 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diagnostic nanoparticle targeting of the EGF-receptor in complex biological conditions using single-domain antibodies†
K.
Zarschler‡
a,
K.
Prapainop‡
b,
E.
Mahon‡
b,
L.
Rocks
b,
M.
Bramini
b,
P. M.
Kelly
b,
H.
Stephan
*a and
K. A.
Dawson
*b
aInstitute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstraße 400, D-01328 Dresden, Germany. E-mail: h.stephan@hzdr.de
bCentre For BioNano Interactions (CBNI), School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: kenneth.a.dawson@cbni.ucd.ie
First published on 16th April 2014
Abstract
For effective localization of functionalized nanoparticles at diseased tissues such as solid tumours or metastases through biorecognition, appropriate targeting vectors directed against selected tumour biomarkers are a key prerequisite. The diversity of such vector molecules ranges from proteins, including antibodies and fragments thereof, through aptamers and glycans to short peptides and small molecules. Here, we analyse the specific nanoparticle targeting capabilities of two previously suggested peptides (D4 and GE11) and a small camelid single-domain antibody (sdAb), representing potential recognition agents for the epidermal growth factor receptor (EGFR). We investigate specificity by way of receptor RNA silencing techniques and look at increasing complexity in vitro by introducing increasing concentrations of human or bovine serum. Peptides D4 and GE11 proved problematic to employ and conjugation resulted in non-receptor specific uptake into cells. Our results show that sdAb-functionalized particles can effectively target the EGFR, even in more complex bovine and human serum conditions where targeting specificity is largely conserved for increasing serum concentration. In human serum however, an inhibition of overall nanoparticle uptake is observed with increasing protein concentration. For highly affine targeting ligands such as sdAbs, targeting a receptor such as EGFR with low serum competitor abundance, receptor recognition function can still be partially realised in complex conditions. Here, we stress the value of evaluating the targeting efficiency of nanoparticle constructs in realistic biological milieu, prior to more extensive in vivo studies.
Introduction
Precise delivery of therapeutics, diagnostics or theranostics to specific tissues represents one of the major challenges in cancer imaging and therapy. Through intensive research in the area of nanomedicine, significant progress has been made in order to address the issue of targeted drug delivery to tumours for cancer treatment.1–3 It is widely proposed that accumulation of nanoparticles at the tumour site can be achieved by passive and active targeting, or frequently by a combination of both.4–10 The former strategy selectively utilizes the unique pathophysiology of tumours, such as the enhanced penetration and retention effect as well as their characteristic tumour microenvironment.11–17 For active targeting, biorecognition molecules (ligands) directed against selected tumour biomarkers are grafted to the nanoparticle surface to increase and specify their delivery through specific ligand–biomarker interactions. The nature of these ligands investigated in clinical and preclinical studies is very diverse ranging from proteins, including antibodies and fragments thereof, through aptamers and glycans to short peptides and small non-proteinaceous molecules.18–20 However, regarding clinical translation, while the limited success of current nanoparticle formulations in achieving highly effective biorecognition can be attributed to various reasons, it is currently incompletely understood.21,22 The fact that actively targeted nanoparticles often fail to show benefit at the (pre-)clinical stage can originate in difficulties these objects encounter in finding their target cells in vivo.23 Dynamic interactions of functionalized nanoparticles with components of complex biological fluids have been identified as only one reason for the dampening, and in some cases even disappearance, of targeting ability and specificity.24–26 Immediately upon exposure of nanoparticle-based agents to a biological environment, macromolecules, such as proteins and lipids, tend to adsorb to their surface and a biomolecular “corona” is formed.27,28 These non-specific binding processes can have a major influence on cellular nanoparticle uptake29,30 as well as on the biorecognition and interaction of surface-grafted targeting moieties with their corresponding receptors.25,26 We stress that this loss of specificity in targeting capacity need not necessarily diminish the overall uptake into cells. This would lead to an inability to discriminate between non-cancerous cells and tumour cells based on receptor profiles. This issue is significant, since avoiding deposition in non-targeted tissues and organs is particularly critical for radiolabelled nanoparticle-based diagnostic agents and other potentially toxic drugs. Different ligands may be affected in different ways by the biological environment, ranging from complete loss of specificity to partial loss. Here we stress the value of preliminary targeting studies in realistic milieu, prior to more extended (for example, in vivo) studies.We begin by comparing the targeting capabilities of two peptides and a small single-domain antibody exemplified by the epidermal growth factor receptor (EGFR). This 170 kDa transmembrane glycoprotein is involved in critical cellular processes such as proliferation, differentiation and apoptosis.31,32 In a variety of solid tumours, including head and neck, breast, non-small-cell lung and glioblastomas, EGFR is constitutively activated as a result of receptor overexpression, mutation or deregulation.33–35 As other members of the ErbB-family, EGFR represents a validated target for anti-cancer therapy.36–39 The current successful approaches include inhibitory antibodies such as Cetuximab and Panitumumab, which prevent EGFR ligands from interacting and activating the receptor as well as receptor–ligand internalisation.40 However, the large size and long half-life of full monoclonal antibodies represent serious disadvantages for the application of monoclonal antibodies in imaging and therapy. They are taken up by various normal tissues, especially accumulating in the liver, and are cleared relatively slowly from the blood stream. Additionally, the diffusion through and penetration into solid tumours is rather poor.41 The optimal probe for multimodal imaging is characterised by fast tissue penetration and rapid circulation clearance as well as high tumour and low liver uptake. Ultrasmall nanoparticles have been proposed to comply with these requirements and thus represent promising next-generation tumour-targeting nanotracers. To maintain their small size, targeting moieties with low spatial dimensions such as peptides, aptamers and antibody fragments are needed.
In the present investigation, the preparation of EGFR-targeted fluorescent silica nanoparticles by conjugation of specific peptides or single-domain antibodies, respectively, is reported. The latter targeting moieties are antagonistic camelid-derived variable domains binding the extracellular domain of the receptor.42,43 Both peptide ligands, GE11 (GYHWYGYTPQNVI)44–47 and D4 (LARLLT),47,48 have been recently reported to bind EGFR-positive cells in vitro and in vivo, GE11 interacts with the EGF binding pocket whereas D4 binds to a distant epitope of the extracellular domain.
