Diverse modifications of the 4-methylphenyl moiety of TAK-779 by late-stage Suzuki–Miyaura cross-coupling†
Anna
Junker
a,
Dirk
Schepmann
a,
Junichiro
Yamaguchi
b,
Kenichiro
Itami
bc,
Andreas
Faust
d,
Klaus
Kopka
e,
Stefan
Wagner
f and
Bernhard
Wünsch
*a
aInstitut für Pharmazeutische und Medizinische Chemie der Universität Münster, Corrensstr. 48, D-48149 Münster, Germany. E-mail: wuensch@uni-muenster.de; Fax: +49-251-8332144; Tel: +49-251-8333311
bDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa-ku, Nagoya, 464-8602, Japan
cInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: itami.kenichiro@a.mbox.nagoya-u.ac.jp; Tel: +81-52-788-6098
dEuropean Institute for Molecular Imaging (EIMI), Mendelstr. 11, D-48149 Münster, Germany
eRadiopharmaceutical Chemistry, German Cancer Research Center (dkfz), Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany. E-mail: k.kopka@dkfz.de; Tel: +49 6221 42 2432
fKlinik für Nuklearmedizin, Albert-Schweitzer-Campus 1, Gebäude A1, D-48149 Münster, Germany
First published on 28th October 2013
Abstract
Chemokine receptor 5 (CCR5) antagonists provide a new therapeutic approach in the treatment of HIV-1 (AIDS). TAK-779 displays high affinity and selectivity for the CCR5 receptor and serves as a lead compound for the development of further antagonists. In order to increase the oral bioavailability replacement of the quaternary ammonium structure by a tertiary amine and modification of the 4-methylphenyl moiety were envisaged. Herein, a new synthetic strategy for the development of TAK-779 analogs by late stage diversification is reported. The Suzuki–Miyaura cross-coupling reactions allowed various modifications of the central amide building block 3 at the end of the synthesis leading to compounds 2f and 2h with a promising CCR5 binding affinity.
1. Introduction
The chemokine receptor 5 (CCR5) is a 7-transmembrane G-protein coupled receptor, which plays a crucial role as the second co-receptor for HIV-1 entry into target cells.1,2 CCR5 antagonists represent a novel class of drugs for the treatment of infections with HIV-1 (AIDS).3,4 Additionally the participation of the CCR5 receptor in the development and progression of certain inflammatory processes, e.g., atherosclerosis,5 rheumatoid arthritis6–8 and asthma,9,10 was observed. TAK-779 (1) developed by Takeda Inc. is among the most potent and selective CCR5 receptor antagonists reported so far and can serve as a lead compound for the development of novel antagonists (Fig. 1).11–13 Shiraishi and coworkers explored different structural modifications of the lead compound; however, variations of the 4-methylphenyl moiety were not performed due to the introduction of the 4-methylphenyl group at a very early stage of the synthesis, i.e. by arylation of benzo[7]annulen-5-one 6.13 TAK-779 (1) was not designed as a clinical candidate because of its poor oral bioavailability resulting from the quaternary ammonium structure (Fig. 1).14 In order to develop TAK-779 analogs with increased oral bioavailability, the quaternary ammonium structure should be replaced by a tertiary amine. With the intention of increasing the CCR5 binding affinity the 4-methylphenyl moiety of the lead compound should be modified by the introduction of different substituents at the phenyl moiety, which are not accessible by the reported synthetic strategy. In this research program, we are highly interested in the development of TAK-779 analogs 2 with diverse and polar functional groups at the phenyl moiety. Compounds 2 with various aryl residues should be synthesized by a late-stage diversification strategy, i.e., introduction of substituted aryl residues at the end of the synthesis. | ||
| Fig. 1 TAK-779, 1. | ||
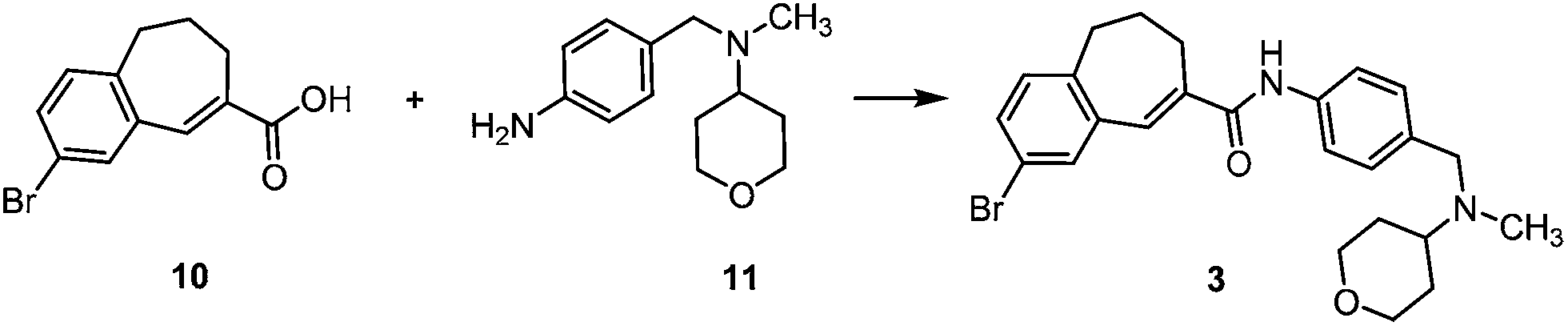
Therefore, a general synthetic method, which allows the introduction of substituted aryl moieties, was required. The palladium-catalyzed Suzuki–Miyaura cross-coupling reaction is one of the most powerful and versatile methods for the formation of C–C bonds, which allows coupling of diverse aryl halides with different arylboronic acids and high functional group tolerance.15,16 Amide 3 was chosen as the central building block for the synthesis of TAK-779 analogs 2, because it allows the introduction of diverse aryl substituents at the final step (Fig. 2).
 | ||
| Fig. 2 Strategy for the synthesis of TAK-779 analogs 2. | ||
2. Results and discussion
Synthesis
The synthesis of the building block 3 started with a Friedel–Crafts acylation of bromobenzene with glutaric acid monomethyl ester chloride, and a subsequent Wolff–Kishner reduction of the resulting δ-keto acid 4 yielded acid 5. An intramolecular Friedel–Crafts acylation of 5 afforded cyclic ketone 6 after a careful optimization (Scheme 1). | ||
| Scheme 1 Synthesis of brominated carboxylic acid 10. Reagents and reaction conditions: (a) H3OCO(CH2)3COCl, AlCl3 (2.2 eq.), rt, overnight, then MeOH, 5 M NaOH, reflux, 3 h, 84%; (b) hydrazine monohydrate (5.5 eq.), KOH (4.4 eq.), triethylene glycol, 200 °C, overnight, 87%; (c) PPA, 90 °C, 1 h, 60%; (d) NaH (10 eq.), dimethyl carbonate, 80 °C, 1 h, 80%; (e) NaBH4 (1.3 eq.), MeOH, CH2Cl2, −10 °C, 1 h, 75%; (f) MeSO2Cl (1.5 eq.), Et3N (3.0 eq.), CH2Cl2, rt, overnight, then DBU (4.0 eq.), CH2Cl2, rt, 1 h, 83%; (g) MeOH, 5 M NaOH, 94%. | ||
The ring closure of arylpentanoic acid 5 was performed in polyphosphoric acid (PPA) as a Brønsted acid at 100 °C. However, in contrast to the study of Shiraishi and coworkers who reported a yield of 67%13 of ketone 6, only 35% yield of ketone 6 resulted following the given reaction conditions. The yield even dropped to 25% after 23 h at 100 °C. Due to the early step of the planned synthesis route, an optimization of the intramolecular Friedel–Crafts acylation of 5 was required. Acid 5 was stirred with trifluoroacetic anhydride (TFAA) for 1 h and subsequently Lewis acids such as AlCl3 or SnCl4 were added at −30 °C. However, only with the weaker Lewis acid SnCl4 the formation of a small amount of the desired product was observed. An elevation of the reaction temperature from −30 °C to +45 °C resulted in a total yield of 40%, which could not be further improved. Eaton's reagent (P4O10 in MeSO3H) and Nafion®, a perfluorinated sulfonic acid resin, did not improve the reaction. The acid 5 was also transformed into an acid chloride by treatment with cyanuric chloride or oxalyl chloride and DMF and an intramolecular acylation was initiated by addition of AlCl3. This strategy led to 25% yield of benzosuberone 6. Neither an increase of the reaction temperature nor extension of the reaction time did further increase the yield. Since all attempts to improve the yield by stronger conditions failed, a PPA promoted cyclization under milder conditions was performed: the temperature during the conversion was kept at 80–95 °C, the amount of PPA was 300 g per 0.1 mol (0.006 M) and the reaction was stopped after 30 min. This protocol led to the reproducible formation of benzosuberone 6 in 60% yield, even on a large scale (up to 20 g).
