
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectrochemical synthesis, optical properties and plasmon-induced charge separation behaviour of gold nanodumbbells on TiO2
Yu
Katagi
,
Emiko
Kazuma†
and
Tetsu
Tatsuma
*
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: tatsuma@iis.u-tokyo.ac.jp
First published on 7th October 2014
Abstract
Chemically synthesized, commercially available Au nanorods were adsorbed on a TiO2 thin film, and photoelectrochemically transformed to Au nanodumbbells by photoelectrochemical deposition of Au at both ends of the NRs under UV irradiation. The nanodumbbells show about fourfold greater light absorption than the nanorods based on localized surface plasmon resonance (LSPR) in the visible to near infrared region. The absorption intensities and wavelengths of the Au nanodumbbells depend on the size of their spheroidal caps, which can be controlled by UV exposure time. The nanodumbbells can be applied to LSPR sensors, as their absorption peak redshifts with increasing local refractive index near the metal surface. The Au nanodumbbells on TiO2 are also suitable for photofunctional materials and devices based on plasmon-induced charge separation (PICS) at the Au–TiO2 interface, because of their higher photoabsorption intensity, better wavelength tunability and greater PICS efficiency than nanorods.
Introduction
Metal nanoparticles (NPs), which exhibit unique properties different from those of bulk metals, have been studied enthusiastically along with improvements of nanostructure processing technologies. One of those properties is the localized surface plasmon resonance (LSPR), which is the coherent oscillations of conduction electrons in metal NPs.1 LSPR is induced by light of a particular wavelength, and optical near fields are generated around the NPs. This phenomenon can be applied to, for instance, surface-enhanced Raman scattering (SERS),1 chemical sensing and biosensing (LSPR sensors),1–3 photoabsorption and fluorescence enhancement,4,5 nanoscale light focusing6 and plasmon-induced charge separation (PICS).7,8 LSPR properties depend on the composition, size, shape and geometric arrangement of NPs, as well as local refractive index near the metal surface. Therefore, tuning and optimization of these factors are important issues for the applications mentioned above. In particular, the particle shape strongly affects the resonance wavelength and intensity, hence they have investigated metal NPs with various different shapes, such as nanorods (NRs)9,10 and nanoprisms.11 Among those NPs, Au NRs have been well studied because some of them are commercially available and their resonance wavelength can be redshifted largely in the visible to near infrared region by increasing their aspect ratio. In the present work, we developed a photoelectrochemical method to transform commercially available, chemically synthesized Au NRs to nanodumbbells (NDs) on a TiO2 thin film.Au is reductively deposited at both ends of a Au NR by a photocatalytic effect of TiO2. The ND thus obtained shows light absorption ∼4 times greater than that of the NR, without a large shift in the absorption wavelength. Besides, Au NDs are advantageous to applications to PICS at the Au–TiO2 interface, because of good electrical contact with the TiO2 substrate. Although there are some reports on synthesis of metal NDs in solutions by chemical or electrochemical reduction,12–20 light-assisted synthesis has never been reported to the best of our knowledge. In this method, the size of the spheroidal caps and optical properties of NDs are easily controlled by changing the photoirradiation time. In addition, the synthesized NDs, which have already been immobilized onto the substrates, are ready to be applied to LSPR sensors and PICS-based devices.
Experimental
Preparation
A polycrystalline anatase TiO2 thin film (∼40 nm thick) was prepared on a Pyrex glass sheet or an indium–tin oxide (ITO)-coated glass plate (Kinoene Kogaku) by a dip-coating method from a titanium alkoxide ethanol solution (NDH-510C, Nippon Soda) at 1 mm s−1 followed by drying at 120 °C for 40 min and sintering at 500 °C for 1 h.Suspension of Au NRs protected with cetyltrimethyl-ammonium bromide (CTAB) (Dai Nippon Toryo, aspect ratio ∼4, 1 mL)21 was diluted 10 times with ultra-pure water and centrifuged at 7000g for 10 min to remove excess CTAB. The precipitate thus obtained was separated and resuspended in water (5 mL). The Au NR suspension was mixed with 0.5 M H2SO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume). The mixture (pH ∼ 1) was cast on TiO2 (∼20 μL cm−2) and left for 3 h in the dark. The substrate was rinsed thoroughly with pure water and water was evaporated.