Results and discussion
Characterization of synthesised nanoparticles
Fluorescently labelled silica nanoparticles (SiO2) were successfully functionalized with EGFR-specific peptides D4 (SiO2–D4) and GE11 (SiO2–GE11) as well as with the single-domain antibody 7C12 (SiO2–sdAb). The initial amine functionalized nanoparticles consistently displayed a surface density of 6 NH2 per nm2, measured by ninhydrin assay, while bifunctional PEG linkers attached with a density of around 1 SMPEG per nm2, according to thermogravimetric analysis. Bioconjugation was then confirmed, following extensive centrifugal cleaning, by micro BCA protein assay against PEG controls. Characterization of nanoparticle conjugates by dynamic light scattering (DLS) and differential centrifugal sedimentation (DCS) showed a shift in apparent particle size after functionalisation with targeting moieties (Table 1 and Fig. 1). The increase in the hydrodynamic diameter upon peptide/protein conjugation without substantial alteration in the polydispersity indices indicated the formation of relatively monodisperse nanoparticle conjugates.| Nanoparticles | Binding affinity Kd of monomeric ligand | Calculated molecular weight of monomeric ligand | Coupled targeting ligand per mg NPa (est. num./NPa) | DCS Wt distribution mean diameter | DLS Z-ave hydrodynamic diameter DH in water | DLS polydispersity index (PDI) of nanoparticles |
|---|---|---|---|---|---|---|
| a Assuming spherical 53 nm core size. | ||||||
| SiO2 | — | — | — | 53 nm | 66 nm | 0.13 |
| SiO2–sdAb | 2.3–3.7 nM (ref. 42 and 43) | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 984.5 g mol−1 (ref. 49) 984.5 g mol−1 (ref. 49) |
25.0 μg/1.7 nmol (140) | 64 nm | 97 nm | 0.13 |
| SiO2–GE11 | 22.3 nM (ref. 44) | 1540.7 g mol−1 (ref. 44) | 12.6 μg/8.2 nmol (710) | 75 nm | 86 nm | 0.12 |
| SiO2–D4 | n.d. | 685.8 g mol−1 (ref. 48) | 6.4 μg/9.5 nmol (810) | 63 nm | 89 nm | 0.15 |
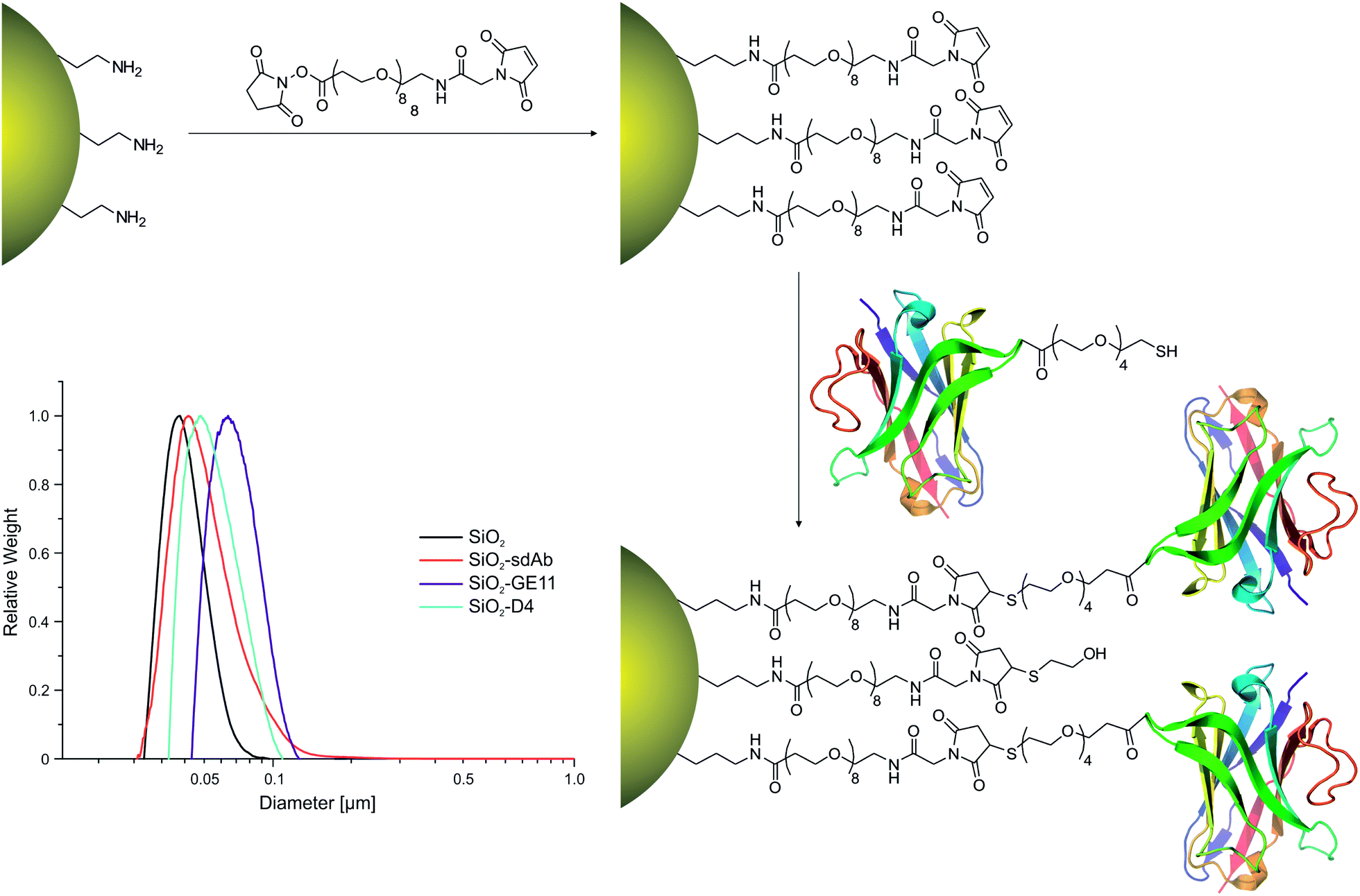 | ||
| Fig. 1 Surface functionalisation of fluorescently labelled silica nanoparticles (SiO2). Fluorescently labelled SiO2 (50 nm) were functionalized with EGFR-affine peptides (D4, GE11) or single-domain antibodies (sdAbs). The insert shows nanoparticle characterisation by differential centrifugal sedimentation (DCS). Black: silica cores, turquoise: SiO2–GE11, purple: SiO2–D4, red: SiO2–sdAbs. | ||
Binding and uptake of fluorescent nanoparticles
In order to investigate EGFR-specific targeting of functionalized nanoparticles, we analysed binding and uptake in the epithelial cell line FaDu originating from a squamous cell carcinoma of the hypopharynx.50 These human head and neck tumour cells express approximately 7 × 105 EGFR molecules per cell, which represents a moderate expression level.51,52 Moreover, RNA interference (RNAi) was used to knockdown the expression of the receptor in these cells to determine the effect of the targeting moieties on nanoparticle uptake. It has been shown recently that the absolute uptake level does not simply give information on the specificity of the targeting moiety on nanoparticles to relevant receptors; however, the difference of particle uptake in silenced and non-silenced cells can be used to indicate the relative contribution made by the specific pathway.26 Two validated small interfering RNA (siRNA) duplexes referred to as siEGFR-1 and siEGFR-2, both targeting different regions of the target mRNA, were separately introduced into FaDu cells. The efficiency of the gene silencing was determined by measuring the reduction of EGFR-encoding mRNA using quantitative real time PCR (Fig. S1A†). Furthermore, the uptake of fluorescently labelled EGF by silenced and non-silenced FaDu cells was analysed by flow cytometry (Fig. S1B†) and confocal microscopy (Fig. S1C and D†). Successful knockdown of EGFR was observed using either of the siRNA duplexes as seen by the reduction of about 90% of mRNA after 48 h post-transfection (Fig. S1A†). In addition, reduction of cell surface located EGFR was confirmed by a decrease in Alexa Fluor® 488-EGF binding by siEGFR-2 silenced FaDu cells from both flow cytometry and confocal microscopy.Cell binding and uptake of peptide functionalized nanoparticles SiO2–D4 and SiO2–GE11 were determined by flow cytometry in EGFR-positive FaDu cells as well as in EGFR-negative MDA-MB-435S cells originally isolated from a ductal adenocarcinoma of the breast (Fig. 2).53