Claisen ester condensation of ketone 6 with dimethyl carbonate and NaH led to the formation of the β-keto ester 7 in 80% yield. Reduction of 7 was performed with NaBH4 in MeOH at −20 °C to −10 °C13 which afforded β-hydroxy ester 8 in 75% yield. Elimination of H2O from secondary alcohol 8 by reaction with methanesulfonyl chloride and subsequent treatment with DBU gave α,β-unsaturated ester 9 in 83% yield, which was hydrolyzed to give acid 10. Synthesis of the central building block 3 required a coupling of acid 10 with the aniline derivative 11, which was synthesized by the reported reductive amination of p-nitrobenzylamine-HCl with tetrahydropyran-4-one, formaldehyde and NaBH(OAc)3 and subsequent reduction of the aromatic nitro group with Zn0 in concentrated HCl.13
In order to optimize the amide formation various coupling reagents were tested (Table 1).17,18 DCC and EDC normally promoting coupling reactions afforded only low yields (entries 1–3) and required high reaction temperatures and long reaction times (entry 4). HATU and COMU coupling reagents gave high yields (entries 5 and 6) under mild conditions and therefore they were employed for the synthesis of amide 3.
|
|
||||
|---|---|---|---|---|
| Entry | Coupling reagent | Temp. | Time | Yield [%] of 3 |
| 1 | DCC, DMAP | rt | 10 h | 10 |
| 2 | EDC, HOBt, NMM | 0 °C | 15 h | 33 |
| 3 | EDC, HOBt, NMM | Reflux | 6 h | 42 |
| 4 | EDC, HOBt, NMM | Reflux | 3 d | 58 |
| 5 | HATU, Et3N | rt | 12 h | 72 |
| 6 | COMU, NMM | rt | 12 h | 77 |
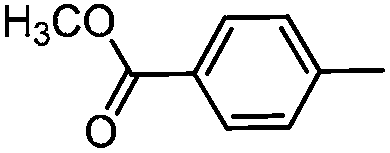
The Suzuki–Miyaura cross-coupling reaction of aryl bromide 3 was carefully investigated using electron rich 4-methylphenylboronic acid (Table 2). Initially the coupling reaction was performed with 5 mol% of Pd(PPh3)4 and 2 M aqueous K2CO3 solution (5 eq.) in an ethanol–toluene mixture (Table 2, entry 1). Biaryl product 2a was isolated in 19% yield together with a large amount of starting material 3 (70%). In order to increase the conversion a stronger base (KOH) in THF was used. These conditions provided a yield of 60% (entry 2). However, despite prolongation of the reaction time the conversion was incomplete (entry 3). The use of DME as a solvent and solid K2CO3, known as Gronowitz conditions,19–21 and the change of the catalyst to PdCl2(dppf), with the 1,1′-bis(diphenylphosphino)ferrocene (dppf) ligand, one of the most efficient stabilizing agents for transition metal-catalyzed cross-coupling reactions, led finally to complete transformation of bromo derivative 3 to biaryl compound 2a (entry 4). The use of various bases such as K2CO3, KOAc and NaOCH3 led to comparable yields (entries 4–6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (5 mol%) | Base | Solvent | Time, temp. | Yield [%] of 2a |
| DME = dimethoxyethane. | |||||
| 1 | Pd(PPh3)4 | 2 M K2CO3 (5 eq.) | Ethanol–toluene = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 :3 |
12 h, 115 °C | 19 |
| 2 | Pd(PPh3)4 | KOH (10 eq.) | THF | 12 h, 70 °C | 60 |
| 3 | Pd(PPh3)4 | KOH (5 eq.) | THF | 20 h, 70 °C | 73 |
| 4 | PdCl2(dppf) | K2CO3 (3 eq.) | DME | 12 h, reflux | 90 |
| 5 | PdCl2(dppf) | KOAc (2 eq.) | DME | 12 h, reflux | 82 |
| 6 | PdCl2(dppf) | NaOCH3 (2 eq.) | DME | 12 h, reflux | 88 |

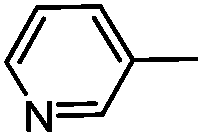
Electron rich (e.g.2a, 2b), electron deficient (e.g.2c, 2d) as well as heterocyclic boronic acids (e.g.2g, 2h) were employed for the arylation of 3 in order to generate a broad variety of test compounds. Moreover, these optimized conditions allowed the synthesis of compounds with different functional groups (e.g. phenol 2b, aldehyde 2c and ester 2d), which are not accessible by other reported methods (see Table 3).







CCR5 receptor binding affinity
The influence of structural modifications of the lead compound TAK-779 on the CCR5 receptor binding affinity was investigated. For this purpose the affinity of compounds 2a–h to the CCR5 receptor was examined by competition receptor binding studies using [3H]TAK-779 as the radioligand (Table 4). In the in vitro assay an IC50-value of 8.8 nM was recorded for the unlabeled TAK-779. Replacement of the quaternary ammonium structure by a tertiary amine 2a (IC50 = 67 nM) resulted in an 8-fold decreased CCR5 affinity. Introduction of polar functional groups at the 4-position of the aryl substituent, e.g. 4-hydroxy (2b), 4-formyl (2c) and 4-acetylamino (2e), was not tolerated by the CCR5 receptor. However, the weakly basic dimethylamino derivative 2f shows a moderate CCR5 receptor affinity (IC50 = 138 nM). The bioisosteric replacement of the 4-methylphenyl moiety of 2a by the 5-methylthienyl moiety (2h) was accepted by the CCR5 receptor, but the affinity was lower (IC50 = 167 nM). The introduction of a 3-pyridinyl group of 2g as an aryl moiety was not tolerated and led to a loss of affinity (IC50 = 5900 nM).3. Conclusion
In order to develop new TAK-779 analogs with increased oral bioavailability the quaternary ammonium structure was replaced by the corresponding tertiary amine and the 4-methylphenyl moiety of the TAK-779 lead compound was modified. Suzuki–Miyaura cross-coupling of the key building block 3 with various arylboronic acids allowed the synthesis of a diverse set of test compounds, which were not accessible using previously reported synthetic strategies. Introduction of polar substituents turned out to be detrimental in terms of CCR5 receptor affinity. However the promising CCR5 affinity of the thiophene bioisostere 2h and of the slightly basic dimethylamine derivative 2f stimulated us to follow this approach in more detail.4. Experimental
Chemistry general
Unless otherwise noted, moisture-sensitive reactions were conducted under dry nitrogen. THF and 1,4-dioxane were dried with sodium/benzophenone and were freshly distilled before use. Flash column chromatography (fc): silica gel 60, 40–64 μm; parentheses include: diameter of the column, length of the column, fraction size, eluent, Rf value. Melting point: melting point apparatus Stuart Scientific® SMP 3, uncorrected. IR: IR spectrophotometer FT-ATR-IR (Jasco®). 1H NMR (400 MHz): Unity Mercury Plus 400 spectrometer (Varian®), AV400 (Bruker®), JEOL JNM-ECA-400. 13C NMR (100 MHz): Unity Mercury plus 400 spectrometer (Varian®) JEOL JNM-ECA-400; δ in ppm relative to tetramethylsilane; coupling constants are given with 0.5 Hz resolution, the assignments of 13C and 1H NMR signals were supported by 2D NMR techniques; MS: APCI = atmospheric pressure chemical ionization, EI = electron impact, ESI = electro-spray ionization: MicroTof (Bruker Daltronics, Bremen), calibration with sodium formate clusters before measurement. The HPLC method for the determination of the product purity: Merck Hitachi Equipment; UV detector: L-7400; autosampler: L-7200; pump: L-7100; degasser: L-7614; Method: column: LiChrospher® 60 RP-select B (5 μm), 250 × 4 mm2 column; flow rate: 1.00 mL min−1; injection volume: 5.0 μL; detection at λ = 210 nm; solvents: A: water with 0.05% (v/v) trifluoroacetic acid; B: acetonitrile with 0.05% (v/v) trifluoroacetic acid: gradient elution: (A%): 0–4 min: 90%, 4–29 min: gradient from 90% to 0%, 29–31 min: 0%, 31–31.5 min: gradient from 0% to 90%, 31.5–40 min: 90%.5. Synthetic procedures