:1 by volume). The mixture (pH ∼ 1) was cast on TiO2 (∼20 μL cm−2) and left for 3 h in the dark. The substrate was rinsed thoroughly with pure water and water was evaporated.
An aqueous solution of HAuCl4 (Kanto Chemical, 10 mM) was mixed with ethanol (1:9 by volume). The mixture was cast on the substrate with Au NRs (∼20 μL cm−2) and irradiated with UV light from a Hg lamp (HB-25103BY-C, Ushio, 26 mW cm−2) for 3, 10 or 60 s for transformation of the NRs to NDs. The substrate was rinsed thoroughly with pure water and dried.
Measurement
The visible-near infrared absorption spectra were collected by a spectrophotometer (V-670, Jasco) (%absorption = 100 − %transmittance − %reflectance − %scattering; forward and backward scattering were measured using an integrating sphere). Particle morphologies were observed by a field emission scanning electron microscope (FE-SEM, JSM-7500FA, JEOL) and an atomic force microscope (AFM, SPA400, SII Nanotechnology). Photopotential responses of the ITO/TiO2/Au ND photoelectrode in 0.1 M aqueous LiClO4 were measured under visible-near infrared light from a Xe lamp (119 mW cm−2, 700–1000 nm, LAX-103, Asahi Spectra) using a potentiostat (SI 1280 B, Solartron) and a Ag|AgCl reference electrode.Simulation of spectra and electric field distribution
Extinction spectra of a Au NR and NDs on an anatase TiO2 film of 40 nm thick on a glass substrate and spatial distributions of electric field and polarization around the NR and NDs were calculated on the basis of a finite-difference time-domain (FDTD) method via FDTD Solutions (Lumerical Solutions). The simulation domain consisted of 10 nm cubic cells, and the central region overlying the nanoparticle was further meshed with a 3D grid of 0.5 nm spacing. The dielectric functions of Au and TiO2 were extracted from the data of Johnson and Christy22 and Jellison,23 respectively.Results and discussion
Morphological changes from NR to ND
The Au NRs were well adsorbed onto the TiO2 surface in the presence of H2SO4 (Fig. 1a and e). The protective agent CTAB on a Au NR is partly dissociated (CTA+ + Br−), and is anchored to the TiO2 surface, which is positively charged under acidic conditions, via SO42− (–OH2+–SO42−–CTA+). The adsorption was also observed in a (NH4)2SO4 or Na2SO4 aqueous solution. After irradiation of the TiO2 film, on which Au NRs are adsorbed, with UV light for 3–60 s in a HAuCl4 aqueous solution containing ethanol, dumbbell-shaped NPs (NDs) were observed instead of the NRs (Fig. 1b–d and f). Each ND has two spheroidal caps at both ends of the NR. Those data, which are obtained reproducibly, indicate that most of the NRs transformed into NDs. Some less anisotropic, spherical NPs were seen both before and after the light irradiation. | ||
| Fig. 1 (a–d) SEM and (e, f) AFM images of (a, e) Au nanorods (NRs) and (b–d, f) Au nanodumbbells (NDs) on TiO2. Au NDs were prepared by UV irradiation for (b, f) 3, (c) 10 and (d) 60 s in an aqueous solution of HAuCl4 containing ethanol. Scale bars are (a–d) 40 nm and (e, f) 200 nm. | ||
The transformation of the Au NR to ND can be explained in terms of preferential growth of the NR at both ends, as shown in Fig. 2a. First, electrons in the TiO2 valence band are excited to the conduction band by UV light. It is known that excited electrons in the TiO2 conduction band tend to be injected into Au NPs on TiO2.24,25 Although the NRs are coated with CTAB, the length of which is 2.1 nm or less, electron tunneling through an insulating organic layer of this thickness is possible.26 The injected electrons are used for the reduction of Au3+ in the solution at the NR surface. Au is deposited at the whole NR surface, but preferentially at both ends of the Au NR ({111} facets), where the surface density of CTAB is lower than that at the rod sides ({110} facets).27 The localized deposition results in transformation of a NR into a ND. The simultaneously generated holes in the TiO2 valence band must be consumed through ethanol oxidation. The size of the spheroidal caps of NDs can be controlled by changing the UV irradiation time. As the SEM images in Fig. 1a–d indicate, the spheroidal caps grow gradually during the UV exposure, and the Au ND looks like a dimer of two spherical NPs at 60 s.