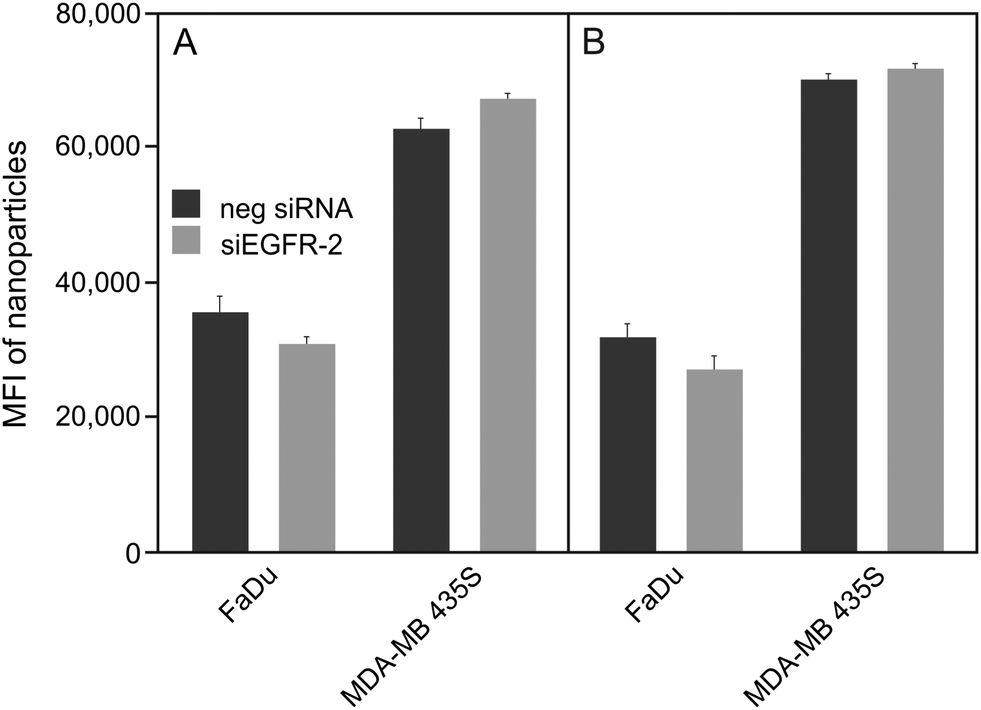 | ||
| Fig. 2 Uptake of peptide-functionalized nanoparticles by different cancer cell lines. EGFR-positive FaDu and EGFR-negative MDA-MB 435S cells were silenced for 48 h with negative silencer control (neg siRNA) and siRNA for EGFR (siEGFR-2) prior to exposure to 100 μg mL−1 SiO2–D4 (A) or SiO2–GE11 (B). Median cell fluorescence intensity was measured by flow cytometry, showing that the uptake is not reduced in cells silenced for EGFR. | ||
For both types of peptide functionalized nanoparticles, a high cellular uptake into EGFR-positive and EGFR-negative cells was observed. This, together with the fact that uptake rates are almost equal between silenced and non-silenced FaDu cells provides evidence that SiO2–D4 as well as SiO2–GE11 were largely not taken up by EGFR-specific pathway. Interestingly, it has been shown previously that GE11 conjugated to cationic polyethylenimine, fluorescein isothiocyanate or polar liposomes showed uptake into EGFR-expressing cells, but no internalization into EGFR-negative cells.44–46 However, Ongarora et al. observed only poor uptake of phthalocyanine–GE11 conjugates, whereas phthalocyanine–D4 derivatives accumulated in different tumour cell lines.47 These partially contradictory outcomes illustrate that the chemical nature of the conjugates and their characteristics such as charge and polarity may have a substantial influence on their specific tumour targeting abilities. Since both peptides, D4 as well as GE11, appeared to be incompatible with the herein utilised nanoparticle platform, single-domain antibodies (sdAbs) representing alternative EGFR-specific targeting moieties were attached to the surface of silica nanoparticles (SiO2–sdAb). Exposure of silenced and non-silenced FaDu cells to sdAb-conjugated nanoparticles reveals substantial disparities in the level of uptake between both cell populations (Fig. 3).
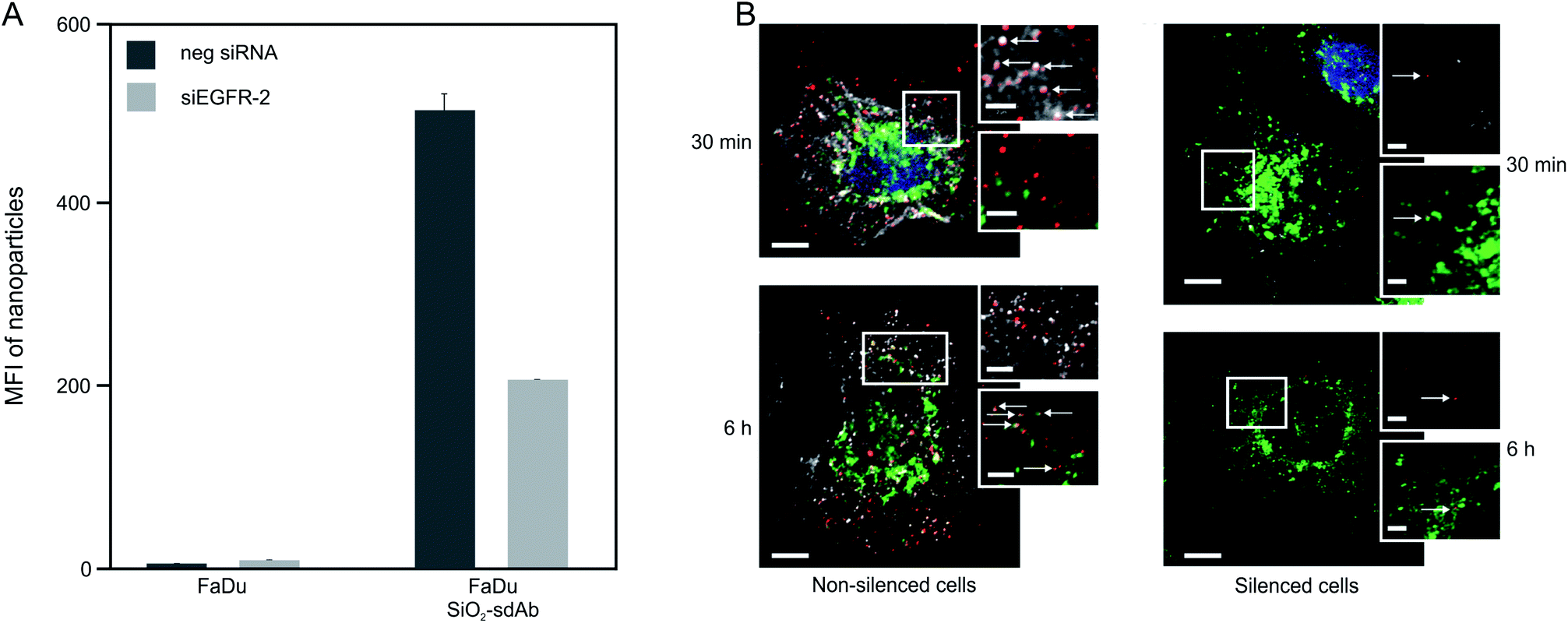 | ||
| Fig. 3 Uptake of sdAb-functionalized nanoparticles by FaDu cells. Median cell fluorescence intensity determined by flow cytometry of FaDu cells exposed to 10 μg mL−1 of SiO2–sdAb showing that the uptake is strongly affected by EGFR knockdown (A). Confocal microscopy images of non-silenced and silenced FaDu cells exposed to SiO2–sdAb nanoparticles for 30 min and 6 h in serum free DMEM (B). Nanoparticles in red, LAMP-1 in green and EGFR in white. Scale bars of 10 μm for the main images and 2 μm for the zoomed images. | ||
Knockdown of EGFR expression leads to a reduction of uptake of about 65% suggesting a predominant receptor dependent binding and internalisation of SiO2–sdAb (Fig. 3A). Moreover, confocal imaging of EGFR-positive FaDu cells shows co-localization of sdAb-conjugated nanoparticles with EGFR after 30 min exposure and internalisation as well as accumulation in the lysosomes after 6 h. Almost no interaction of SiO2–sdAb was observed by confocal microscopy of silenced FaDu cells even after 6 h of exposure (Fig. 3B). Similar results were obtained for the epidermoid carcinoma cell line A431 (Fig. S2†), which is characterised by strong overexpression of EGFR with 1–3 × 106 receptors per cell.54,55 Although these results prove EGFR-specific binding and uptake of sdAb-functionalized silica nanoparticles in buffer or serum-free medium, efficient targeting in more realistic biological environments