A step toward polytwistane: synthesis and characterization of C2-symmetric tritwistane†
Martin
Olbrich
,
Peter
Mayer
and
Dirk
Trauner
*
Department of Chemistry, Ludwig-Maximilians-Universität München, and Munich Center for Integrated Protein Science, Butenandtstrasse 5-13, 81377 München, Germany. E-mail: dirk.trauner@lmu.de
First published on 31st October 2013
Abstract
Twistane is a classic hydrocarbon with fascinating stereochemical properties. Herein we describe a series of oligomers of twistane that converges on a chiral nanorod, which we term polytwistane. A member of this series, C2-symmetric tritwistane, has been synthesized for the first time.
Twistane (tricyclo[4.4.0.03,8]decane) (1) is a tricyclic hydrocarbon that has played an important role in the development of organic stereochemistry.1 The D2-symmetrical molecule consists of three interwoven twist boat rings, all of which have approximate D2-symmetry with identical handedness. It can be formed, conceptually, through bridging of a cis-decalin with a single bond or by adding an ethano (ethane-1,2-diyl) bridge to a bicyclo[2.2.2]octane. In reality, twistane (1) has been synthesized numerous times, in both enantiomeric forms1,2 and as a racemate,3 starting with Howard Whitlock's seminal work in 1962.4 A large variety of substituted twistanes, including unsaturated derivatives, are now known.5
In a “Gedankenexperiment”, twistane (1) can be further extended by bridging its six-membered rings with additional ethano bridges mounted in a 1,4-fashion (Scheme 1). Due to geometrical constraints, the newly formed six-membered rings continue to adopt a twist boat conformation with the same handedness as the existing rings. Provided quaternary carbons are avoided, the first substitution can only proceed in one way to yield the C2-symmetrical ditwistane (2). Further addition of an ethano bridge to ditwistane in a linear fashion produces C2-symmetric tritwistane (3). Linear extension of C2-tritwistane (3) affords tetratwistane (4), pentatwistane (5), hexatwistane (6), heptatwistane (7), and so on. All of these helical compounds are chiral molecules with C2 symmetry.
 | ||
| Scheme 1 Twistane (1), oligotwistanes and polytwistane (8). | ||
Further extension of these oligotwistanes in a linear fashion, ideally to infinity, affords a polymer with fascinating geometrical properties, which we propose to call “polytwistane” (8) (Fig. 1). Ideal polytwistane has the same molecular formula, CnHn, as polyacetylene but it is fully saturated with all of its sp3 hybridized carbons and hydrogens in an identical chemical environment. It is a chiral nanorod of high rigidity since none of its C,C-bonds can rotate freely and it can exist in two enantiomorphic forms, i.e. as a P or M helix. A more detailed description of polytwistane as a geometrical object and a prediction of its physical properties will be described elsewhere.6
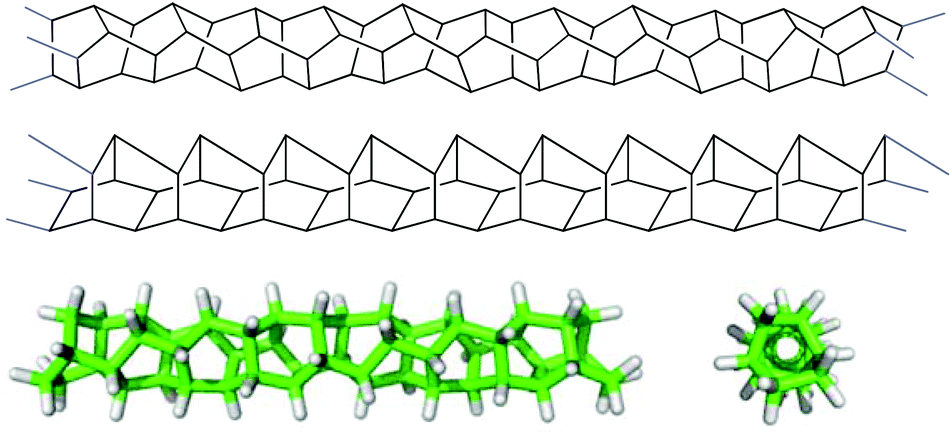 | ||
| Fig. 1 Aspects of polytwistane (8). Top: M helical polytwistane (8) drawn in a manner that emphasizes the structural unit of twistane. Middle: M helical polytwistane (8) drawn in a manner that emphasizes the incorporated bicyclo[2.2.2]octane units. Bottom: side view (left) and top view (right) of a C52H58-fragment of M helical polytwistane calculated at the B3LYP/6-31 g(d) level.7 | ||