5-(4-Bromophenyl)-5-oxo-pentanoic acid (4)13
Anhydrous AlCl3 (48 g, 0.36 mol, 2.2 eq.) was added portionwise to vigorously stirred bromobenzene (200 mL) at 0 °C. After stirring for 30 min, methyl 5-chloro-5-oxopentanoate (27 g, 0.16 mol) was added over a period of 30 min followed by stirring for 1 h at 0 °C and overnight at rt. Then crushed ice (200 g) and conc. HCl (150 mL) were added and the mixture was warmed until the suspended material was dissolved. The layers were separated; the aqueous layer was extracted with EtOAc (4 × 200 mL). The combined organic layers were washed thoroughly with water (400 mL) and brine (300 mL), dried (Na2SO4) and concentrated in vacuo to give an orange oil. The oil was dissolved in a mixture of MeOH (200 mL) and 2 M NaOH (200 mL), and the mixture was refluxed for 6 h. The mixture was concentrated in vacuo, the residue was acidified with conc. HCl and extracted with CH2Cl2 (4 × 200 mL). The combined organic layers were washed with water (300 mL) and brine (300 mL), dried (Na2SO4) and evaporated in vacuo to give the crude product as a slightly yellow solid, which was finally recrystallized from an EtOAc–cyclohexane mixture to give 5-(4-bromophenyl)-5-oxo-pentanoic acid as a colorless solid. Rf = 0.24, (MeOH–CH2Cl2 = 5:95), mp 128–129 °C, yield 38 g (84%). C11H1179BrO3 (271.1 g mol−1). Purity (HPLC): 99%, tR = 17.45 min. Exact MS (APCI): m/z = calcd for C11H1279BrO3 [MH+] 270.9964, found 270.9959. 1H NMR (CDCl3): δ (ppm) = 2.07 (quint, J = 7.1 Hz, 2H, 3-CH2), 2.50 (t, J = 7.1 Hz, 2H, 2-CH2), 3.04 (t, J = 7.1 Hz, 2H, 4-CH2), 7.60 (d, J = 8.6 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.82 (d, J = 8.6 Hz, 2H, 2-Hphenyl, 6-Hphenyl). FT-IR (neat): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2938 (C–Halkyl), 1705 (C
(cm−1) = 2938 (C–Halkyl), 1705 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
5-(4-Bromophenyl)pentanoic acid (5)13
A solution of 4 (69.3 g, 0.25 mol), KOH (63 g, 1.12 mol, 4.4 eq.) and hydrazine monohydrate (49 g, 1.4 mol, 5.5 eq.) in triethylene glycol was heated to reflux (200 °C) in a Dean–Stark apparatus for 12 h. The mixture was poured into cold water and washed with Et2O. The aqueous layer was acidified with 6 M HCl and extracted with CH2Cl2 (4 × 200 mL). The combined organic layers were washed with water (2 × 200 mL) and brine (300 mL), dried (Na2SO4), and concentrated in vacuo to give the crude product as a yellow solid, which was recrystallized from EtOAc to give the acid 2 as a colorless solid. Rf = 0.37 (MeOH–CH2Cl2 = 5:95), mp 91–93 °C, yield 57 g (87%). C11H1379BrO2 (257.1 g mol−1). Purity (HPLC): 97%, tR = 19.37 min. Exact MS (APCI): m/z = calcd for C11H1479BrO2 [MH+] 257.0172, found 257.0165. 1H NMR (CDCl3): δ (ppm) = 1.61–1.72 (m, 4H, 3-CH2, 4-CH2), 2.38 (t, J = 7.0 Hz, 2H, 2-CH2), 2.58 (t, J = 7.0 Hz, 2H, 5-CH2), 7.03 (d, J = 8.4 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.40 (d, J = 8.4 Hz, 2H, 3-Hphenyl, 5-Hphenyl). FT-IR (neat): (cm−1) = 2917 (C–Halkyl), 2360 (COOH), 1698 (CO).
3-Bromo-6,7,8,9-tetrahydrobenzo[7]annulen-5-one (6)13
Acid 5 (20 g, 77.8 mmol) was added portionwise to polyphosphoric acid (300 g) at 90 °C. The mixture was stirred for 1 h during which it turned brown. Next it was poured into ice (350 g) and the mixture was warmed until the suspended material was dissolved. The mixture was extracted with CH2Cl2 (4 × 200 mL). The combined organic layers were washed with water (300 mL) and brine (300 mL), dried (Na2SO4) and concentrated in vacuo to give the crude product as a dark brown oil, which was distilled to give the ketone 6 as a slightly yellow oil. Rf = 0.16 (EtOAc–cyclohexane = 1:9), 185 °C (1.8 × 10−2 mbar), yield 11 g (60%). C11H1179BrO (239.1 g mol−1). Purity (HPLC): 98%, tR = 20.68 min. Exact MS (APCI): m/z = calcd for C11H1279BrO [MH+] 239.0066, found 239.0043. 1H NMR (CDCl3): δ (ppm) = 1.75–1.98 (m, 4H, 7-CH2, 8-CH2), 2.72 (t, J = 6.0 Hz, 2H, 6-CH2), 2.88 (t, J = 6.1 Hz, 2H, 9-CH2), 7.08 (d, J = 8.1 Hz, 1H, 1-CH), 7.52 (dd, J = 8.1/2.2 Hz, 1H, 2-CH), 7.83 (d, J = 2.2 Hz, 1H, 4-CH). FT-IR (neat): (cm−1) = 2938 (C–H), 1675 (CO).
Methyl 2-bromo-6,7-dihydro-5H-benzo[7]annulene-8-carboxylate (9)13
The suspension of NaH in mineral oil was washed three times with petroleum ether and filtered. NaH (3.3 g, 140 mmol, 10 eq.) was suspended in dimethyl carbonate (50 mL), and the mixture was heated to 80 °C for 30 min. Then a solution of 6 (3.34 g, 14 mmol) in dimethyl carbonate (50 mL) was added dropwise to the suspension. The mixture was refluxed for 3 h under a N2 atmosphere. Next the reaction mixture was diluted with MeOH (100 mL) and poured into 1 M HCl (150 mL) under ice cooling. The aqueous mixture was concentrated in vacuo and extracted with EtOAc (2 × 200 mL). The combined organic layers were washed successively with water (150 mL) and brine (150 mL), dried (Na2SO4) and evaporated in vacuo to give an orange oil which was purified by fc (petroleum ether–EtOAc = 8:2, ∅ = 6 cm, l = 10 cm, V = 65 mL) to give the β-keto ester 7 in a yield of 3.33 g (80%) as light yellow prism-like crystals.
A solution of the β-keto ester 7 (3.33 g, 11.2 mmol) in CH2Cl2 (50 mL) was cooled down to −20 °C, and then NaBH4 (560 mg, 14.5 mmol, 1.3 eq.) and abs. MeOH (2 mL) were added. The mixture was stirred at −10 °C for 1 h. The reaction mixture was poured into water and acidified with 1 M HCl. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed successively with water (100 mL) and brine (100 mL), dried (Na2SO4) and evaporated in vacuo to give the crude product as a yellow oil, which was purified by fc (cyclohexane–EtOAc = 4:1 → 2:1, ∅ = 4 cm, l = 14 cm, V = 65 mL) to give the β-hydroxy ester 8 in a yield of 2.48 g (75%) as a yellow oil.
