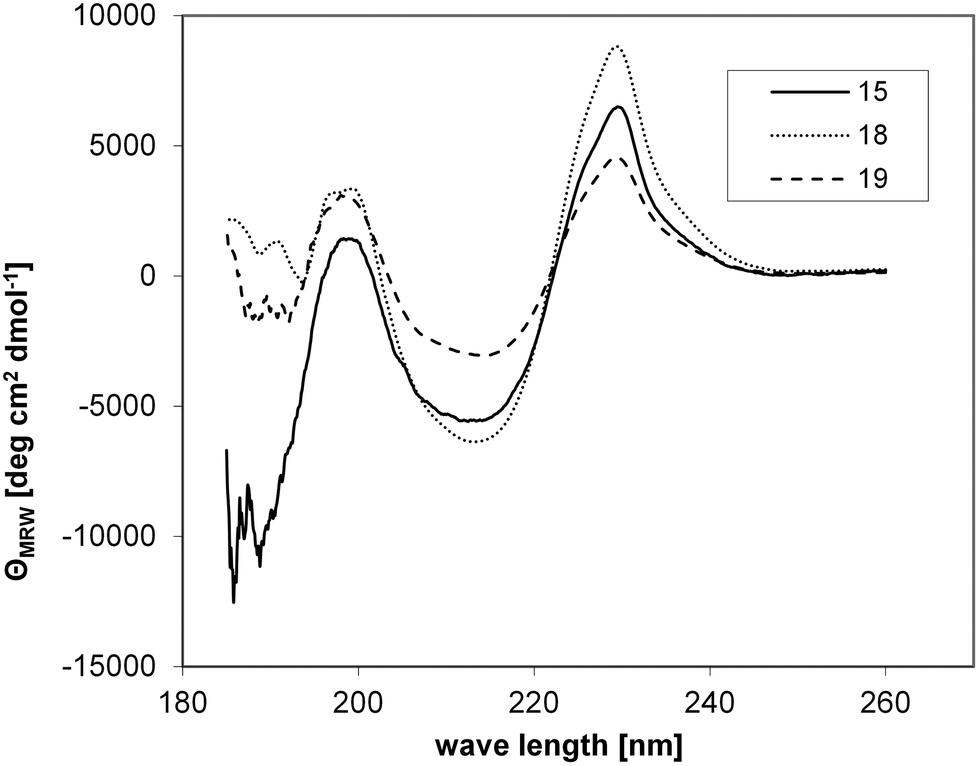 | ||
| Fig. 4 CD spectra of the non-covalent foldon trimer (15), as well as covalently stabilized trimers 18 and 19. | ||
The notion of structural similarity between wt foldon (15) and the two foldon–scaffold conjugates was corroborated by solving the crystal structures of 18 and 19, and comparing it with the structure of wt foldon. Conjugates 18 and 19 were subjected to a number of crystallization trials with various salts at different concentrations. The conjugates spontaneously crystallized in more than 70% of the tested buffers (approximately 500). The most promising crystals were obtained in 0.2 M ammonium sulfate, 30% PEG 4000 and 1.4 M Na/K-phosphate, pH 5. Despite the relatively small size of the crystals, high resolution data could be collected up to 1.1 Å resolutions. Interestingly, the X-ray data revealed a high tendency of the foldon to crystallize in different space groups. Even crystals harvested from the same drop showed different symmetries. We determined the structures of conjugates 18 and 19, which crystallized in the space groups p21 (1.1 Å) and C2 (1.3 Å). Both proteins present a native-like fold, comprising a 310-helix at the C-terminus, which is preceded by a β-hairpin and an extended structure at the N-terminus (Fig. 5). Superposition of 18 and 19 with wt foldon (PDB ID 4NCU) confirmed the overall similarity of the three structures, as local heterogeneity is only observed at the flexible spacer region (root mean square deviation of Cα < 0.3 Å) (Fig. 5a and S27†). Furthermore, the structures are largely congruent with the previously solved NMR structure.15 The calculated size of the interface between the three foldon monomers (approximately 3000 Å2) was very similar in wt foldon and the covalently stabilized trimers 18 and 19. The scaffold and linker regions of 18 and 19, however, are resolved differently in the two structures. The electron density map of 18 merely displays the amide part of the linker that is directly attached to the foldon N-terminus (Fig. 5b). In the structure of 19, on the other hand, the trimesic acid scaffold is well defined and positioned at the center of the tri-fold non-crystallographic symmetry axis atop the C-terminal leucine residues, whereas the first three N-terminal foldon residues, as well as the linker region composed of 6-aminohexanoic acid, the triazole ring and DOOA, are structurally distorted (Fig. 5c). In summary, the crystallographic data confirm that the structure of the foldon trimer is largely unaffected by covalent linking of the three monomers to the trimesic acid scaffold.