Among the hydrocarbons shown in Scheme 1, only twistane (1) and ditwistane (2) are known compounds. Both enantiomers of ditwistane (2) have been synthesized8 and dozens of compounds that incorporate the ditwistane skeleton have been described.8 Structures containing the carbon skeleton of C2-tritwistane9 and C2-hexatwistane10 have also been reported. None of these compounds, however, have been identified as oligotwistanes, and polytwistane (8) does not seem to have been mentioned in the literature so far.
We now report our studies on linear oligotwistanes, which were undertaken to extend the series of known oligotwistanes and obtain spectral data that might help in the identification of polytwistane. Our initial studies were based on additions to laticyclic polyenes, which are known to yield twistane motifs.10 To this end, we synthesized the diene 11 and the triene 13 following a strategy developed by Gleiter and colleagues (Scheme 2).11
 | ||
| Scheme 2 Synthesis of the laticyclic conjugated precursors. | ||
Addition of ethylene to triene 9 at high pressure gave tetracyclic tetrachloro diene 10, which was dechlorinated to afford 11. Similarly, Diels–Alder addition of dihydrobarrelene12 to 9 gave 12, the dechlorination of which yielded the hexacyclic triene 13.
Our first attempts to synthesize tritwistane from dienes 10 or 11 are shown in Scheme 3. Epoxidation of tetrachloro diene 10 gave 14. Treatment of 14 with a Lewis acid in the presence of a hydride source, however, did not yield a twistane but exclusively gave 16. Presumably, this compound is the product of a “U-type” (instead of “N-type”) nucleophilic attack, yielding intermediate 15, followed by an intramolecular hydride shift, as evidenced by the stereochemistry of the product 16.13 Epoxidation of diene 11 with m-CPBA yielded the analogous ketone 19 as the only isolable product, due to the instability of the intermediary epoxide 17 under the conditions. The X-ray structures of ketones 16 and 19, which bear a novel carbon skeleton, as well as epoxide 14 are shown in the ESI.†
 | ||
| Scheme 3 Initial attempts to synthesize tritwistane via epoxidation of 10 and 11. | ||
Our attempts to access the tritwistane skeleton, via halogen addition, using conditions reported by Chou and coworkers9 were more successful (Scheme 4). Exposure of tetrachloro diene 10 to bromine in chloroform gave the halogenated tritwistanes 22a,b together with the interesting syn-adduct 24. The former presumably stem from an “N-type” nucleophilic attack onto bromonium ion 20, followed by quenching of the intermediary carbocation 21 by bromide, which yielded a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of diastereomers 22a and b, respectively. The formation of 24 can be rationalized by a “U-type” nucleophilic attack, followed by an SN2-type displacement with overall double inversion. The X-ray structures of the major diastereomer 22a and the syn-dibromide 24 are shown in Fig. 2. We next investigated the bromination on laticyclic hydrocarbon 11, which gave the dibromo tritwistane 27 as a single diastereomer, together with the unusual dibromide 30. The former is again the product of an “N-type” cyclization, whereas the latter presumably arises from a twofold Wagner–Meerwein rearrangement of the initial “U-type” cation 28, followed by nucleophilic interception by bromide. The X-ray structures of 27 and the rearranged 30 are also shown in Fig. 2.