Methanesulfonyl chloride (1.43 g, 12.5 mmol, 1.5 eq.) was added to a solution of β-hydroxy ester 8 (2.48 g, 8.3 mmol) and triethylamine (2.52 g, 25 mmol, 3 eq.) in CH2Cl2 (30 mL) under ice cooling. The reaction mixture was stirred overnight at rt. Then DBU (5.05 g, 33.2 mmol, 4 eq.) was added dropwise under ice cooling. The reaction mixture was stirred for 1 h at rt and then poured into water, 1 M HCl was added (pH 3) and the mixture was extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with water (100 mL) and brine (100 mL), dried (Na2SO4) and evaporated in vacuo to give the crude product, which was purified by fc (petroleum ether–EtOAc = 9:1, ∅ = 4 cm, l = 12 cm, V = 65 mL) to give 9 as a colorless solid. Rf = 0.90 (EtOAc–cyclohexane = 9:1), yield 1.94 g (83%). C13H1379BrO2 (281.1 g mol−1). Purity (HPLC): 98%, tR = 22.77 min. Exact MS (APCI): m/z = calcd for C13H1479BrO2 [MH+] 281.0172, found 281.0181. 1H NMR (CDCl3): δ (ppm) = 1.98–2.08 (m, 2H, 6-CH2), 2.78 (t, J = 6.0 Hz, 2H, 7-CH2), 2.71–2.79 (m, 2H, 5-CH2), 3.81 (s, 3H, CO2CH3), 7.02 (d, J = 8.1 Hz, 1H, 4-CH), 7.32 (dd, J = 8.1/2.1 Hz, 1H, 3-CH), 7.44 (d, J = 2.1 Hz, 1H, 1-CH), 7.61 (s, 1H, 9-CH). FT-IR (neat): (cm−1) = 2970, 2924 (C–Halkyl), 1705 (CO).
2-Bromo-6,7-dihydro-5H-benzo[7]annulene-8-carboxylic acid (10)
In a round bottom flask the ester 9 (1.45 g, 5 mmol) was dissolved in MeOH (50 mL), and 5 M NaOH (50 mL) was added. The mixture was heated to reflux for 3 h. After cooling down to rt, the mixture was concentrated in vacuo and acidified with conc. HCl to give a precipitate. The mixture was cooled in an ice bath to complete the precipitation and the solid was filtered off, washed with a 1 M HCl solution and water and dried overnight to give the acid 10 as a colorless solid. Rf = 0.70 (EtOAc–MeOH = 95:5), mp 211–213 °C (dec.), yield 1.25 g (94%). C12H1179BrO2 (267.1 g mol−1). Purity (HPLC): 99%, tR = 20.09 min. Exact MS (APCI): m/z = calcd for C12H1279BrO2 [MH+] 267.0015, found 267.0022. 1H NMR (CDCl3): δ (ppm) = 2.02–2.10 (m, 2H, 6-CH2), 2.65 (t, J = 6.0 Hz, 2H, 7-CH2), 2.76–2.80 (m, 2H, 5-CH2), 7.04 (d, J = 8.1 Hz, 1H, 4-CH), 7.35 (dd, J = 8.1/2.1 Hz, 1H, 3-CH), 7.47 (d, J = 2.0 Hz, 1H, 1-CH), 7.72 (s, 1H, 9-CH). FT-IR (neat): (cm−1) = 2970, 2866 (C–Halkyl), 2534 (COOH), 1662 (CO).
2-Bromo-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (3)
The amine 11 (815 mg, 3.7 mmol, 1 eq.) was added to a mixture of acid 10 (1.0 g, 3.7 mmol), N-methylmorpholine (750 mg, 7.4 mmol, 2 eq.) and COMU® (1.75 g, 4.1 mmol, 1.1 eq.) in acetonitrile (10 mL). The reaction mixture was stirred at rt overnight. Next, water (20 mL) was added and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried (Na2SO4) and concentrated in vacuo to give the crude product as an orange oil, which was purified by fc (EtOAc–MeOH = 95:5, ∅ = 4 cm, l = 14 cm, V = 30 mL) to give the product as a faint yellow oil. Addition of acetonitrile induced crystallization of the oil, which was then recrystallized from acetonitrile to give 3 as a colorless solid. Rf = 0.11 (EtOAc–MeOH = 95:5), mp 147 °C, yield 1.33 g (77%). C25H2979BrN2O2 (469.4 g mol−1). Purity (HPLC): 97%, tR = 19.34 min. Exact MS (APCI): m/z = calcd for C25H3079BrN2O2 [MH+] 469.1485, found 469.1495. 1H NMR (CDCl3): δ (ppm) = 1.60–1.83 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.06–2.18 (m, 2H, 6-CH2), 2.23 (s, 3H, N-CH3), 2.63–2.72 (m, 3H, 7-CH2, 4-Hpyran), 2.72–2.82 (m, 2H, 5-CH2), 3.37 (td, J = 11.5/2.4 Hz, 2H, CH2axial–O–CH2axial), 3.60 (s, 2H, Ph–CH2–N), 4.04 (dd, J = 10.7/3.7 Hz, 2H, CH2equat.–O–CH2equat.), 7.04 (d, J = 5.1 Hz, 1H, 4-CH), 7.23 (s, 1H, 9-CH), 7.33 (m, 3H, 3-CH, 3-Hphenyl, 5-Hphenyl), 7.43 (d, J = 2.0 Hz, 1H, 1-CH), 7.55 (d, J = 8.4 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.60 (s, 1H, N-H). 13C NMR (CDCl3): δ (ppm) = 28.1 (C-6), 29.3 (C-3pyran, C-5pyran), 30.4 (C-7), 34.4 (C-5), 37.6 (N–CH3), 57.4(Ph–CH2–N), 59.7 (C-4pyran), 67.8 (C-2pyran, C-6pyran), 119.8 (C-2), 120.2 (C-2phenyl, C-6phenyl), 129.5 (C-3phenyl, C-5phenyl), 131.1 (C-3), 131.2 (C-4), 132.7 (C-9), 134.4 (C-1), 136.2 (C-4a), 136.6 (C-4phenyl), 136.8 (C-1phenyl), 139.5 (C-9a), 141.4 (C-8), 167.8 (OC–NH). FT-IR (neat): (cm−1) = 3313 (N–H), 2966, 2920 (C–Halkyl), 1647 (CO), 1512 (CC).
2-(4-Methyphenyl)-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2a)
A 50 mL Schlenk flask was equipped with a Dimroth condenser and a magnetic stirring bar and then closed. The flask was flame-dried under vacuum and filled with N2. Under a permanent flow of N2, amide 3 (200 mg, 0.42 mmol), PdCl2(dppf) (20 mg, 0.02 mmol, 5 mol%), K2CO3 (174 mg, 1.26 mmol, 3 eq.) and 4-methylphenylboronic acid (86 mg, 0.63 mmol, 1.5 eq.) were suspended in dry dimethoxyethane (15 mL). The flask was sealed and heated to reflux for 12 h. After cooling to rt, the mixture was filtered through a short silica pad (EtOAc). The filtrate was concentrated in vacuo to give the crude product as a brown oil, which was purified by fc (EtOAc–MeOH = 95:5, ∅ = 3 cm, l = 8 cm, V = 30 mL) to give the purified product as a colorless oil. Recrystallization from acetonitrile afforded the amide 2a as a colorless solid. Rf = 0.28 (CH2Cl2–MeOH = 95:5), mp 160–162 °C, yield 74 mg (90%). C32H36N2O2 (480.6 g mol−1). Purity (HPLC): 99%, tR = 21.31 min. Exact MS (APCI): m/z = calcd for C32H37N2O2 [MH+] 481.2850, found 481.2835. 1H NMR (CDCl3): δ (ppm) = 1.52–1.79 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.03–2.11 (m, 2H, 6-CH2), 2.15 (s, 3H, N-CH3), 2.33 (s, 3H, CH3tolyl), 2.51–2.68 (m, 3H, 4-Hpyran, 7-CH2), 2.74–2.89 (m, 2H, 5-CH2), 3.30 (td, J = 11.5/2.5 Hz, 2H, CH2axial–O–CH2axial), 3.52 (s, 2H, N–CH2), 3.89–4.02 (m, 2H, CH2equat.–O–CH2equat.), 7.13–7.18 (m, 3H, 4-CH, 3-Htolyl, 5-Htolyl), 7.25 (d, J = 8.4 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.31–7.39 (m, 2H, 3-CH, 9-CH), 7.41 (d, J = 8.1 Hz, 2H, 2-Htolyl, 6-Htolyl), 7.44 (d, J = 1.9 Hz, 1H, 1-CH), 7.50 (d, J = 8.5 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.65 (s, 1H, N–H). 13C NMR (CDCl3): δ (ppm) = 21.6 (CH3tolyl), 28.4 (C-6), 29.6 (C-3pyran, C-5pyran), 30.9 (C-7), 35.1 (C-5), 38.0 (N–CH3), 57.8 (Ph–CH2), 60.0 (C-4pyran), 68.2 (C-2pyran, C-6pyran), 120.5 (C-2phenyl, C-6phenyl), 127.2 (C-2tolyl, C-6tolyl), 127.3 (C-3), 129.8 (C-3tolyl, C-5tolyl), 130.0 (C-3phenyl, C-5phenyl), 130.3 (C-4), 130.3 (C-4tolyl), 131.1 (C-1), 134.7 (C-9), 135.1 (C-4a), 137.3 (C-4phenyl), 137.6 (C-1tolyl), 138.0 (C-2), 138.4 (C-1phenyl), 139.6 (C-9a), 141.9 (C-8), 168.5 (OC–NH). FT-IR (neat): (cm−1) = 3298 (N–H), 2970, 2920 (C–Halkyl), 1644 (CO), 1095, 1014 (C–O).