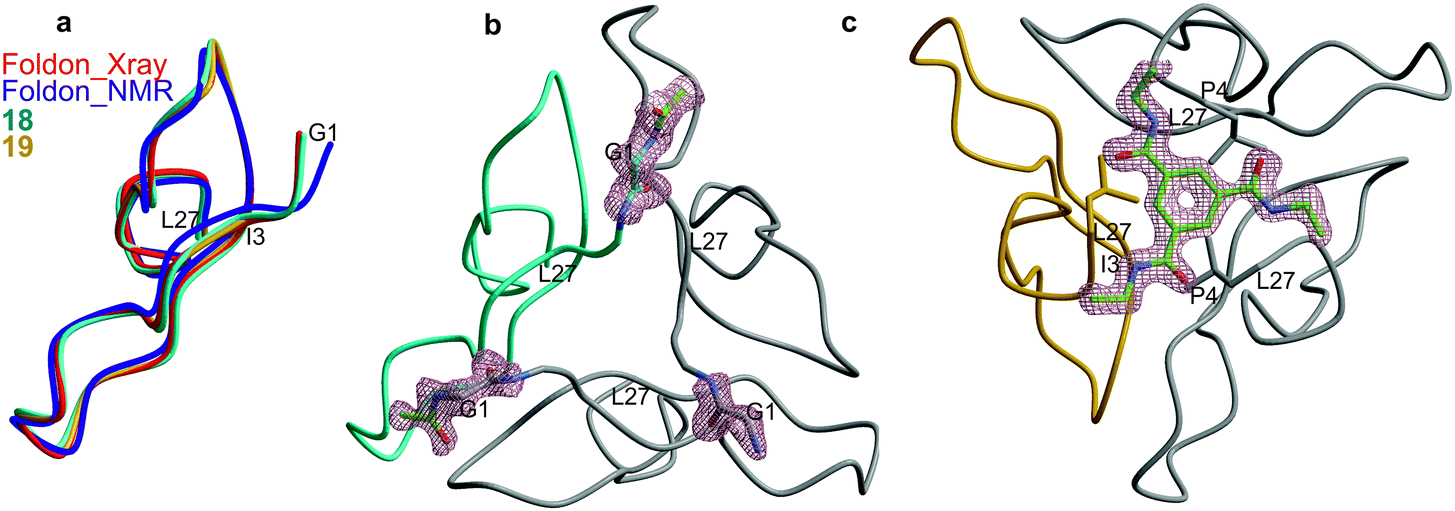 | ||
| Fig. 5 (a) Backbone superposition of one monomer of wt foldon (red: crystal structure (PDB ID 4NCU), purple: NMR structure15) with the covalent conjugates 18 (cyan) and 19 (gold). The structurally distorted N-termini of 19 are labelled. (b) Trimer structure of 18. The acetamide of the linker is well defined in the electron density map. (c) Trimer structure of 19 (rotated about the x-axis by 180°). The electron density (blue) represents a 2FO–FC-omit map contoured at 1.0 σ of the trimesic acid scaffold. | ||
Thermal unfolding of wt and covalently stabilized foldon trimers was assessed by monitoring the temperature-dependent changes in CD signals at 228 nm.35
As shown in Fig. 6, the stability of the trimer was greatly enhanced by covalently attaching the three monomers to the trimesic acid scaffold, as evidenced by a strong shift in Tm of 18 and 19, compared to 15. Even under denaturing conditions (5 M urea), the covalently stabilized trimers (18, Tm = 77 °C and 19, Tm = 78 °C) were substantially more stable, with Tm approximately 30 degrees higher than that of the non-covalent foldon (15, Tm = 48 °C) (Fig. 6). In a buffer without detergent, on the other hand, the trimers of 18 and 19 did not start to unfold until temperatures as high as 80 °C were reached, so that no melting points could be determined (Fig. S26†). These results demonstrate a strong stabilization of the foldon trimer by covalently attaching the three monomers to a C3-symmetric scaffold, regardless of the orientation of the foldon sequence with respect to the scaffold (N- and C-terminal attachment, respectively).