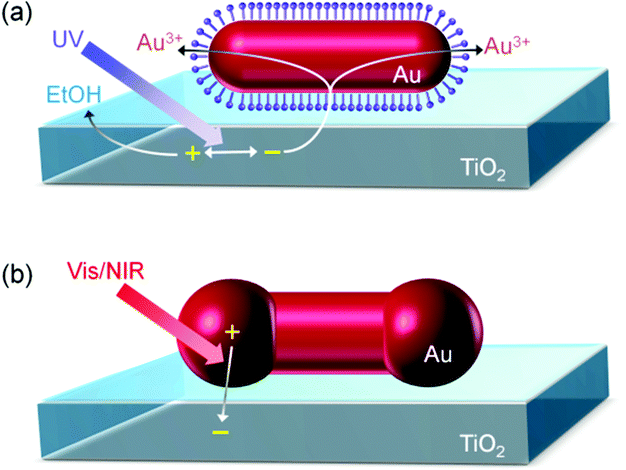 | ||
| Fig. 2 Schematic illustrations for (a) photoelectrochemical transformation of a Au NR to Au ND on TiO2 and (b) plasmon-induced charge separation (PICS) at the interface between Au ND and TiO2. | ||
Changes in optical properties
Fig. 3a shows absorption spectra of the Au NRs and Au NDs on TiO2 in air. A plasmonic NR has two LSPR modes, transverse and longitudinal modes.9,10 In the former mode, electron oscillation is perpendicular to the rod axis, while in the latter mode it is parallel. A ND should also have at least two modes equivalent to them. To analyze the plasmon peaks of the NDs, we calculated theoretical spectra by a FDTD method (Fig. 3b and c). The models used for the simulation, which are shown in Fig. 3d, are basically based on SEM and AFM images, and tuned to fit the calculated main peak wavelengths to the experimentally observed ones. The NR model is uniformly covered with a CTAB layer of 2 nm thickness with a refractive index of 1.38.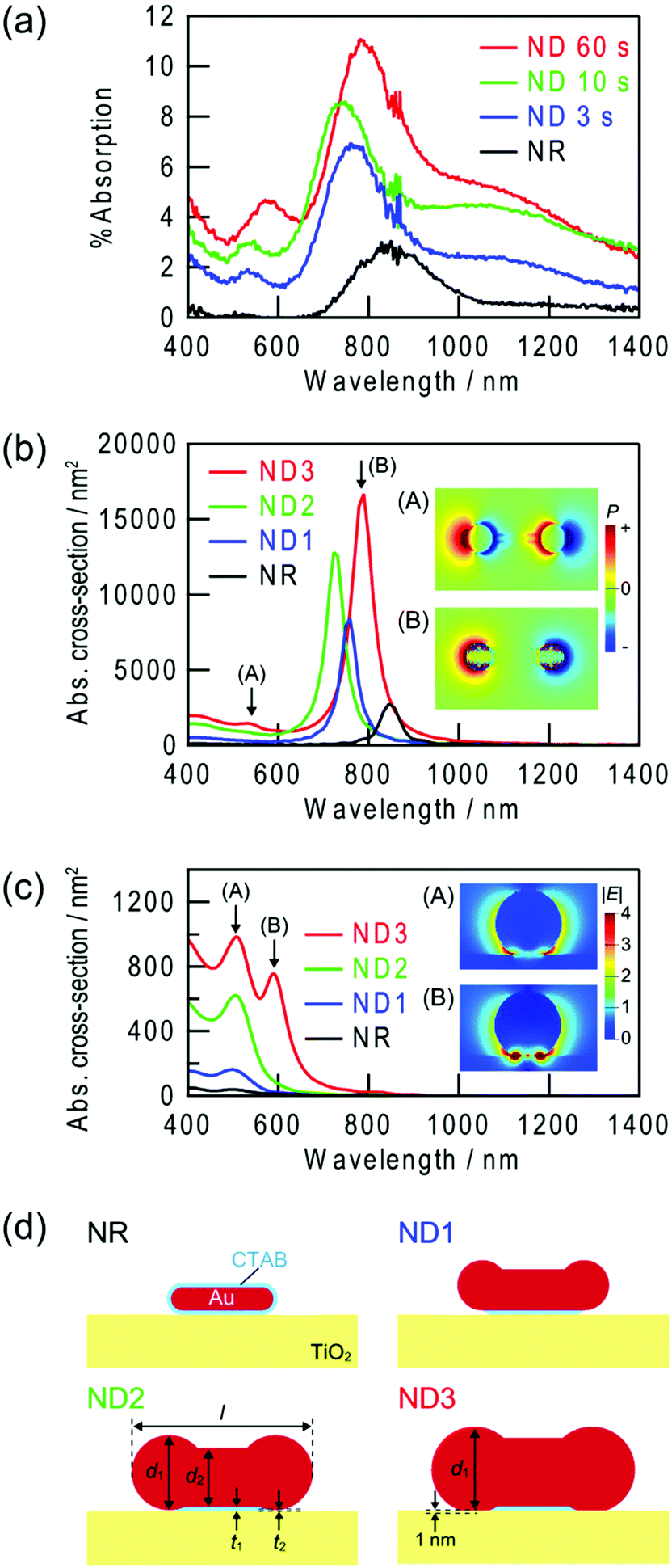 | ||
| Fig. 3 (a) Experimentally obtained absorption spectra of Au NRs and Au NDs on TiO2. Au NDs were prepared by UV irradiation for 3, 10 and 60 s. (b, c) Theoretically calculated spectra of absorption cross-section for (b) longitudinal and (c) transverse modes. (d) Cross sections of the simulation models ([l, d1, d2, t1, t2] (in nm) = [45, 10, 10, 2, 2], [67, 22, 18, 2, 2], [80, 33, 26, 2, 0.5], and [90, 38, 30, 2, 0] for NR, ND1, ND2 and ND3, respectively). Inset: spatial distributions of (b) polarization at the interface with TiO2 and (c) electric field at the transverse cross-section of the spherical part of the ND at the peak wavelengths. | ||