2-(4-Hydroxyphenyl)-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2b)
As described for 2a, amide 3 (938 mg, 2 mmol), PdCl2(dppf) (75 mg, 0.1 mmol, 5 mol%), KOAc (400 mg, 0.4 mmol, 2 eq.) and 4-hydroxyphenylboronic acid (305 mg, 2.2 mmol, 1.1 eq.) in dry dimethoxyethane (20 mL) were heated to reflux for 12 h. After cooling to rt, the mixture was filtered through a short silica pad (EtOAc). Purification by fc (EtOAc–MeOH = 95:5, ∅ = 5 cm, l = 10 cm, V = 30 mL) afforded a colorless oil, which was recrystallized from acetonitrile to give the amide 2b as a colorless solid. Rf = 0.15 (CH2Cl2–MeOH = 95:5), mp 248 °C (dec.), yield 782 mg (81%). C31H34N2O3 (482.6 g mol−1). Purity (HPLC): 99%, tR = 18.57 min. Exact MS (APCI): m/z = calcd for C31H35N2O3 [MH+] 483.2642, found 483.2617. 1H NMR (CDCl3): δ (ppm) = 1.60–1.86 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.02–2.18 (m, 2H, 6-CH2), 2.23 (s, 3H, N-CH3), 2.61–2.77 (m, 3H, 4-CHpyran, 7-CH2), 2.79–2.91 (m, 2H, 5-CH2), 3.37 (td, J = 11.5/1.7 Hz, 2H, CH2axial–O–CH2axial), 3.59 (s, 2H, Ph–CH2–N), 4.04 (dd, J = 11.1/3.8 Hz, 2H, CH2equat.–O–CH2equat.), 6.87 (d, J = 8.6 Hz, 2H, 3-Hhydroxyphen., 5-Hhydroxyphen.), 7.18 (d, J = 7.8 Hz, 1H, 4-CH), 7.32 (d, J = 8.2 Hz, 2H, 3-Hphenyl., 5-Hphenyl.), 7.36 (d, J = 1.9 Hz, 1H, 1-CH), 7.39 (s, 1H, 9-CH), 7.42 (m, 3H, 3-CH, 2-Hhydroxyphen., 6-Hhydroxyphen.), 7.56 (d, J = 8.3 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.72 (s, 1H, N–H). The signal for the OH moiety is not seen in the spectrum. 13C NMR (CDCl3): δ (ppm) = 28.0 (C-6), 29.2 (C-3pyran, C-5pyran), 30.7 (C-7), 34.8 (C-5), 37.6 (N–CH3), 57.5 (Ph–CH2–N), 59.8 (C-4pyran), 67.8 (C-2pyran, C-6pyran), 116.1 (C-3hydroxyphen., C-5hydroxyphen.), 120.4 (C-2phenyl, C-6phenyl), 126.8 (C-3), 128.3 (C-2hydroxyphen., C-6hydroxyphen), 129.9 (C-3phenyl, C-5phenyl), 130.1 (C-4), 130.7 (C-1), 132.9 (C-9a), 134.7 (C-4phenyl), 134.9 (C-9), 135.4 (C-4a), 137.2 (C-2), 137.7 (C-1hydroxyphen.), 139.1 (C-1phenyl), 141.1 (C-8), 155.8 (C-4hydroxyphen.), 168.5 (OC–NH). FT-IR (neat): (cm−1) = 3351 (N–H), 2950 (C–Halkyl), 1647 (CO), 1132 (C–O), 812, 652 (out of plane).
2-(4-Formylphenyl)-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2c)
As described for 2a, amide 3 (100 mg, 0.21 mmol), PdCl2(dppf) (8 mg, 0.01 mmol, 5 mol%), KOAc (14 mg, 0.42 mmol, 2 eq.) and 4-formylphenylboronic acid (36 mg, 0.23 mmol, 1.1 eq.) in dry dimethoxyethane (5 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–MeOH = 95:5, ∅ = 2 cm, l = 7 cm, V = 10 mL) afforded a pale yellow oil, which was recrystallized from acetonitrile to give the amide 2c as a colorless solid. Rf = 0.28 (CH2Cl2–MeOH = 95:5), mp 188–190 °C, yield 72 mg (68%). C32H34N2O3 (494.6 g mol−1). Purity (HPLC): >99%, tR = 19.34 min. Exact MS (APCI): m/z = calcd for C32H35N2O3 [MH+] 495.2642, found 495.2669. 1H NMR (CDCl3): δ (ppm) = 1.56–1.82 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.13–2.22 (m, 2H, 6-CH2), 2.21 (s, 3H, N-CH3), 2.56–2.71 (m, 1H, 4-CHpyran) 2.74 (t, J = 6.6 Hz, 2H, 7-CH2), 2.87–2.97 (m, 2H, 5-CH2), 3.37 (td, J = 11.6/2.3 Hz, 2H, CH2axial–O–CH2axial), 3.57 (s, 2H, Ph–CH2–N), 4.04 (dd, J = 11.5/4.2 Hz, 2H, CH2equat.–O–CH2equat.), 7.29 (d, J = 8.1 Hz, 1H, 4-CH), 7.32 (d, J = 8.6 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.43 (s, 1H, 9-CH), 7.50 (dd, J = 7.8/1.9 Hz, 1H, 3-CH), 7.53–7.59 (m, 3H, 1-CH, 2-Hphenyl, 6-Hphenyl), 7.64 (s, 1H, N–H), 7.75 (d, J = 8.3 Hz, 2H, 2-Hformylphen, 6-Hformylphen.), 7.96 (d, J = 8.3 Hz, 2H, 3-Hformylphen, 5-Hformylphen), 10.06 (s, 1H, CHO). 13C NMR (CDCl3): δ (ppm) = 28.3 (C-6), 29.7 (C-3pyran, C-5pyran), 30.1 (C-7), 35.1 (C-5), 38.0 (N–CH3), 57.7 (Ph–CH2–N), 60.0 (C-4pyran), 68.2 (C-2pyran, C-6pyran), 120.5 (C-2phenyl, C-6phenyl), 127.6 (C-3), 127.9 (C-3formylphen., C-5formylphen), 129.9 (C-3phenyl, C-5phenyl), 130.6 (C-4), 130.8 (C-2formylphen., C-6formylphen), 131.4 (C-1), 134.3 (C-9), 135.5 (C-9a), 135.6 (C-4phenyl), 138.2 (C-2), 138.9 (C-8), 140.7 (C-1phenyl), 143.3 (C-4formylphen), 146.9 (C-1formylphen), 168.2 (OC–NH), 192.3 (CHO). The signal for the carbon atom C-4a is not visible. FT-IR (neat): (cm−1) = 3305 (N–H), 2966, 2947 (C–Halkyl), 1697 (HCO), 1647 (CO), 1099, 1010, (C–O).