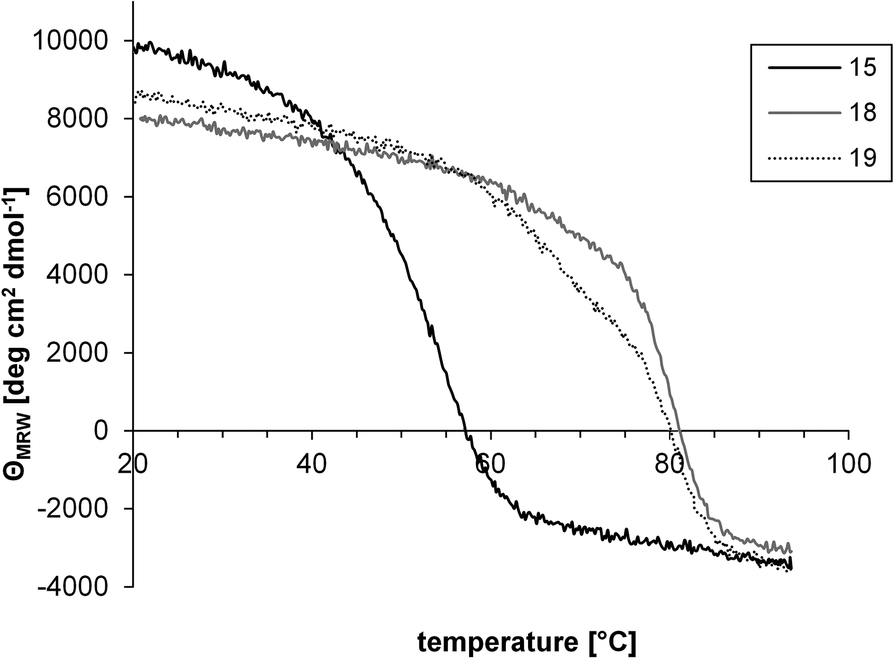 | ||
| Fig. 6 Thermal unfolding of the non-covalent foldon trimer (15), as well as the covalently stabilized trimers 18 and 19, in the presence of 5 M urea. | ||
Experimental section
Materials and methods
:2:3; 30 min). The Fmoc group was removed using 20% piperidine–DMF (20 min). Fmoc-propargyl glycine (peptides 10, 16 and 17) as well as 4-formylbenzoic acid (peptide 11) were coupled overnight using 3 and 10 eq., respectively, of activated acid. To avoid aspartimide formation in peptides 15, 16 and 17, Fmoc-L-Asp(OMpe)-OH was used to introduce aspartic acid residues in these peptides. Peptide 9, 10, 15, 16 and 17 were cleaved from the resin using a mixture of TFA–thioanisole–phenol–water–EDT (82.5:5:5:5:2.5). Peptide 11 was cleaved with a mixture of TFA–TIPS–4-formylbenzoic acid–water (85:5:5:5) to avoid reduction of the aldehyde group. The cleaved peptides were precipitated in a cold 1:1 mixture of cyclohexane and t-butyl methyl ether (10 mL), extracted with water (10 mL), lyophilized twice, purified by preparative HPLC, and characterized by LC-MS and high resolution ESI mass spectrometry, respectively.
:1)) were mixed with 200 μL 0.1 M carbonate buffer pH 9.6, resulting in a final concentration of 1 mM of peptide 9. After an overnight reaction, the product was analyzed by LC-MS, lyophilized, purified by preparative HPLC, and analyzed by LC-MS.
:1)), were mixed with 100 μL of 4 mM scaffold 6 solution, 100 μL of 24 mM CuSO4 in isopropanol–water (1:1), as well as with 100 μL of 72 mM sodium ascorbate in isopropanol–water (1:1), resulting in a final concentration of 1 mM of peptides. Isopropanol can be replaced with DMF. After an overnight reaction, the product was analyzed by LC-MS, lyophilized, purified by preparative HPLC, and analyzed by LC-MS and high resolution ESI mass spectrometry (18 and 19).
:1)) were mixed with 200 μL of 0.1 M citrate buffer pH 2.5, resulting in a final concentration of 1 mM of peptide 14. After an overnight reaction, the product was analyzed by LC-MS, lyophilized, purified by preparative HPLC, and analyzed by LC-MS.