is an essential prerequisite for later in vivo application. It has been shown recently, that the transfer of nanoparticles into a complex biological environment, e.g. serum, leads to the formation of a dynamic protein corona on the surface of nanoparticles.56,57 These corona components may block the interactions of targeting moieties conjugated to the nanoparticle surface with their putative target and cause a loss of targeting specificity.25,26,58 In order to verify SiO2–sdAb targeting to EGFR of FaDu cells in a biological milieu, we investigated their cellular binding and uptake in presence of different concentration of both human serum and foetal calf serum (Fig. 4). Increasing concentrations of human serum interfere with overall SiO2–sdAb uptake (Fig. 4A), however, the fraction of uptake via EGFR does not decrease substantially (Fig. 4B). In the presence of foetal calf serum, FaDu cells internalise sdAb-functionalized silica nanoparticles to a greater extent compared to human serum. For both sera, the reduction of overall uptake levels can be related the formation of a protein corona.29 To further investigate this, nanoparticles were exposed to 50 mg mL−1 of human serum and the associated biomolecular corona was isolated as described previously.59 As shown in Fig. 5, grafting of a PEG linker interlayer and sdAbs on the surface of nanoparticles obviously reduces the non-specific adsorption of serum proteins. Such a functionalisation strategy has been shown recently to largely but not completely suppress serum protein adsorption.26
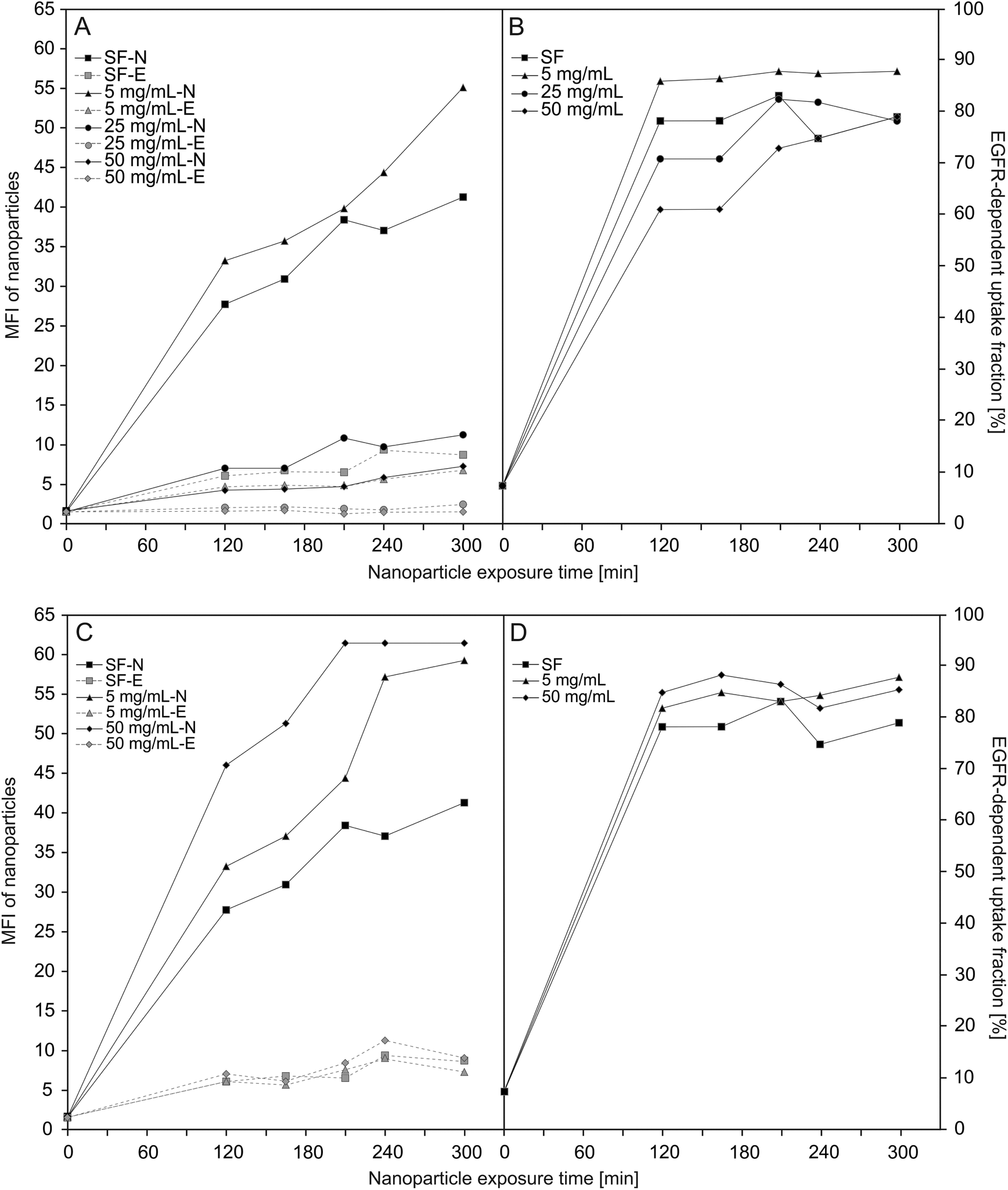 | ||
| Fig. 4 Uptake of SiO2–sdAb in different concentration of human (A/B) and foetal calf (C/D) serum. Median cell fluorescence intensity measured by flow cytometry of silenced (-E) and non-silenced (-N) FaDu cells exposed to 10 μg mL−1 of SiO2–sdAb in serum-free medium (SF) and medium supplemented with human (A) or foetal calf (C) serum, respectively, showing that the uptake is strongly dependent on the present concentration of serum. The EGFR-dependent fractions were calculated using the difference in fluorescence between non-silenced (neg siRNA) and silenced (siEGFR-2) cells divided by the fluorescence of non-silenced cells from the uptake curves in (A) or (C), e.g. ((non-silenced – silenced)/non-silenced). This allows quantifying that, in spite of increasing serum concentrations, the fraction of uptake depending on EGFR remains high (B/D). | ||
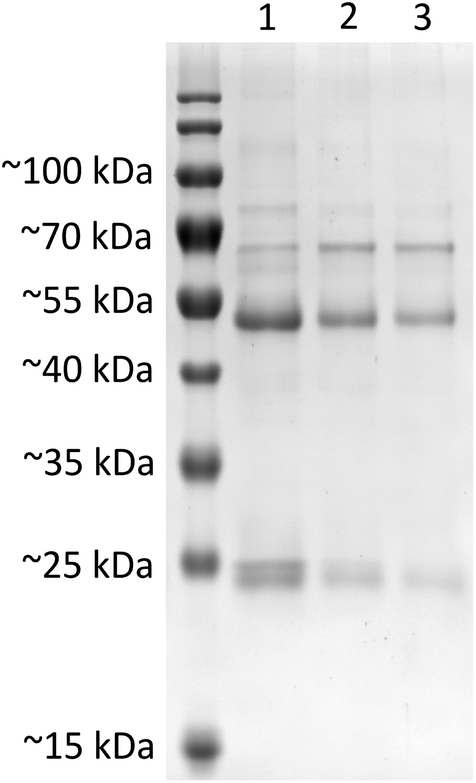 | ||
| Fig. 5 SDS-PAGE analysis of protein corona composition on SiO2 nanoparticles upon incubation in 50 mg mL−1 of human serum. Nanoparticle surface associated proteins were isolated after incubation of SiO2 (lane 1), SiO2–sdAb (lane 2) or SiO2–sdAb–NOTA (lane 3) with 50 mg mL−1 of “off the clot” human serum. Attachment of sdAbs on the surface of nanoparticles obviously reduces the unspecific adsorption of serum proteins, whereas further functionalisation with the copper-64 chelator 1,4,7-triazacyclononane-triacetic acid (NOTA) shows minimal influence on corona composition. | ||
The observed differences in cellular internalisation between human and foetal calf serum in spite of similar protein concentrations might be caused by characteristic components of the particular serum. These include soluble, serum-resident forms of EGFR,60 which bind and block the antigen binding regions of the sdAbs conjugated to silica nanoparticles. Such EGFR analogs lack the cytoplasmic and transmembrane domains of the receptor and originate either from alternative splicing of primary mRNAs or from proteolytic cleavage of full-length EGFR isoforms.61 Also human EGF representing an endogenous competitor for sdAb-mediated EGFR binding of nanoparticle conjugates may contribute to the identified effect, that FaDu cells internalise SiO2–sdAb to a lesser extent in human compared to foetal calf serum.