The main peaks at 700–900 nm are assigned to the longitudinal mode across the whole NR and NDs (Fig. 3b, inset B). As a Au NR transforms into a ND, the peak height increases in both experiments and calculations. Similar behaviour has been observed in other studies on Au NDs,15,16,18,20 and the behaviour can be explained in terms of increased particle volume.10 The shifts in the peak wavelength are somewhat complicated; the peak blueshifts until 10 s, then redshifts. In previous studies, both blueshifts15,16,18,20 and redshifts were observed.16,18,19 The different behaviours may be explained in terms of different initial NR shapes and different transformation mechanisms. In the present case, NRs and NDs are adsorbed on TiO2. The initial blueshift of the peak must be caused by a decrease in the effective aspect ratio. It is known that a decrease in the aspect ratio of NRs results in a blueshift of the longitudinal peak.9,10 The following redshift can be simulated only when the spherical parts of the ND is in direct contact with the TiO2 substrate. Initially the NR is in contact with TiO2via the CTAB layer. It is reasonable to assume that the growth of the spherical parts at the both ends of the NR finally results in the direct contact with TiO2. The local refractive index around a plasmonic particle strongly affect the resonance, and an increase in the refractive index redshifts the resonance wavelength.28 Since the refractive index of TiO2 is very high (∼2.523 for anatase), the direct contact results in the redshift. The direct contact is also confirmed by an increased photoinduced current described below.