:3 mixture of diastereomers 22a and b, respectively. The formation of 24 can be rationalized by a “U-type” nucleophilic attack, followed by an SN2-type displacement with overall double inversion. The X-ray structures of the major diastereomer 22a and the syn-dibromide 24 are shown in Fig. 2. We next investigated the bromination on laticyclic hydrocarbon 11, which gave the dibromo tritwistane 27 as a single diastereomer, together with the unusual dibromide 30. The former is again the product of an “N-type” cyclization, whereas the latter presumably arises from a twofold Wagner–Meerwein rearrangement of the initial “U-type” cation 28, followed by nucleophilic interception by bromide. The X-ray structures of 27 and the rearranged 30 are also shown in Fig. 2.
 | ||
| Scheme 4 Synthesis of tritwistanes 22a, b and 27via bromination of 10 and 11. | ||
 | ||
| Fig. 2 X-ray structures of 22a, 24, 27 and 30. | ||
Attempts to synthesize hexatwistane derivatives via bromination of laticyclic triene 13 were less successful and gave complex mixtures. So far, we have been unable to separate and cleanly characterize the major components of this mixture. Global dehalogenation of tetrachloro dibromo tritwistane 22 also failed under a variety of conditions. By contrast, dehalogenation of dibromo tritwistane 27 using Chatgilialoglu's reagent14 was more successful and gave the parent hydrocarbon 3 (Scheme 5). C2-Symmetric tritwistane (3) was easy to identify due to its simplified 1H- and 13C-NMR spectra (see ESI†).
 | ||
| Scheme 5 Synthesis of tritwistane (3). | ||
In summary, we have described the first synthesis of racemic tritwistane (3), which also yielded a variety of interesting byproducts. Several of these, such as compounds 16, 19 and 30, bear a novel carbon skeleton. Our results indicate that N-type additions across laticyclic polyenes are not a viable way to access longer oligotwistanes or polytwistane (8) due to a variety of possible side reactions and difficulties associated with the procurement of precursors. Attempts to synthesize polytwistane in a more direct fashion from acetylene itself are ongoing in our laboratory and will be reported in due course. The relationship of polytwistane to recently reported diamond nanowires15 and ultra-small carbon nanotubes16 is also under investigation.
Experimental section
Pentacyclo[6.2.2.23,6.02,7.05,9]tetradecan-4-one (19)
To a solution of tetracyclotetradecadiene 11 (30.0 mg, 0.161 mmol, 1.00 eq.) in CH2Cl2 (2.50 mL) at 0 °C was added m-CPBA (70–75 wt%, 37.5 mg, 0.161 mmol, 1.00 eq.). The reaction mixture was stirred at 0 °C for 1 h, was then allowed to warm to room temperature and stirred at room temperature for an additional 2 h. After dilution with CH2Cl2 (15 mL) the reaction mixture was poured into 10% aqueous NaOH (10 mL) and the organic layer was washed with H2O (2 × 7.5 mL), brine (7.5 mL), dried (Na2SO4) and concentrated in vacuo. Purification of the residue by flash column chromatography (hexanes–EtOAc = 9:1) afforded ketone 19 (20.0 mg, 61%) as a colorless solid. Recrystallization from hexanes afforded crystals suitable for X-ray analysis.
R
f 0.36 (hexanes–EtOAc = 9:1); mp 95–97 °C; 1H NMR (300 MHz, CDCl3) δ 2.80–2.71 (m, 1H), 2.40–2.29 (m, 1H), 2.27–2.19 (m, 1H), 2.17–2.10 (m, 1H), 2.07–2.01 (m, 1H), 2.00–1.88 (m, 2H), 1.85–1.72 (m, 4H), 1.67–1.53 (m, 5H), 1.52–1.37 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 222.7, 58.9, 47.1, 42.5, 40.6, 39.1, 38.3, 37.4, 29.5, 28.6, 28.3, 27.2, 19.8, 19.8; IR νmax 2925, 2859, 1721, 1260, 1090, 1063, 1025, 994, 909, 886, 858, 812, 798, 732; HRMS (EI) m/z: [M]+ calcd for C14H18O+ 202.1352; found: 202.1348.