Characterisation of radiolabelled nanoparticles
The sdAb-functionalized silica nanoparticles were further modified with 1,4,7-triazacyclononane-triacetic acid (NOTA) in order to achieve the attachment of a 64Cu radiolabel for positron emission tomographic (PET) imaging.62,63 Grafting this bifunctional chelator did not affect the biorecognition of EGFR-targeted nanoparticles by FaDu cells, as shown in Fig. 6, where following NOTA conjugation to the corresponding batch uptake behaviour remains unchanged.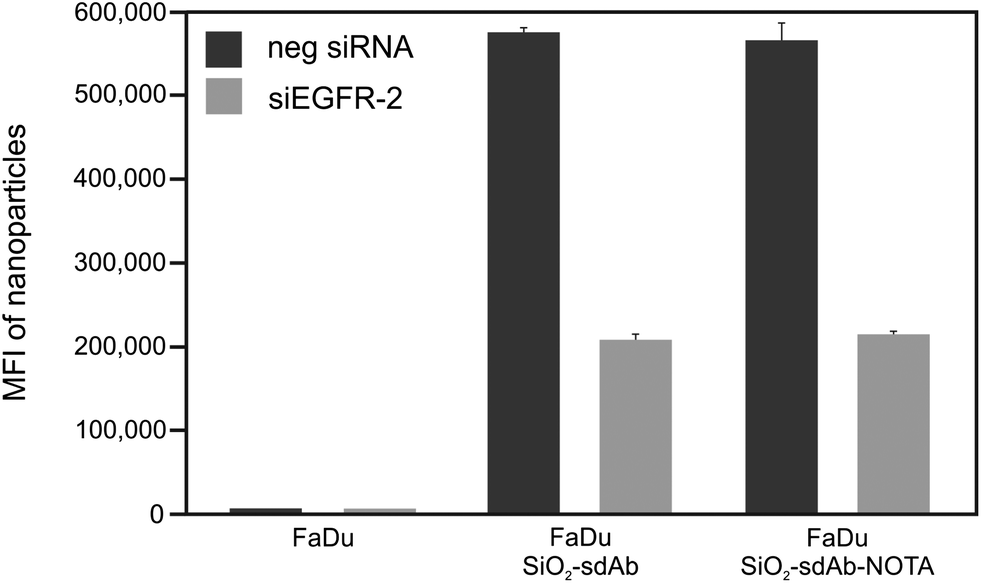 | ||
| Fig. 6 Uptake of SiO2–sdAb and SiO2–sdAb–NOTA by flow cytometry in FaDu cells. Median cell fluorescence intensities determined by flow cytometry of FaDu cells exposed to 10 μg mL−1 SiO2–sdAb or SiO2–sdAb–NOTA, respectively, showing that the uptake is not affected by nanoparticle modification with 1,4,7-triazacyclononane-triacetic acid (NOTA). | ||
Moreover, NOTA-functionalisation of SiO2–sdAb has no influence on the formation of the biomolecular corona (Fig. 5). NOTA-conjugated nanoparticles were radiolabelled by incubation with [64Cu]CuCl2 solution at room temperature for up to 1 h. Within this time period, a radiochemical yield of >98% (as analysed by radio-TLC) was obtained and longer incubation times did not improve the radiochemical yield (Fig. 7A).
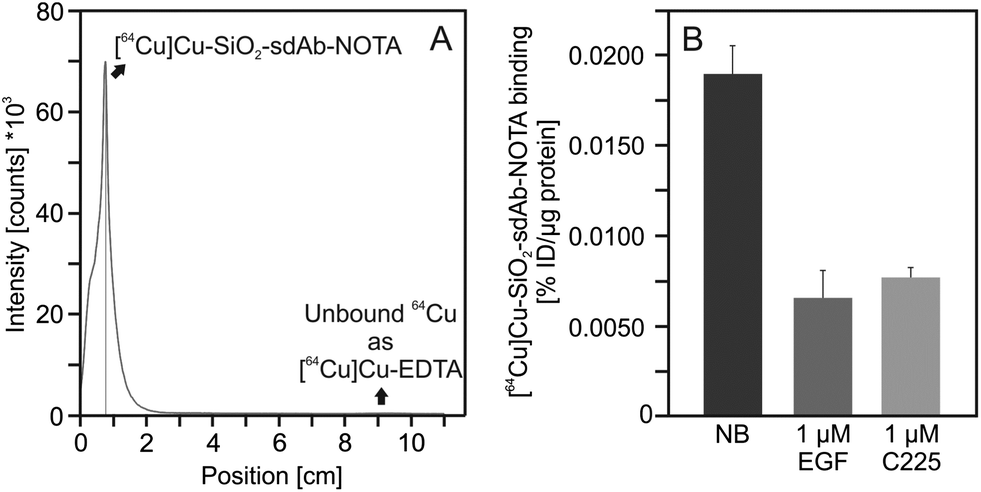 | ||
| Fig. 7 Radiolabelling and cellular binding of SiO2–sdAb–NOTA. After modification with 1,4,7-triazacyclononane-triacetic acid (NOTA), sdAb-functionalized silica nanoparticles were labelled with 64Cu until a radiochemical purity of >98% was obtained as analysed by radio-TLC (A). A 1 μM excess of human epidermal growth factor (EGF) or of the EGFR-inhibitory antibody Cetuximab (C225), respectively, blocks binding of radiolabelled sdAb-functionalized silica nanoparticles to EGFR-presenting FaDu cells. Binding data are expressed as % of injected dose per μg protein (%ID per μg protein). Each point represents the mean ± SD of three samples. | ||
In order to investigate the competition of free human EGF with radiolabelled SiO2–sdAb–NOTA for EGFR binding, we analysed nanoparticle binding to FaDu cells in the presence of an excess of this endogenous ligand (Fig. 7B). Upon incubation of FaDu cells with free human EGF, targeting of SiO2–sdAb–NOTA to EGFR is lost. Furthermore, the therapeutic antibody Cetuximab competes for the binding to EGFR, suggesting that sdAb-functionalized nanoparticles bind epitopes overlapping with or in close proximity to EGF and Cetuximab binding sites. To investigate EGF competition in more detail, we determined cellular binding of radiolabelled SiO2–sdAb–NOTA to FaDu in the presence of increasing EGF concentrations (Fig. 8).