Methyl 4-[8-(N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}carbamoyl)-6,7-dihydro-5H-benzo[7]annulen-2-yl]benzoate (2d)
As described for 2a, amide 3 (100 mg, 0.21 mmol), PdCl2(dppf) (8 mg, 0.01 mmol, 5 mol%), KOAc (14 mg, 0.42 mmol, 2 eq.) and 4-methoxycarbonylphenylboronic acid (43 mg, 0.23 mmol, 1.1 eq.) in dry dimethoxyethane (5 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–MeOH = 95:5, ∅ = 2 cm, l = 8 cm, V = 10 mL) afforded a colorless oil, which was recrystallized from acetonitrile to give the amide 2d as a colorless solid. Rf = 0.17 (CH2Cl2–MeOH = 95:5), mp 191 °C, yield 82 mg (73%). C33H36N2O4 (524.6 g mol−1). Purity (HPLC): >97%, tR = 22.19 min. Exact MS (APCI): m/z = calcd for C33H37N2O4 [MH+] 525.2748, found 525.2776. 1H NMR (CDCl3): δ (ppm) = 1.59–1.79 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.08–2.21 (m, 2H, 6-CH2), 2.21 (s, 3H, N–CH3), 2.64 (tt, J = 11.0/3.8 Hz, 1H, 4-CHpyran) 2.73 (t, J = 6.6 Hz, 2H, 7-CH2), 2.85–2.97 (m, 2H, 5-CH2), 3.37 (td, J = 12.0/1.8 Hz, 2H, CH2axial–O–CH2axial), 3.57 (s, 2H, Ph–CH2-N), 3.94 (s, 3H, CO2CH3), 4.04 (dd, J = 11.0/4.0 Hz, 2H, CH2equat.–O–CH2equat.), 7.27 (d, J = 8.9 Hz, 1H, 4-CH), 7.31 (d, J = 7.4 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.42 (s, 1H, 9-CH), 7.49 (dd, J = 7.9/1.6 Hz, 1H, 3-CH), 7.50–7.60 (m, 3H, 1-CH, 2-Hphenyl, 6-Hphenyl), 7.62 (s, 1H, N–H), 7.65 (d, J = 8.5 Hz, 2H, 2-Hmethylbenzoate, 6-Hmethylbenzoate), 8.11 (d, J = 8.4 Hz, 2H, 3-Hmethylbenzoate., 5-Hmethylbenzoate). 13C NMR (CDCl3): δ (ppm) = 28.3 (C-6), 29.6 (C-3pyran, C-5pyran), 30.9 (C-7), 35.1 (C-5), 38.0 (N–CH3), 52.6 (CO2CH3), 57.7 (Ph–CH2–N), 60.0 (C-4pyran), 68.2 (C-2pyran, C-6pyran), 120.5 (C-2phenyl, C-6phenyl), 127.2 (C-3methylbenzoate., C-5methylbenzoate.), 127.6 (C-3), 129.4 (C-1methylbenzoate.), 129.9 (C-3phenyl, C-5phenyl), 130.5 (C-4), 130.6 (C-2methylbenzoate, C-6methylbenzoate), 131.4 (C-1), 134.3 (C-9), 134.4 (C-9a), 135.4 (C-4a), 137.2 (C-4phenyl), 138.5 (C-2), 138.9 (C-8), 142.9 (C-1phenyl), 145.3 (C-4methylbenzoate), 167.4 (CO2CH3), 168.4 (OC–NH). FT-IR (neat): (cm−1) = 3286 (N–H), 2951, 2920 (C–Halkyl), 1701 (COester), 1647 (CO), 1076, 1014, (C–O).
2-[4-(Acetamido)phenyl]-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2e)
As described for 2a, amide 3 (100 mg, 0.21 mmol), PdCl2(dppf) (8 mg, 0.01 mmol, 5 mol%), KOAc (14 mg, 0.42 mmol, 2 eq.) and 4-acetamidophenylboronic acid (43 mg, 0.24 mmol, 1.1 eq.) in dry dimethoxyethane (5 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–MeOH = 95:5, ∅ = 2 cm, l = 6 cm, V = 30 mL) afforded a pale yellow oil, which was recrystallized from acetonitrile to give the amide 2e as a colorless solid. Rf = 0.10 (CH2Cl2–MeOH = 95:5), mp 164–166 °C, yield 88 mg (78%). C33H37N3O3 (523.6 g mol−1). Purity (HPLC): 99%, tR = 18.41 min. Exact MS (APCI): m/z = calcd for C33H38N3O3 [MH+] 524.2908, found 524.2919. 1H NMR (CDCl3): δ (ppm) = 1.57–1.79 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.12–2.20 (m, 2H, 6-CH2), 2.21 (s, 6H, N–CH3, COCH3), 2.56–2.70 (m, 1H, 4-CHpyran) 2.72 (t, J = 6.7 Hz, 2H, 7-CH2), 2.83–2.94 (m, 2H, 5-CH2), 3.37 (td, J = 11.6/2.3 Hz, 2H, CH2axial–O–CH2axial), 3.57 (s, 2H, Ph–CH2–N), 4.04 (dd, J = 11.4/4.2 Hz, 2H, CH2equat.–O–CH2equat.), 7.23 (d, J = 7.8 Hz, 1H, 4-CH), 7.31 (d, J = 8.5 Hz, 3H, 3-Hphenyl, 5-Hphenyl, N–Hacetamidophenyl), 7.38–7.44 (m, 2H, 9-CH, 3-CH), 7.48 (d, J = 2.0 Hz, 1H, 1-CH), 7.51–7.62 (m, 6H, 2-Hphenyl, 6-Hphenyl, 2-, 3-, 5-, 6-Hacetamidophenyl), 7.66 (s, 1H, N–H). 13C NMR (CDCl3): δ (ppm) = 25.1 (COCH3), 28.4 (C-6), 29.7 (C-3pyran, C-5pyran), 30.9 (C-7), 35.0 (C-5), 38.0 (N–CH3), 57.7 (Ph–CH2–N), 60.0 (C-4pyran), 68.2 (C-2pyran, C-6pyran), 120.5 (C-2phenyl, C-6phenyl), 120.6 (C-3acetamidophenyl, C-5acetamidophenyl.), 127.2 (C-3), 127.9 (C-2acetamidophenyl, C-6acetamidophenyl), 129.9 (C-3phenyl, C-5phenyl), 130.4 (C-4), 130.9 (C-1), 134.7 (C-9), 135.2 C-9a), 137.3 (C-1acetamidophenyl), 137.7 (C-4phenyl), 138.5 (C-2), 138.9 (C-8), 141.9 (C-1phenyl), 146.3 (1C, C-4acetamidophen), 168.5 (OC–NH), 170.4 (COCH3). The signal for the quaternary carbon atom C-4a is not visible. FT-IR (neat): (cm−1) = 3305 (N–H), 2947, 2920 (C–Halkyl), 1670, 1697 (CO), 1111, 1010, (C–O).
2-[4-(Dimethylamino)phenyl]-N-{4-[N-methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2f)
As described for 2a, amide 3 (200 mg, 0.42 mmol), PdCl2(dppf) (16 mg, 0.02 mmol, 5 mol%), KOAc (84 mg, 0.84 mmol, 2 eq.) and 4-(dimethylamino)phenylboronic acid (106 mg, 0.63 mmol, 1.5 eq.) in dimethoxyethane (10 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–CH2Cl2 + 5% MeOH = 1:2, ∅ = 3 cm, l = 8 cm, V = 30 mL) afforded a pale yellow solid, which was recrystallized from acetonitrile to give the amide 2f as a colorless solid. Rf = 0.19 (CH2Cl2–MeOH = 95:5), mp 174–176 °C, yield 202 mg (93%). C33H39N3O2 (509.7 g mol−1). Purity (HPLC): 99%, tR = 18.43 min. Exact MS (ESI): m/z = calcd for C33H40N3O2 [MH+] 510.3115, found 510.21. 1H NMR (CDCl3): δ (ppm) = 1.61–1.80 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.10–2.21 (m, 2H, 6-CH2), 2.21 (s, 3H, N–CH3), 2.64 (tt, J = 11.4/4.2 Hz, 1H, 4-CHpyran) 2.71 (t, J = 6.0 Hz, 2H, 7-CH2), 2.82–2.92 (m, 2H, 5-CH2), 3.00 (s, 6H, N(CH3)2), 3.37 (td, J = 11.6/2.4 Hz, 2H, CH2axial–O–CH2axial), 3.57 (s, 2H, Ph–CH2–N), 3.96–4.09 (m, 2H, CH2equat.–O–CH2equat.), 6.80 (d, J = 8.8 Hz, 2H, 3-Haminophenyl, 5-Haminophenyl), 7.20 (d, J = 7.8 Hz, 1H, 4-CH), 7.31 (d, J = 8.4 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.38–7.45 (m, 2H, 9-CH, 3-CH), 7.46–7.52 (m, 3H, 1-CH, 2-Haminophenyl, 6-Haminophenyl), 7.56 (d, J = 8.4 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.62 (s, 1H, N–H). 13C NMR (CDCl3): δ (ppm) = 28.4 (C-6), 29.6 (C-3pyran, C-5pyran), 30.9 (C-7), 35.0 (C-5), 37.9 (N–CH3), 41.0 (N(CH3)2), 57.7 (Ph–CH2–N), 59.9 (C-4pyran), 68.2 (C-2pyran, C-6pyran), 113.2 (C-3aminophenyl, C-5aminophenyl), 120.4 (C-2phenyl, C-6phenyl), 126.7 (C-3), 127.9 (C-2aminophenyl, C-6aminophenyl), 128.8 (C-1aminophenyl), 129.8 (C-3phenyl, C-5phenyl), 130.3 (C-4), 130.5 (C-1), 134.9 (C-9a), 135.0 (C-4phenyl), 136.2 (C-9), 138.1 (C-2), 139.7 (C-8), 140.7 (C-1phenyl), 150.4 (C-4aminophenyl), 168.6 (OC–NH). A signal for the quaternary carbon atom C-4a is not visible. FT-IR (neat): (cm−1) = 3332 (N–H), 2785 (C–Halkyl), 1647 (CO), 1199, 1138, (C–O).