Assignment of the small peaks at 500–600 nm may be more complicated, because some different peaks are observed in the calculated spectra. The small peaks at 500–600 nm in the calculated longitudinal mode spectra for NDs (Fig. 3b) are ascribed to dipole oscillation localized at the spherical parts or quadrupole oscillation over the whole particle (Fig. 3b, inset A). For the calculated transverse mode spectra (Fig. 3c), a peak splits into two peaks when the spherical parts of the ND are in contact with TiO2. The peak at a longer wavelength is more like the interface plasmon mode (Fig. 3c, inset B), in which electron oscillation is localized at the interface with the substrate of a high refractive index, in comparison with the peak at a shorter wavelength (Fig. 3c, inset A). This mode has been observed for spherical gold29 and silver30 NPs placed on TiO2. The interface mode resonance may contribute to the redshift of the small peak when UV irradiation time is extended from 10 to 60 s.
On the other hand, the shoulders observed in the experimental spectra at 1000–1400 nm are not observed in the spectra calculated for the single NDs. Therefore, those must be explained in terms of interparticle plasmon coupling, which are caused by electrostatic interaction between the adjacent plasmonic particles. It is known that resonance peaks are redshifted by plasmon coupling.31
The experimentally observed peaks are much broader than the calculated peaks, probably because the NRs and NDs are not completely monodisperse. Background of the spectra seems to increase gradually during the UV irradiation. This may also be explained in terms of overlapping of the broad peaks with each other.
Optical responses to refractive index changes
Plasmonic NPs are often used as chemical sensors and biosensors, because their absorption peak redshifts as the refractive index around the NPs increases.1–3 The NPs are used for, for instance, monitoring of specific adsorption or antigen–antibody binding at the NP surface. Here we examined absorption peak wavelengths of the Au NRs and NDs on TiO2 in aqueous solutions with different refractive indices, which were adjusted by glycerol. Both the small peaks at 500–600 nm (Fig. 4a) and the large peaks at 700–900 nm (Fig. 4b) redshifted as the refractive index of the solution increased. The straight lines in the graph are the linear fits, and their slopes reflect refractive index sensitivities (in nm RIU−1, RIU = refractive index unit).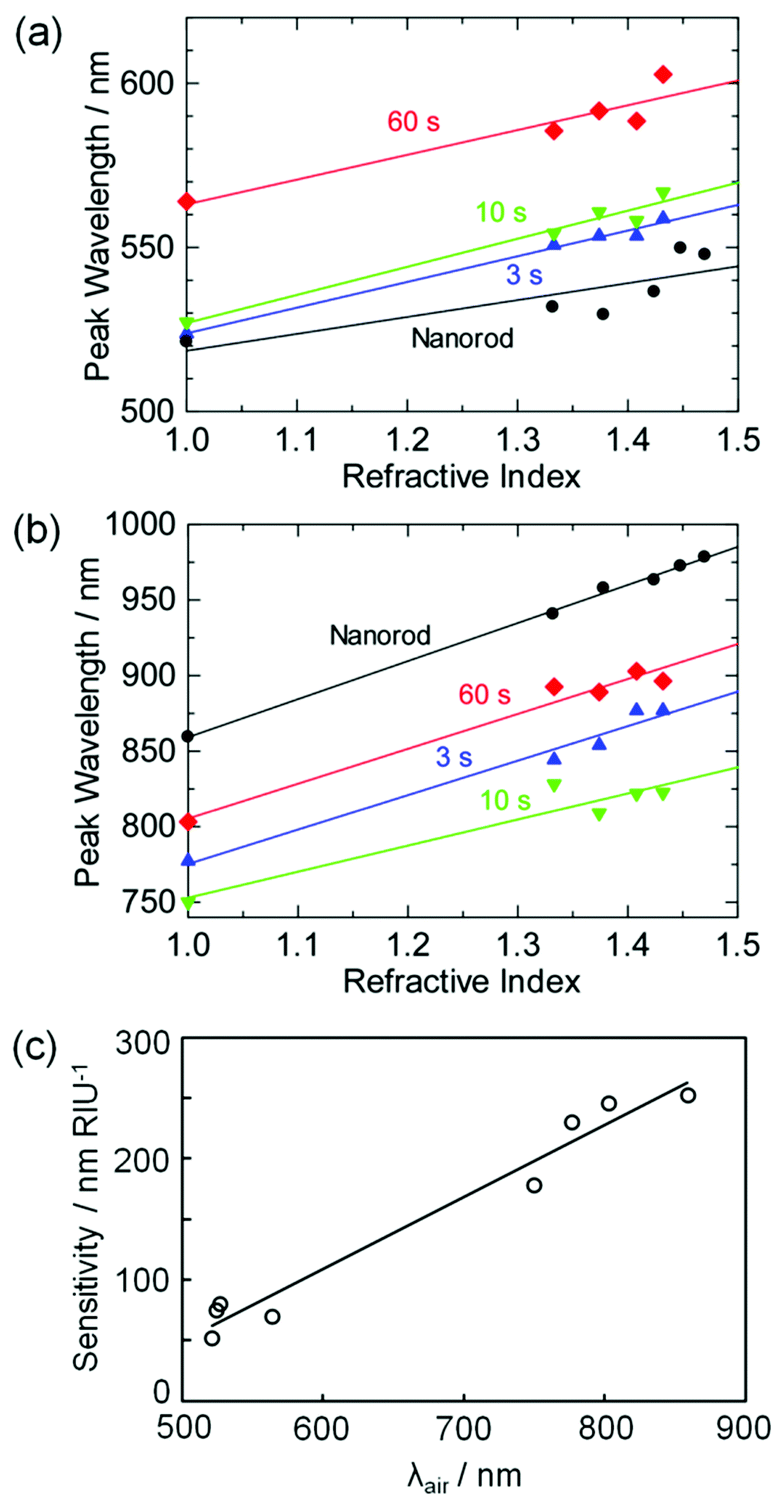 | ||