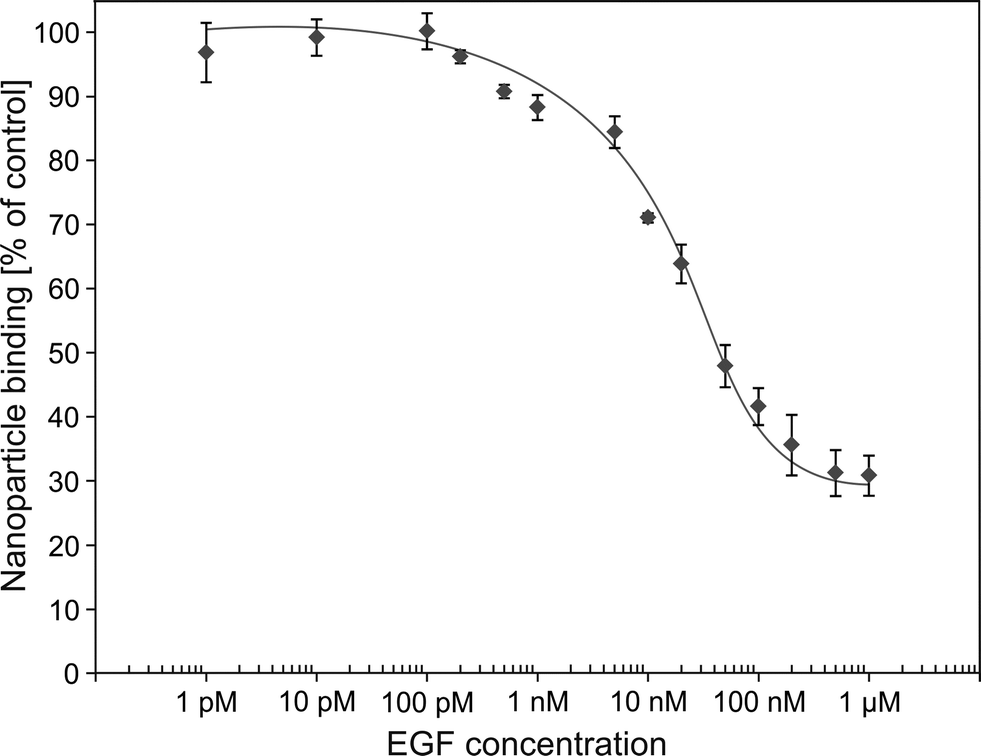 | ||
| Fig. 8 Competition curves of human epidermal growth factor versus [64Cu]Cu–SiO2–sdAb–NOTA using FaDu cells. Binding of radiolabelled sdAb-functionalized silica nanoparticles to EGFR-presenting FaDu cells was investigated in the presence of increasing concentrations of EGF. Percentage of bound activity was calculated in the way that the mean counts of a triplicate data point were related to the counts of data points without competitor. All counts were decay corrected. Each point represents the mean ± SD of three samples. | ||
No reduction of nanoparticle binding was observed up to 200 pM EGF, whereas higher concentrations of the endogenous EGFR ligand substantially decrease receptor-specific nanoparticle interaction. An EGF concentration of 500 nM completely blocks the corresponding receptor and remaining nanoparticle binding occurs by EGFR non-specific nanoparticle–cell interaction. However, at physiological EGF serum concentrations ranging from 10 pM to 190 pM,64,65 no impairment of SiO2–sdAb–NOTA binding to their molecular target was observed. Concentration of EGF in the human serum used here was determined by either dilution of serum (1280 pg mL−1) or by serum spiking (1145 pg mL−1). These values correspond to ∼180 to 200 pM and are in good agreement with EGF levels of other commercially available pooled serum samples (Fig. S3†).
Overall, the presented results clearly illustrate the strong influence of the corresponding biological context on the efficiency of receptor-specific nanoparticle targeting. Recently we have shown that targeting specificity of transferrin-conjugated nanoparticles is lost upon transfer to a complex biological environment. Furthermore, we found that proteins in the cell culture media restrain NP surface bound transferrin from interacting with its receptor.26 The results presented herein confirm these findings, since in both cases we observed that the efficiency of receptor-specific nanoparticle targeting is affected by the biological context. However, for the sdAb–EGFR ligand–receptor pair we see that the specificity is reduced, but not obscured completely. These observations clearly illustrate, that results obtained in biologically irrelevant conditions (e.g. simple buffer systems, serum-free conditions) are not very meaningful. As a minimal prerequisite we suggest to carry out cellular binding and uptake studies in the biological fluids in which the particles will be applied. However, currently no prediction can be made as to if a certain ligand–nanoparticle conjugate maintains its specificity in complex biological context. This means that targeting ability has to be checked for every single ligand–receptor pair.
Experimental
Nanoparticles synthesis
Tetraethyl orthosilicate (TEOS; #86578), (3-aminopropyl)trimethoxysilane (APTMS; #281778), fluorescein isothiocyanate isomer I (FITC; #F7250), rhodamine B isothiocyanate (RITC; #283924), tris(2-carboxyethyl)phosphine hydrochloride (#C4706) were all purchased from Sigma-Aldrich. Succinimidyl-([N-maleimidoproprionamido]-octylethyleneglycol)ester(SM(PEG)8) and N-succinimidyl-S-acetyl(thiotetraethylene glycol) (SAT(PEG)4) were purchased from Thermo Scientific. S-2-(4-Isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (SCN-Bn-NOTA; #B-605) was purchased from Macrocyclics.Dye conjugate solution
N-1-(3-Trimethoxysilylpropyl)-N′-fluoresceyl thiourea (FITC-APTMS) or (RITC-APTMS) conjugate solutions were prepared by dissolving 4 mg of reactive dye in 2 mL of anhydrous ethanol. Twenty μL of APTMS (about 11× molar excess) was then added immediately to this solution, with the mixture then shaken at room temperature in darkness for 4 h. The reaction time course was initially monitored by 1H NMR (CD3OD).Nanoparticle preparation
To 25 mL of EtOH (99.9%) was added 0.91 g of aq. ammonia (28.0–30.0% NH3 basis) in a polypropylene container. To this mixture, under rapid stirring, was added 500 μL of the prepared conjugate solution. The reaction was stirred for 15 min, upon which TEOS (940 μL) was added. The reaction was then stirred at 600 rpm at 25 °C for further 20 h in darkness. The resulting nanoparticle suspension was centrifuged down at 14000 rpm for 20 min, with the pellet then resuspended in fresh EtOH aided by bath sonication. This washing procedure was repeated twice more, followed by three water washes and a final resuspension in water at a total volume of 12 mL.
Surface amination
The FITC–SiO2 particles were suspended in water at a concentration of 10 mg mL−1 and to this suspension APTES was added to a final concentration of 1 vol%. The reaction which proceeded with gradual agglomeration visible, was shaken at 600 rpm for 2 h at room temperature followed by incubation at 90 °C for 1 h. The particles were cleaned by centrifugation and resuspension in water four times, giving a final clear suspension. The number of amines presented at the NP surface was measured by ninhydrin assay. Following centrifugal washing of NPs into pure ethanol (×3) they were then incubated with ninhydrin reagent (0.7 mg mL−1) in absolute ethanol at 60 °C for 30 minutes and measured against APTES standard curves.Protein conjugation to pegylated nanoparticles
To 0.12 μmol of protein (per 10 mg nanoparticles) dissolved at a concentration of 2 mg mL−1 in PBS (pH 7.4) was added SAT(PEG)4 dissolved in dimethyl sulfoxide (DMSO) (76 μL of 1 mg mL−1, 0.18 μmol). After 30 min shaking slowly at room temperature, 100 μL (mL−1 reaction) of deacetylation buffer composed of 0.5 M hydroxylamine and 25 mM ethylenediaminetetraacetic acid (EDTA) in PBS, pH 7.4 was added. The reaction was allowed to continue for 2 h, followed by cleaning on a Sephadex G25 column with exchange into deoxygenated 20 mM HEPES buffer (pH 7.4). The collected protein fraction was then incubated for five minutes with tris(2-carboxyethyl)phosphine (TCEP) (0.24 μmol) before mixing with PEG modified NPs.Nanoparticle pegylation
The aminated particles were washed twice with 20 mM HEPES buffer (pH 7.4) by centrifugation, before resuspension in the same buffer at a concentration of 10 mg mL−1. They were added to an equal volume solution of freshly diluted 5 mg mL−1 SM-PEG8-Mal, which corresponds to around 10× close packed monolayer in 20 mM HEPES (pH 7.4), with mixing. The clear suspension reaction was shaken for 2 h followed by centrifugation at 14000 rpm and two washes with 20 mM HEPES buffer (pH 7.4) and then finally resuspended in deoxygenated 20 mM HEPES buffer (pH 7.4) to a final concentration of 10 mg mL−1 nanoparticles. The work was timed so that the modified protein solution and modified particle dispersion would be ready simultaneously and were then combined in a ratio of 0.12 μmol proteins per 10 mg particles with a nanoparticle reaction concentration of 5 mg mL−1 and shaken gently together for 2 h at RT before incubating at 4 °C overnight. The solution was then cleaned of unreacted protein by centrifugation and resuspension three times in filtered 20 mM HEPES (pH 7.4). The number of bound proteins was measured by micro BCA assay against their corresponding preserved PEG control samples.