N-{4-[N-Methyl-N-(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl}-2-(pyridin-3-yl)-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2g)
As described for 2a, amide 3 (100 mg, 0.21 mmol), PdCl2(dppf) (8 mg, 0.01 mmol, 5 mol%), KOAc (40 mg, 0.42 mmol, 2 eq.) and pyridine-3-ylboronic acid (30 mg, 0.23 mmol, 1.1 eq.) in dimethoxyethane (5 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–MeOH = 95:5, ∅ = 2 cm, l = 8 cm, V = 10 mL) afforded a colorless oil, which was recrystallized from acetonitrile to give the amide 2g as a colorless solid. Rf = 0.06 (CH2Cl2–MeOH = 95:5), mp 153–154 °C, yield 59 mg (60%). C30H33N3O2 (467.6 g mol−1). Purity (HPLC): >98%, tR = 14.97 min. Exact MS (APCI): m/z = calcd for C30H34N3O2 [MH+] 468.2646, found 468.2654. 1H NMR (CDCl3): δ (ppm) = 1.60–1.87 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.12–2.23 (m, 2H, 6-CH2), 2.21 (s, 3H, N–CH3), 2.64 (tt, J = 10.7/4.4 Hz, 1H, 4-CHpyran), 2.74(t, J = 6.5 Hz, 2H, 7-CH2), 2.85–3.01 (m, 2H, 5-CH2), 3.37 (td, J = 11.6/2.4 Hz, 2H, CH2axial–O–CH2axial), 3.58 (s, 2H, Ph–CH2–N), 4.04 (dd, J = 11.5/3.7 Hz, 2H, CH2equat.–O–CH2equat.), 7.26–7.28 (m, 1H, 4-CH), 7.32 (d, J = 8.4 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.37 (ddd, J = 7.9/4.9/0.9 Hz, 1H, 5-Hpyridine), 7.43 (s, 1H, 0.9-CH), 7.45 (dd, J = 7.8/2.0 Hz, 1H, 3-CH), 7.52 (d, J = 1.7 Hz, 1H, 1-CH), 7.56 (d, J = 8.4 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.62 (s, 1H, N–H), 7.82–7.91 (m, 1H, 4-Hpyridine), 8.60 (dd, J = 4.9/1.6 Hz, 1H, 6-Hpyridine), 8.85 (dd, J = 2.4/0.9 Hz, 1H, 2-Hpyridine). 13C NMR (CDCl3): δ (ppm) = 28.1 (C-6), 29.3 (C-3pyran, C-5pyran), 30.6 (C-7), 34.7 (C-5), 37.7 (N–CH3), 57.4 (Ph–CH2–N), 59.7 (C-4pyran), 67.9 (C-2pyran, C-6pyran), 120.2 (C-2phenyl, C-6phenyl), 123.7 (C-5pyridine), 127.1 (C-3), 129.6 (C-3phenyl, C-5phenyl), 130.4 (C-4), 130.8 (C-1), 133.9 (C-9), 134.3 (C-4pyridine), 135.3 (C-4a), 136.1 (C-4phenyl), 136.1 (C-2), 136.2 (C-3pyridine), 136.9 (C-1phenyl), 138.7 (C-9a), 142.6 (C-8), 148.3 (C-2pyridine), 148.7 (C-6pyridine), 168.3 (OC–NH). FT-IR (neat): (cm−1) = 3302 (N–H), 2939, 2924 (C–Halkyl), 1643 (CO), 1141, 1010 (C–O).
N-{4-[Methyl(tetrahydro-2H-pyran-4-yl)aminomethyl]phenyl-2-(5-methylthiophen-2-yl)-6,7-dihydro-5H-benzo[7]annulene-8-carboxamide (2h)
As described for 2a, amide 3 (60 mg, 0.13 mmol), PdCl2(dppf) (10 mg, 0.01 mmol, 10 mol%), NaOCH3 (14 mg, 0.25 mmol, 2 eq.) and 5-methylthiophen-2-ylboronic acid (20 mg, 0.14 mmol, 1.1 eq.) in dimethoxyethane (5 mL) were heated to reflux for 12 h. Purification by fc (EtOAc–MeOH = 95:5, ∅ = 2 cm, l = 6 cm, V = 10 mL) afforded an orange oil, which was recrystallized from acetonitrile to give the amide 2h as an orange solid. Rf = 0.28 (CH2Cl2–MeOH = 95:5), mp 161–163 °C, yield 59 mg (94%). C30H34N2O2S (486.6 g mol−1). Purity (HPLC): 96%, tR = 21.46 min. Exact MS (APCI): m/z = calcd for C30H35N2O2S [MH+] 487.2414, found 487.2437. 1H NMR (CDCl3): δ (ppm) = 1.54–1.80 (m, 4H, 3-CH2pyran, 5-CH2pyran), 2.05–2.19 (m, 2H, 6-CH2), 2.22 (s, 3H, N–CH3), 2.61–2.78 (m, 3H, 4-CHpyran, 7-CH2), 2.79–2.89 (m, 2H, 5-CH2), 3.37 (td, J = 11.7/2.2 Hz, 2H, CH2axial–O–CH2axial), 3.63 (s, 2H, Ph–CH2–N), 4.05 (dd, J = 11.8/2.1 Hz, 2H, CH2equat.–O–CH2equat.), 3.59 (s, broad, 3H, CH3thiophene), 6.72 (dt, J = 3.5/1.1 Hz, 1H, 4-Hthiophene), 7.08 (d, J = 3.5 Hz, 1H, 3-Hthiophene), 7.15 (d, J = 7.9 Hz, 1H, 4-CH), 7.31 (s, 1H, 0.9-CH), 7.34 (d, J = 8.8 Hz, 2H, 3-Hphenyl, 5-Hphenyl), 7.39 (dd, J = 7.8/2.0 Hz, 1H, 3-CH), 7.47 (d, J = 2.0 Hz, 1H, 1-CH), 7.57 (d, J = 8.4 Hz, 2H, 2-Hphenyl, 6-Hphenyl), 7.64 (s, 1H, N–H). 13C NMR (CDCl3): δ (ppm) = 16.0 (CH3thiophene), 28.4 (C-6), 29.6 (C-3pyran, C-5pyran), 30.9 (C-7), 35.1 (C-5), 37.9 (N–CH3), 57.7 (Ph–CH2–N), 60.0 (C-4pyran), 68.1 (C-2pyran, C-6pyran), 120.5 (C-2phenyl, C-6phenyl), 123.2 (C-3thioph.,), 125.8 (C-2thioph.,), 126.7 (C-3), 129.5 (C-5thioph.,), 129.9 (C-4thiophene, C-3phenyl, C-5phenyl), 130.4 (C-4), 133.3 (C-1), 134.5 (C-9), 135.2 (C-4a), 137.4 (C-4phenyl), 138.6 (C-1phenyl), 139.2 (C-2), 141.7 (C-9a), 141.7 (C-8), 168.4 (OC–NH). FT-IR (neat): (cm−1) = 3294 (N–H), 2924, 2843 (C–Halkyl), 1643 (CO), 1141, 1010 (C–O).