| Fig. 4 Peak wavelengths of Au NRs and Au NDs on TiO2 for the (a) small and (b) large peaks plotted against the refractive index around the particles. (c) Correlation between the refractive index sensitivity and the peak wavelength in air (λair) for the Au NRs and Au NDs. Au NDs were prepared by UV irradiation for 3, 10 and 60 s. | ||
The refractive index sensitivities are plotted in Fig. 4c as a function of the peak wavelength in air, λair. Although the sensitivity exhibits a certain error, it virtually shows linear dependence on λair, as reported for most NPs.3,32 The sensitivity is in agreement with that for previously reported Au NR ensembles on a solid substrate (170 nm RIU−1 for λair = 660 nm).3 The results indicate that at least the main peak of the Au NDs can be utilized for LSPR sensing. If a sample solution is coloured or turbid, the peak wavelength may be optimized by changing the photoirradiation time so that the interference from the absorption and scattering of the solution is minimized. The NDs are thus favorable in terms of fine tunability of the peak wavelength, as well as high absorption intensity.
Plasmon-induced charge separation efficiency
At the interface between plasmonic Au,33,34 Ag33,35 or Cu36 NP and semiconductors such as TiO2,33–36 ZnO,37 CeO238 or Si,39 plasmon-induced charge separation (PICS) occurs; electrons are injected from the resonant NP to the conduction band of the semiconductor due to the external photoelectric effect or hot electron injection.25,34,40 PICS is applied to photovoltaics,34,41 photosensing,39 photocatalysis,34,38 biosensing,42 photochromism35,43 and photoactuation.44 Since the resonant wavelength of the longitudinal mode of a plasmonic NR redshifts with increasing aspect ratio, PICS is possible under red or near infrared light.25,39,40,43 However, in general, chemically synthesized NRs are protected with a CTAB layer,21,27 which retards electron transfer from the NR to TiO2.