Bromination of syn-3,4,5,6-tetracyclo[6.2.2.23,6.02,7]tetradeca-4,9-diene (11)
To a solution of diene 11 (49.9 mg, 268 μmol, 1.00 eq.) in CHCl3 (3.00 mL) at 0 °C was added a solution of bromine in CHCl3 (1.253 mL of a 1.37 vol% solution, 335 μmol, 1.25 eq.). The reaction mixture was stirred at 0 °C for 1 h and the reaction was then quenched by addition of diluted aqueous Na2S2O3. The organic layer was separated and the aqueous layer was extracted with CHCl3 (3 × 15 mL). The combined organic layer was washed with NaHCO3 (30 mL), brine (30 mL), dried (Na2SO4) and concentrated in vacuo. Purification of the residue by flash column chromatography (hexanes–Et2O = 95:5) afforded rearranged dibromide 30 (15 mg, 14%) and dibromotritwistane 27 (35 mg, 38%) as colorless solids. Recrystallization from hexanes afforded crystals suitable for X-ray diffraction experiments for both compounds.
C 2-Tritwistane (3)
To a solution of dibromide 27 (34.6 mg, 0.100 mmol, 1.00 eq.) in toluene (3.00 mL) was added TTMSS (0.075 mL, 59.7 mg, 0.240 mmol, 2.40 eq.) and one crystal of AIBN (2.50 mg, 0.015 mmol, 0.150 eq.). The resulting mixture was heated to 90 °C for 3 h and was then allowed to cool to room temperature and was concentrated in vacuo. The residue was purified by flash column chromatography (2 × 20 cm, n-pentane, 8 mL) to afford tritwistane (3) contaminated with some silicon species as a colorless oil. A solution of this oil in CDCl3 (1.50 mL) was stirred over a fluoride polymer at room temperature for 8 d. The solution was filtered over a short silica column and the solvent was removed in vacuo to afford tritwistane (3) (6 mg, 32%) as a waxy amorphous solid.R f 0.92 (hexanes); 1H NMR (600 MHz, CDCl3) δ 1.72 (m, 2H), 1.70–1.63 (m, 6H), 1.52–1.44 (m, 10H), 1.36 (dd, J = 12.1, 5.0 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 35.4, 33.6, 29.1, 28.6, 27.3, 24.8, 22.8; IR νmax 2921, 2908, 2871, 2860; HRMS (EI) m/z: [M]+ calcd for C14H20+ 188.1560; found: 188.1549.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (SFB 749) for financial support. We thank Prof. Benjamin List and Lars Winkel (MPI für Kohlenforschung, Mülheim) for assistance with high-pressure ethylene Diels–Alder reactions.Notes and references
- M. Tichý, Tetrahedron Lett., 1972, 13, 2001 CrossRef.
- (a) K. Adachi, K. Naemura and M. Nakazaki, Tetrahedron Lett., 1968, 9, 5467 CrossRef; (b) M. Nakazaki, H. Chikamatsu and M. Taniguchi, Chem. Lett., 1982, 1761 CrossRef CAS; (c) M. Tichý and J. Sicher, Tetrahedron Lett., 1969, 10, 4609 CrossRef.
- (a) J. Gauthier and P. Deslongchamps, Can. J. Chem., 1967, 45, 297 CrossRef CAS; (b) A. Bélanger, J. Poupart and P. Deslongchamps, Tetrahedron Lett., 1968, 9, 2127 CrossRef; (c) E. Osawa, P. v. R. Schleyer, L. W. K. Chang and V. V. Kane, Tetrahedron Lett., 1974, 15, 4189 CrossRef; (d) D. P. G. Hamon and R. N. Young, Aust. J. Chem., 1976, 29, 145 CAS.