Chelator conjugation to nanoparticles
Five mg (8.9 μmol) of SCN-Bn-NOTA was dissolved in DMSO (1000 μL). Seven μL (60 nmol) of this solution was then added to 0.5 mL of NP suspension (5 mg NP, 12.6 nmol sdAb) giving a reaction ratio of approx. 5:1 (reactive macrocycle: sdAb), with immediate mixing by inversion. The dispersion was then slowly shaken for 30 min followed by washing by three cycles of centrifugation (12000 rpm for 15 min) and resuspension in 20 mM HEPES (pH 7.4).
Differential centrifugal sedimentation (DCS) and dynamic light scattering (DLS)
Nanoparticle dispersion was measured by DLS performed on a Malvern Nanosizer ZS. Particles were suspended at a concentration of 100 μg mL−1 in the relevant buffer. Size measurements were averaged results from 3 × 11 runs. DCS experiments were performed with a CPS Disc Centrifuge DC24000 (CPS Instruments). Particles were injected at a concentration of 500 μg mL−1 into a 24–8% sucrose-suspension medium (water or PBS) gradient spinning at 20000 rpm.
Radiolabelling and instant thin-layer chromatography
The production of 64Cu was performed at Cyclone® 18/9 (Helmholtz-Zentrum Dresden-Rossendorf) in a 64Ni(p, n) 64Cu nuclear reaction with specific activities of 150–250 GBq μmol−1 Cu diluted in HCl (10 mM).66 To 100 μg of SiO2–sdAb–NOTA nanoparticles in 100 μL 10 mM MES, pH 6.0, 1 MBq [64Cu]CuCl2 was added and incubated at room temperature for 60 min. A 5 μL aliquot of the reaction was combined with 2 nmol EDTA, pH 7.0 and the labelling process of the nanoparticles (Rf = 0) was monitored by radio-TLC using ITLC-SA plates (Merck Millipore) in combination with a mobile phase of 0.9% NaCl in dH2O. As control, separate radio-TLC analysis of [64Cu]Cu–EDTA (Rf = 1) was performed in the same mobile phase. Evaluation of radio-TLC was carried out using a radioactivity thin layer analyser (Rita Star, Raytest).Heterologous expression and purification of sdAb
Single-domain antibodies were expressed and purified as described recently.49Cell culture
Tissue culture reagents were purchased from Biochrom AG and GIBCO Invitrogen Corporation/Life Technologies Life Sciences unless otherwise specified. The adherent human tumour cell lines A431 (ATCC® number: CRL-1555), FaDu (ATCC® number: HTB-43) and MDA-MB 435S (ATCC® number: HTB-129) were maintained as monolayer cultures in DMEM supplemented with 10% foetal calf serum (FCS), respectively, and incubated in a humidified atmosphere of 95% air/5% CO2 at 37 °C. All cell lines were confirmed to be mycoplasma negative using the LookOut mycoplasma PCR detection kit (Sigma-Aldrich) and were tested monthly.Cell silencing and flow cytometry
A total of 30000 cells were seeded in 24 well plates (Greiner), and incubated for 24 h before silencing of the gene coding for epidermal growth factor receptor (EGFR). Cells were then transfected with 15 pmol of Silencer Select siRNA siEGFR-1 (#s563) or siEGFR-2 (#s564) using Oligofectamine™ according to the manufacturer's instructions (Life Technologies). Neg1 silencer was used as a negative control. Cells were transfected with siRNAs in all experiments 48 h before exposure to nanoparticles or labelled EGF. After 48 h silencing, cells were washed for 10 min in serum-free DMEM. The medium was then replaced by the nanoparticle dispersions, freshly prepared by diluting the nanoparticle stock in serum-free DMEM, or medium supplemented with different concentration of FCS or human serum, for different times, depending on the experiment. Similar experiments were performed by exposing cells to 200 ng mL−1 Alexa Fluor® 488-labelled human EGF in serum-free DMEM. For flow cytometry, cells were washed once with DMEM supplement with 10% FCS and twice with PBS and harvested with trypsin. Cell pellets were then fixed at room temperature with 4% formalin (Sigma-Aldrich) for 20 min, and resuspended in PBS before cell-associated fluorescence (15000 cells per sample) was measured using an Accuri C6 reader (BD Accuri Cytometers). The results are reported as the median of the distribution of cell fluorescence intensity, averaged over two to three independent replicates. Error bars represent the standard deviation between replicates. Each experiment was performed at least three times.
Confocal microscopy
For confocal microscopy, 104 cells were seeded onto 35 mm plates with 15 mm diameter glass coverslips and grown for 24 h prior to silencing. After 48 h silencing, both silenced cells and non-silenced cells (controls) were exposed to fluorescently labelled EGF protein (Alexa Fluor® 488-conjugated, at a concentration of 200 ng mL−1 for 2 h) and to SiO2–sdAb nanoparticles at a concentration of 10 μg mL−1 for 30 min and for 6 h. For organelle and protein staining, samples were then washed three times with 1 mL PBS, fixed for 20 min with 1 mL of 4% formalin at room temperature. The cell-membrane was permeabilised using 1 mL of 0.1% saponin (Sigma Aldrich) solution for 5 min at room temperature and cell were then incubated for 30 min at room temperature with a blocking solution of 1% bovine serum albumin fraction V (Sigma Aldrich) in PBS–Tween to prevent antibody non-specific binding. Samples were then incubated for 1 h at room temperature with a primary antibody 1:200 rabbit polyclonal to LAMP-1 (Abcam) and with a primary antibody 1:200 mouse monoclonal antibody to EGFR (Abcam), washed three times with 1 mL PBS, and then incubated at room temperature for 1 h with 1:400 dilution of Alexa Fluor® 488 goat anti-rabbit IgG and with 1:400 dilution of Alexa Fluor® 647 goat anti-mouse IgG as secondary antibodies (Molecular Probes, Life Technologies). Samples were washed three times with 1 mL PBS and incubated for 5 min with DAPI (Sigma Aldrich) before mounting with MOWIOL (Polysciences Inc.) on slides for imaging. The cells were observed using a Carl Zeiss LSM 510 Meta laser scanning confocal microscope with lasers at 364 nm and long pass filter LP 385 nm (DAPI), 488 nm and band pass filter 505–530 nm (fluorescently labelled EGF protein and LAMP-1 antibody), 543 nm and band pass filter 558–612 nm (nanoparticles) and 633 nm and band pass filter 644–719 nm filter (EGFR antibody).