The electron transfer from the UV-excited TiO2 to Au NRs need not to be so fast in the transformation of Au NRs to NDs, which takes only 60 s or less. However, regarding the PICS-based photofunctional materials and devices, the rate of electron transfer from Au NPs to TiO2 directly affect the efficiencies of photovoltaic cells, sensitivities and response time of photosensors and reaction rates of photocatalysts. The photoelectrochemically synthesized NDs are expected to be more suitable to the electron transfer than the chemically synthesized NRs because the spheroidal caps are close to the TiO2 surface (Fig. 2b).
To verify this, we coated ITO electrodes with TiO2 films and loaded them with the Au NRs. Some of the ITO/TiO2/Au NR electrodes thus obtained were further irradiated with UV for 60 s in the HAuCl4 solution containing ethanol so that ITO/TiO2/Au ND electrodes were prepared. Fig. 5 shows the photopotential responses of the electrodes to red-near infrared light irradiation (700–1000 nm, 119 mW cm−2). The ND-modified electrode exhibited photopotential responses of ca. −0.4 V in 60 s, indicating that electrons are injected from the NDs to the TiO2 conduction band. On the other hand, the NR-modified electrodes exhibited lower and slower responses of ca. −0.03 V in 1200 s. The initial rate of the potential shift of the former is more than two orders of magnitude higher than that of the latter. UV irradiation for 10 min during the loading of an electrode with the Au NRs did not improve the response of the NR electrode; the CTAB layer was not removed by the UV irradiation. The dramatic acceleration by the transformation from the Au NRs to NDs reflects a significant improvement in the PICS efficiency as well as the increased photoabsorption intensity.
 | ||
| Fig. 5 Photopotential responses of the ITO/TiO2/Au NR and ITO/TiO2/Au ND electrodes to 700–1000 nm light (119 mW cm−2). Au NDs were prepared by UV irradiation for 60 s. | ||
Conclusions
Au NRs loaded on a TiO2 thin film were photoelectrochemically transformed to Au NDs, based on the electron excitation of TiO2 by UV light. The transformation results in enhancement of the LSPR-based absorption intensity in the visible to near infrared region. The absorption intensities and wavelengths of the Au NDs depend on the size of their spheroidal caps, which can be controlled by UV exposure time. The Au NDs can be applied to LSPR sensors like other plasmonic metal NPs, as the absorption peak redshifts with increasing local refractive index. The Au NDs on TiO2 are also suitable to PICS-based photofunctional materials and devices, because of their higher photoabsorption intensity, better wavelength tunability and greater PICS efficiency than NRs.Acknowledgements
The authors are grateful to Prof. S. Yamada for useful discussion and Dai Nippon Toryo for supply of Au NRs. This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Area “Artificial Photosynthesis” (Area No. 2406) No. 25107511, a Grant-in-Aid for Scientific Research No. 25288063 and a Grant-in-Aid for Challenging Exploratory Research No. 25600002.Notes and references
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205 CrossRef CAS PubMed.
- J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308 RSC.
- D. K. Gramotnev and S. I. Bozhevolnyi, Nat. Photonics, 2010, 4, 83 CrossRef CAS.
- T. Tatsuma, Bull. Chem. Soc. Jpn., 2013, 86, 1 CrossRef CAS.
- C. Clavero, Nat. Photonics, 2014, 8, 95 CrossRef CAS.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661 CrossRef CAS.
- L. Stephan and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410 CrossRef.
- L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2009, 6, 2060 CrossRef PubMed.
- C. S. Ah, S. D. Hong and D.-J. Jang, J. Phys. Chem. B, 2001, 105, 7871 CrossRef CAS.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2004, 108, 5882 CrossRef CAS.
- C.-C. Huang, Z. Yang and H.-T. Chang, Langmuir, 2004, 20, 6089 CrossRef CAS PubMed.