- (a) H. W. Whitlock, J. Am. Chem. Soc., 1962, 84, 3412 CrossRef CAS; (b) H. W. Whitlock and M. W. Siefken, J. Am. Chem. Soc., 1968, 90, 4929 CrossRef CAS.
- (a) H.-G. Capraro and C. Ganter, Helv. Chim. Acta, 1980, 63, 1347 CrossRef CAS; (b) H. Greuter and H. Schmid, Helv. Chim. Acta, 1972, 55, 2382 CrossRef CAS; (c) A. Bélanger, Y. Lambert and P. Deslongchamps, Can. J. Chem., 1969, 47, 795 CrossRef.
- M. Olbrich, H. Quanz, S. Barua, P. R. Schreiner, D. Trauner and W. D. Allen, Chem.–Eur. J., 2013 DOI:10.1002/chem.201303081.
- The Gaussian 03 reference is included in the ESI.†.
- (a) K.-I. Hirao, T. Iwakuma, M. Taniguchi, E. Abe, O. Yonemitsu, T. Date and K. Kotera, J. Chem. Soc., Chem. Commun., 1974, 22, 691 RSC; (b) K. Hirao, T. Iwakuma, M. Taniguchi, O. Yonemitsu, T. Date and K. Kotera, J. Chem. Soc., Perkin Trans. 1, 1980, 163 RSC; (c) M. Nakazaki, K. Naemura, Y. Kondo, S. Nakahara and M. Hashimoto, J. Org. Chem., 1980, 45, 4440 CrossRef CAS.
- (a) E. LeGoff and S. Oka, J. Am. Chem. Soc., 1969, 91, 5665 CrossRef CAS; (b) E. Osawa, K. Aigami and Y. Inamoto, Tetrahedron, 1978, 34, 509 CrossRef CAS; (c) C.-T. Lin, N.-J. Wang, Y.-L. Yeh and T.-C. Chou, Tetrahedron, 1995, 51, 2907 CrossRef CAS; (d) C.-T. Lin, H.-C. Hsu and T.-C. Chou, J. Org. Chem., 1999, 64, 7260 CrossRef CAS.
- C.-T. Lin, N.-J. Wang, H.-Z. Tseng and T.-C. Chou, J. Org. Chem., 1997, 62, 4857 CrossRef CAS.
- W. Grimme, J. Wortmann, J. Frowein, J. Lex, G. Chen and R. Gleiter, J. Chem. Soc., Perkin Trans. 2, 1998, 1893 RSC.
- D. A. Lightner, J. K. Gawronski and T. D. Bouman, J. Am. Chem. Soc., 1980, 102, 5749 CrossRef CAS.
- S. B. Soloway, A. M. Damiana, J. W. Sims, H. Bluestone and R. E. Lidov, J. Am. Chem. Soc., 1960, 82, 5377 CrossRef CAS.
- C. Chatgilialoglu, D. Griller and M. Lesage, J. Org. Chem., 1988, 53, 3641 CrossRef CAS.
- J. Zhang, Z. Zhu, Y. Feng, H. Ishiwata, Y. Miyata, R. Kitaura, J. E. P. Dahl, R. M. K. Carlson, N. A. Fokina, P. R. Schreiner, D. Tománek and H. Shinohara, Angew. Chem., Int. Ed., 2013, 52, 3717 CrossRef CAS PubMed.
- X. Zhao, Y. Liu, S. Inoue, T. Suzuki, R. O. Jones and Y. Ando, Phys. Rev. Lett., 2004, 92, 125502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 948941–948947, 949955 and 949956. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob42152j |
| This journal is © The Royal Society of Chemistry 2014 |