Conclusions
In conclusion, we have generated three differently functionalized C3-symmetric scaffolds based on trimesic acid for the trivalent presentation of biomolecules. The utility of these versatile scaffolds for trimerization reactions was demonstrated using appropriately functionalized peptides in conjunction with three different ligation strategies, i.e. thioether formation, click chemistry and oxime ligation, respectively. Kinetic analysis of each ligation reaction demonstrated that all three reactions are powerful tools for the synthesis of trivalent peptides. Depending on the desired flexibility of the trivalent molecules, the length and flexibility of the spacer can be varied using other diamines. It should also be noted that the utility of these scaffolds is not limited to the three ligation reactions presented here, but could readily be extended to include other ligation reactions, such as Staudinger ligation42 and native chemical ligation,43 simply by reacting the scaffold amino groups with appropriate chemical moieties. Furthermore, covalent attachment of alkyne-modified foldon peptides to the triazido scaffold using the click reaction distinctly enhanced the stability of the foldon trimer, while maintaining its correct fold. This illustrates an alternative application of multivalent peptide presentation, in which folded, non-covalent peptide and protein oligomers are thermodynamically stabilized by covalently linking the monomers to an appropriate multivalent scaffold.Abbreviations
| ACN | Acetonitrile |
| Boc | t-Butyloxycarbonyl |
| DCM | Dichloromethane |
| DIC | N,N′-Diisopropylcarbodiimide |
| DIPEA | N,N-Diisopropylamine |
| DOOA | 3,6-Dioxaoctane-1,8-diamine |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EDT | 1,2-Ethanedithiol |
| Fmoc | 9-Fluorenylmethoxycarbonyl |
| HOBt | 1-Hydroxybenzotriazole |
| TFA | Trifluoroacetic acid. |
Acknowledgements
This work was supported by SFB 796 (Project A5) from the German Research Foundation (DFG). We thank Astrid König for excellent technical assistance (crystallization experiments), Frank Hampel for high resolution ESI mass spectra and Thomas Kiefhaber for valuable advice regarding thermal unfolding transition measurements. The coordinates and structure factors of the two covalent conjugates 18 and 19, have been deposited in the Protein Databank (PDB accession codes 4NCV and 4NCW).Notes and references
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- B. Stephens and T. M. Handel, in Progress in Molecular Biology and Translational Science, ed. K. Terry, Academic Press, 2013, vol. 115, pp. 375–420 Search PubMed.
- M. G. Tansey and D. E. Szymkowski, Drug Discovery Today, 2009, 14, 1082–1088 CrossRef CAS PubMed.
- J. Liu, A. Bartesaghi, M. J. Borgnia, G. Sapiro and S. Subramaniam, Nature, 2008, 455, 109–113 CrossRef CAS PubMed.
- P. Zhu, J. Liu, J. Bess Jr., E. Chertova, J. D. Lifson, H. Grise, G. A. Ofek, K. A. Taylor and K. H. Roux, Nature, 2006, 441, 847–852 CrossRef CAS PubMed.
- B. J. Doranz, S. S. W. Baik and R. W. Doms, J. Virol., 1999, 73, 10346–10358 CAS.
- J. Eichler, Curr. Opin. Chem. Biol., 2008, 12, 707–713 CrossRef CAS PubMed.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- C. P. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- D. M. Eckert and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11187–11192 CrossRef CAS PubMed.
- E. Bianchi, M. Finotto, P. Ingallinella, R. Hrin, A. V. Carella, X. S. Hou, W. A. Schleif, M. D. Miller, R. Geleziunas and A. Pessi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12903–12908 CrossRef CAS PubMed.
- V. V. Mesyanzhinov, in Advances in Virus Research, Academic Press, Editon edn, 2004, vol. 63, pp. 287–352 Search PubMed.
- Y. Tao, S. V. Strelkov, V. V. Mesyanzhinov and M. G. Rossmann, Structure, 1997, 5, 789–798 CrossRef CAS.
- S. Güthe, L. Kapinos, A. Möglich, S. Meier, S. Grzesiek and T. Kiefhaber, J. Mol. Biol., 2004, 337, 905–915 CrossRef PubMed.