- J. H. Song, F. Kim, D. Kim and P. Yang, Chem. – Eur. J., 2005, 11, 910 CrossRef CAS PubMed.
- C. Wang, T. Wang, Z. Ma and Z. Su, Nanotechnology, 2005, 16, 2555 CrossRef CAS.
- C.-J. Huang, P.-H. Chiu, Y.-H. Wang, W.-R. Chen, T.-H. Meen and C.-F. Yang, Nanotechnology, 2006, 17, 5355 CrossRef CAS.
- Y. Xiang, X. Wu, D. Liu, L. Feng, K. Zhang, W. Chu, W. Zhou and S. Xie, J. Phys. Chem. C, 2008, 112, 3203 CAS.
- M. Grzelczak, A. Sánchez-Iglesias, B. Rodríguez-González, R. Alvarez-Puebla, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Funct. Mater., 2008, 18, 3780 CrossRef CAS.
- P. Wang, M. Liu, G. Gao, S. Zhang, H. Shi, Z. Li, L. Zhang and Y. Fang, J. Mater. Chem., 2012, 22, 24006 RSC.
- Y. Niidome, K. Nishioka, H. Kawasaki and S. Yamada, Chem. Commun., 2003, 2376 RSC.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370 CrossRef CAS.
- G. E. Jellison Jr., L. A. Boatner, J. D. Budai, B.-S. Jeong and D. P. J. Norton, J. Appl. Phys., 2003, 93, 9537 CrossRef PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef CAS PubMed.
- E. Kazuma and T. Tatsuma, Adv. Mater. Interfaces, 2014, 1, 1400066 Search PubMed.
- W. Wang, T. Lee and M. A. Reed, Phys. Rev. B: Condens. Matter, 2003, 68, 035416 CrossRef.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368 CrossRef CAS.
- K. L. Kelly and K. Yamashita, J. Phys. Chem. B, 2006, 110, 7743 CrossRef CAS PubMed.
- I. Tanabe and T. Tatsuma, Chem. Lett., 2014, 43, 931 CrossRef CAS.
- I. Tanabe and T. Tatsuma, Nano Lett., 2012, 12, 5418 CrossRef CAS PubMed.
- A. M. Funston, C. Novo, T. J. Davis and P. Mulvaney, Nano Lett., 2009, 9, 1651 CrossRef CAS PubMed.
- M. M. Miller and A. A. Lazarides, J. Phys. Chem. B, 2005, 109, 21556 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, Chem. Commun., 2004, 1810 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632 CrossRef CAS PubMed.
- Y. Ohko, T. Tatsuma, T. Fujii, K. Naoi, C. Niwa, Y. Kubota and A. Fujishima, Nat. Mater., 2003, 2, 29 CrossRef CAS PubMed.
- T. Yamaguchi, E. Kazuma, N. Sakai and T. Tatsuma, Chem. Lett., 2012, 41, 1340 CrossRef CAS.
- K. Kawahara, K. Suzuki, Y. Ohko and T. Tatsuma, Phys. Chem. Chem. Phys., 2005, 7, 3851 RSC.
- H. Kominami, A. Tanaka and K. Hashimoto, Chem. Commun., 2010, 46, 1287 RSC.
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702 CrossRef CAS PubMed.
- N. Sakai, Y. Fujiwara, Y. Takahashi and T. Tatsuma, ChemPhysChem, 2009, 10, 766 CrossRef CAS PubMed.
- Y. Takahashi and T. Tatsuma, Appl. Phys. Lett., 2011, 99, 182110 CrossRef PubMed.
- E. Kazuma and T. Tatsuma, Nanoscale, 2014, 6, 2397 RSC.
- E. Kazuma and T. Tatsuma, Chem. Commun., 2012, 48, 1733 RSC.
- T. Tatsuma, K. Takada and T. Miyazaki, Adv. Mater., 2007, 19, 1249 CrossRef CAS.
Footnote |
| † Present address: RIKEN, Hirosawa, Wako, Saitama 351-0198, Japan. |
| This journal is © The Royal Society of Chemistry 2014 |