6. CCR5 assay
Materials
The CCR5 receptor containing membrane homogenates was commercially available (MERCK, Millipore, Darmstadt, Germany). Homogenizers: Elvehjem Potter (B. Braun Biotech International, Melsungen, Germany) and Soniprep 150 (MSE, London, UK). Centrifuges: cooling centrifuge model Rotina 35R (Hettich, Tuttlingen, Germany) and high-speed cooling centrifuge model Sorvall RC-5C plus (Thermo Fisher Scientific, Langenselbold, Germany). Multiplates: standard 96-well multiplates (Diagonal, Muenster, Germany). Shaker: self-made device with adjustable temperature and tumbling speed (scientific workshop of the institute). Vortexer: Vortex Genie 2 (Thermo Fisher Scientific, Langenselbold, Germany). Harvester: MicroBeta FilterMate-96 Harvester. Filter: Printed Filtermat Type A and B. Scintillator: Meltilex (Type A or B) solid state scintillator. Scintillation analyzer: MicroBeta Trilux (all from Perkin Elmer LAS, Rodgau-Jügesheim, Germany). Chemicals and reagents were purchased from different commercial sources and were of analytical grade.General protocol for the CCR5 binding assay
The assay was performed with the radioligand [3H]TAK-779 (specific activity 79.1 Ci/mmol, custom synthesis by Perkin Elmer). The CCR5 receptor containing membrane fragments was used according to the instructions of the manufacturer and incubated with various concentrations of the test compound, 2 nM [3H]TAK-779, and the binding buffer (50 mM HEPES pH 7.4, 5 mM MgCl2, 1 mM CaCl2 and 0.2% BSA) at room temperature. The filtermats were washed with a buffer solution (50 mM HEPES pH = 7.4, 500 mM NaCl and 0.1% BSA). The test compound solutions were prepared by dissolving approximately 10 μmol (usually 2–4 mg) of the test compound in DMSO so that a 10 mM stock solution was obtained. To obtain the required test solutions for the assay, the DMSO stock solution was diluted with the respective assay buffer. The filtermats were presoaked in 0.5% aqueous polyethylenimine solution for 2 h at room temperature before use. All binding experiments were carried out in duplicate in 96-well multiplates. The concentrations given are the final concentrations in the assay. Generally, the assays were performed by the addition of 50 μL of the respective assay buffer, 50 μL of test compound solution in various concentrations (10−5, 10−6, 10−7, 10−8, 10−9 and 10−10 mol L−1), 50 μL of the corresponding radioligand solution and 50 μL of the respective receptor preparation into each well of the multiplate (total volume 200 μL). The receptor preparation was always added last. During the incubation, the multiplates were shaken at a speed of 500–600 rpm at the specified temperature. The assays were terminated after 120 min by rapid filtration using the harvester. During the filtration each well was washed five times with 300 μL of water. Subsequently, the filtermats were dried at 95 °C. The solid scintillator was melted on the dried filtermats at a temperature of 95 °C for 5 min. After solidifying of the scintillator at room temperature, the trapped radioactivity in the filtermats was measured with the scintillation analyzer. Each position on the filtermat corresponding to one well of the multiplate was measured for 5 min with the [3H]-counting protocol. The overall counting efficiency was 20%. The IC50 values were calculated using the program GraphPad Prism® 3.0 (GraphPad Software, San Diego, CA, USA) by non-linear regression analysis.Abbreviations
| bp | Boiling point |
| COMU™ | (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate |
| dec. | Decomposed |
| DCC | Dicyclohexyl-carbodiimide |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DMAP | 4-(Dimethylamino)-pyridine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate |
| HOBt | Hydroxybenzotriazole |
| IC50 | Half maximal inhibitory concentration |
| NMM | N-Methylmorpholine |
| PPA | Polyphosphoric acid |
Acknowledgements
This work was performed within the framework of the International Research Training Group ‘Complex Functional Systems in Chemistry: Design, Synthesis and Applications’ in collaboration with the University of Nagoya. Financial support of the IRTG and this project by the Deutsche Forschungsgemeinschaft and the Funding Program for Next Generation World-Leading Researchers from JSPS is gratefully acknowledged. We are also grateful for the financial support from the Collaborative Research Center 656 MoBil.References
- M. Balter, Science, 1996, 272, 1740 CrossRef CAS.
- R. Liu, W. A. Paxton, S. Choe, D. Ceradini, S. R. Martin, R. Horuk, M. E. MacDonald, H. Stuhlmann, R. A. Koup and N. R. Landau, Cell, 1996, 86, 367–377 CrossRef CAS.
- P. Dorr, M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Armour, D. Price, B. Stammen, A. Wood and M. Perros, Antimicrob. Agents Chemother., 2005, 49, 4721–4732 CrossRef CAS PubMed.
- D. Armour, M. J. de Groot, M. Edwards, M. Perros, D. A. Price, B. L. Stammen and A. Wood, ChemMedChem, 2006, 1, 706–709 CrossRef CAS PubMed.
- C. L. Hyde, A. MacInnes, F. A. Sanders, J. F. Thompson, R. A. Mazzarella, O. Faergeman, D. F. van Wijk, L. Wood, M. Lira and S. A. Paciga, Circ.: Cardiovasc. Genet., 2010, 3, 162–168 CrossRef CAS PubMed.
- D. D. Patel, J. P. Zachariah and L. P. Whichard, Clin. Immunol., 2001, 98, 39–45 CrossRef CAS PubMed.
- C. L. Kohem, J. C. Brenol, R. M. Xavier, M. Bredemeier, C. V. Brenol, T. L. Dedavid e Silva, A. de Castilhos Mello, A. D. Canedo, A. G. Neves and J. A. Chies, Scand. J. Rheumatol., 2007, 36, 359–364 CrossRef CAS PubMed.
- H. A. Martens, C. G. Kallenberg and M. Bijl, Autoimmunity, 2009, 42, 260–262 CrossRef CAS PubMed.
- T. J. Mitchell, A. J. Walley, J. E. Pease, P. J. Venables, S. Wiltshire, T. J. Williams and W. O. Cookson, Lancet, 2000, 356, 1491–1492 CrossRef CAS.
- I. P. Hall, A. Wheatley, G. Christie, C. McDougall, R. Hubbard and P. J. Helms, Lancet, 1999, 354, 1264–1265 CrossRef CAS.
- A. Junker, J. Yamaguchi, K. Itami and B. Wünsch, J. Org. Chem., 2013, 78, 5579–5586 CrossRef CAS PubMed.
- A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2002, 17, 69–76 CrossRef CAS PubMed.
- M. Shiraishi, Y. Aramaki, M. Seto, H. Imoto, Y. Nishikawa, N. Kanzaki, M. Okamoto, H. Sawada, O. Nishimura, M. Baba and M. Fujino, J. Med. Chem., 2000, 43, 2049–2063 CrossRef CAS PubMed.
- M. Seto, K. Aikawa, N. Miyamoto, Y. Aramaki, N. Kanzaki, K. Takashima, Y. Kuze, Y. Iizawa, M. Baba and M. Shiraishi, J. Med. Chem., 2006, 49, 2037–2048 CrossRef CAS PubMed.
- A. Suzuki and N. Miyaura, Abstr. Pap. Am. Chem. S, 1995, 209, 1–Orgn Search PubMed.
- N. Arai, T. Miyaoku, S. Teruya and A. Mori, Tetrahedron Lett., 2008, 49, 1000–1003 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- S. Gronowitz, V. Bobosik and K. Lawitz, Chem. Scr., 1984, 23, 120–122 CAS.
- S. Gronowitz and K. Lawitz, Chem. Scr., 1983, 22, 265–266 CAS.
- A. R. Martin and Y. Yang, Acta Chem. Scand., 1993, 47, 221–230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C and gHSQC NMR spectra, HPLC analysis, MS spectra of all compounds. See DOI: 10.1039/c3ob41873a |
| This journal is © The Royal Society of Chemistry 2014 |