- Z. Qi, C. Pan, H. Lu, Y. Shui, L. Li, X. Li, X. Xu, S. Liu and S. Jiang, Biochem. Biophys. Res. Commun., 2010, 398, 506–512 CrossRef CAS PubMed.
- T. Ito, K. Iwamoto, I. Tsuji, H. Tsubouchi, H. Omae, T. Sato, H. Ohba, T. Kurokawa, Y. Taniyama and Y. Shintani, Appl. Microbiol. Biotechnol., 2011, 90, 1691–1699 CrossRef CAS PubMed.
- L. Du, V. H.-C. Leung, X. Zhang, J. Zhou, M. Chen, W. He, H.-Y. Zhang, C. C. S. Chan, V. K.-M. Poon, G. Zhao, S. Sun, L. Cai, Y. Zhou, B.-J. Zheng and S. Jiang, PLoS One, 2011, 6, e16555 CAS.
- D. Seebach, G. F. Herrmann, U. D. Lengweiler, B. M. Bachmann and W. Amrein, Angew. Chem., Int. Ed. Engl., 1996, 35, 2795–2797 CrossRef CAS.
- Y. Nishida, T. Tsurumi, K. Sasaki, K. Watanabe, H. Dohi and K. Kobayashi, Org. Lett., 2003, 5, 3775–3778 CrossRef PubMed.
- H. Kubas, M. Schafer, U. Bauder-Wust, M. Eder, D. Oltmanns, U. Haberkorn, W. Mier and M. Eisenhut, Nucl. Med. Biol., 2010, 37, 885–891 CrossRef CAS PubMed.
- H. Li, Y. Guan, A. Szczepanska, A. J. Moreno-Vargas, A. T. Carmona, I. Robina, G. K. Lewis and L. X. Wang, Bioorg. Med. Chem., 2007, 15, 4220–4228 CrossRef CAS PubMed.
- A. Torres, C. Mas-Moruno, E. Perez-Paya, F. Albericio and M. Royo, Bioconjugate Chem., 2011, 22, 2172–2178 CrossRef CAS PubMed.
- E.-M. Kim, M.-H. Joung, C.-M. Lee, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. W. Kim, Bioorg. Med. Chem. Lett., 2010, 20, 4240–4243 CrossRef CAS PubMed.
- D. J. Hlasta and J. H. Ackerman, J. Org. Chem., 1994, 59, 6184–6189 CrossRef CAS.
- J. Eichler and R. A. Houghten, Biochemistry, 1993, 32, 11035–11041 CrossRef CAS.
- H. Lindley, Biochem. J., 1960, 74, 577–584 CAS.
- M. Monso, W. Kowalczyk, D. Andreu and B. G. de la Torre, Org. Biomol. Chem., 2012, 10, 3116–3121 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A. Brik, J. Alexandratos, Y. C. Lin, J. H. Elder, A. J. Olson, A. Wlodawer, D. S. Goodsell and C. H. Wong, ChemBioChem: a Eur. J. Chem. Biol., 2005, 6, 1167–1169 CrossRef CAS PubMed.
- S. Punna, J. Kuzelka, Q. Wang and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2215–2220 CrossRef CAS PubMed.
- Y. Angell and K. Burgess, J. Org. Chem., 2005, 70, 9595–9598 CrossRef CAS PubMed.
- P. Marceau, C. Bure and A. F. Delmas, Bioorg. Med. Chem. Lett., 2005, 15, 5442–5445 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- J. Habazettl, A. Reiner and T. Kiefhaber, J. Mol. Biol., 2009, 389, 103–114 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- D. Turk, Acta Crystallogr., 2013, D69, 1342–1357 Search PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- A. Perrakis, R. Morris and V. Lamzin, Nat. Struct. Biol., 1999, 6, 458–463 CrossRef CAS PubMed.
- A. A. Vagin, R. A. Steiner, A. A. Lebedev, L. Potterton, S. McNicholas, F. Long and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2184–2195 Search PubMed.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef PubMed.
- C. Haase and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 1553–1556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 3 through 8; LC-MS data/MALDI spectra of 9 through 17; X-ray data collection and refinement statistics of foldon–scaffold conjugates; thermal unfolding of the non-covalent foldon trimer (15), as well as the covalently stabilized trimers 18 and 19, in a buffer without detergent. See DOI: 10.1039/c3ob42251h |
| This journal is © The Royal Society of Chemistry 2014 |