Serum characterisation
Human serum (Biochrom AG) was tested for total protein content using a bicinchoninic acid (BCA) protein assay (Thermo Scientific). The amount of EGF present in human serum was quantified using a Human EGF ELISA Kit (Invitrogen). The ELISA assay was carried out according to manufacturer's specifications. The absorbance at 450 nm was read using a SpectraMAX 190 plate reader. Two approaches were used and compared in order to determine the concentration of EGF. The first method was carried out by serially diluting serum and examining the levels of EGF quantified for each of the diluted samples. The second approach involved spiking a sample of serum with known amounts of EGF and measuring the response observed in the assay.In vitro binding and uptake studies of radiolabelled SiO2–sdAb–NOTA
A total of 50000 cells were seeded in 24 well plates (Greiner) and cultivated for 24 h before exposure to nanoparticles. After 24 h, cells were washed for twice with warm PBS. The buffer was then replaced by the nanoparticle dispersions, freshly prepared by diluting the radiolabelled nanoparticle stock in serum-free DMEM, or medium supplemented with different concentration of FCS or human serum, for different times, depending on the experiment. Following treatment with radiolabelled nanoparticles for certain time periods, cells were washed twice with PBS in order to ensure removal of loosely attached nanoparticles from the cellular membrane. Finally, cell lysis was achieved by the addition of 1% SDS in 0.1 M NaOH and incubation for 30 min at room temperature with vigorous shaking. The radioactivity in the cell extracts was quantified using an automated gamma counter (PerkinElmer Life and Analytical Sciences). Total protein concentration in cell extracts was determined colorimetrically with the DC Protein Assay (Bio-Rad Laboratories) according to the manufacture's microplate assay protocol using bovine serum albumin as protein standard.
Competition assay
A total of 15000 FaDu cells were seeded in 48 well plates (Greiner) and cultivated for 24 h before exposure to nanoparticles. After 24 h, cells were washed twice with ice-cold PBS and incubated on ice for 30 min. Subsequently, different concentrations of human EGF ranging from 1 pM up to 1 μM as well as 10 μg mL−1 radiolabelled SiO2–sdAb–NOTA were added. After further incubation on ice for 2 h, cells were washed twice with ice-cold PBS, lysed by addition of 1% SDS in 0.1 M NaOH and incubated for 30 min at room temperature with vigorous shaking. The radioactivity in the cell extracts was quantified using an automated gamma counter (PerkinElmer Life and Analytical Sciences).
Isolation and characterisation of nanoparticle–protein complexes
Biomolecular corona forming on silica nanoparticles was isolated as described recently with slight modifications.59 Briefly, samples containing 100 μg mL−1 of SiO2, SiO2–sdAb or SiO2–sdAb–NOTA, respectively, were incubated with 50 mg mL−1 of “off the clot” human serum (Biochrom AG) diluted with dH2O for 1 h at 37 °C in protein LoBind vials (Eppendorf) with significantly reduced protein-to-surface binding. After incubation in serum, samples were centrifuged for 20 min at 10000 × g at 4 °C to pellet the nanoparticle–protein complexes and to remove the supernatant serum. The pellet was then washed three times with 1 mL dH2O and centrifuged again for 20 min at 10000 × g at 4 °C to remove proteins with low affinity for the nanoparticle surface. Before the last centrifugation step, the nanoparticle dispersions were transferred into new vials in order to discard proteins bound to the inner surface of the vials. The nanoparticle–protein pellet was resuspended in Laemmli sample buffer (Bio-Rad Laboratories) immediately after the last centrifugation step and incubated for 5 min at 100 °C to denature the proteins. After cooling to room temperature, the samples were finally loaded on a 12% polyacrylamide gel and subjected to electrophoresis until the bromophenol blue dye of the sample buffer reached the end of the gel. On each gel, one lane was used to separate a molecular weight ladder standard, the PageRuler pre-stained protein ladder (Thermo Fisher Scientific). After electrophoresis, proteins were stained with PageBlue protein staining solution (Thermo Fisher Scientific) according to the manufacturer's instructions.
Conclusions
In conclusion, sufficient specific recognition of targeting ligands grafted to the surface of nanoparticles by their corresponding receptors depends on a variety of factors. These include the binding affinity of the ligated nanoparticle to its molecular target as well as the endogenous competitor concentration, and both factors influence the residence time for a ligand at its receptor binding site. The dissociation constant, which describes how tightly a particular ligand binds to its corresponding target, differs by one order of magnitude between the investigated peptide GE11 and the sdAb 7C12. It is not surprising, then, that the fraction of specific EGFR-mediated cellular uptake is substantially increased for sdAb-functionalized nanoparticles compared to their peptide-conjugated counterparts. However in this case, as for all nanoparticle–cell interaction studies there are a range of variables at play such as colloidal stability related to peptide pI, NP surface self-adsorption effects, etc. precluding direct comparison based on dissociation constants. In this study sdAb functionalized platforms were shown to function well in terms of biological recognition specific interactions. We observed a serum species type dependence in overall NP uptake where matching cell and serum protein for species resulted in the greatest diminution of overall nanoparticle uptake, suggesting the possibility of loss of specificity in situ. Our investigations using EGF competition studies suggest that it may not result mainly from endogenous EGF competition. Nevertheless, the sdAb-functionalized nanoparticles retain sufficient efficiency to remain credible candidates for further consideration.We stress here the key overarching point. There is considerable potential for particles in situ to lose, or at least modulate, their specificity, compared to expectations in simple buffers. Even the differences between human and bovine serum, may be significant and clearly demonstrates the need to choose carefully appropriate experimental conditions and combinations in drawing conclusions from in vitro data. While we are not yet in a position to predict which ligands, and which ligation chemistries and nanoparticles lead to modulation of targeting efficiency, we believe that studies such as those presented here should be a basic prerequisite screen prior to more in depth consideration and in vivo study.
Acknowledgements
We thank Utta Herzog for excellent technical assistance. Financial support by the Helmholtz Virtual Institute NanoTracking (Agreement Number VH-VI-421) is gratefully acknowledged. This study is part of a research initiative “Technologie und Medizin – Multimodale Bildgebung zur Aufklärung des in vivo Verhaltens von polymeren Biomaterialien” of the Helmholtz-Portfoliothema. Financial support through Science Foundation Ireland and the Irish Research Council (IRC) are gratefully acknowledged. The work performed was supported by the IRC through an Enterprise Partnership Postdoctoral Fellowship with Intel (Ireland) (Ref no: EPSPD/2012/443) and the IRCSET EMPOWER Postdoctoral Fellowship Scheme. Experimental method development supported through the QualityNano research infrastructure is acknowledged.Notes and references
- K. Bourzac, Nature, 2012, 491, S58–S60 CrossRef.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- B. Y. Kim, J. T. Rutka and W. C. Chan, N. Engl. J. Med., 2010, 363, 2434–2443 CrossRef CAS PubMed.
- A. H. Faraji and P. Wipf, Bioorg. Med. Chem., 2009, 17, 2950–2962 CrossRef CAS PubMed.
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS PubMed.
- K. Cho, X. Wang, S. Nie, Z. G. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- S. Hirsjarvi, C. Passirani and J. P. Benoit, Curr. Drug Discovery Technol., 2011, 8, 188–196 CrossRef CAS.
- N. T. Huynh, E. Roger, N. Lautram, J. P. Benoit and C. Passirani, Nanomedicine, 2010, 5, 1415–1433 CrossRef CAS PubMed.
- A. Mitra, A. Nan, B. R. Line and H. Ghandehari, Curr. Pharm. Des., 2006, 12, 4729–4749 CrossRef CAS.
- C. Minelli, S. B. Lowe and M. M. Stevens, Small, 2010, 6, 2336–2357 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharm., 2010, 7, 1913–1920 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 1992, 19, 315–324 CrossRef CAS.
- H. Maeda, Adv. Drug Delivery Rev., 2001, 46, 169–185 CrossRef CAS.
- S. P. Egusquiaguirre, M. Igartua, R. M. Hernandez and J. L. Pedraz, Clin. Transl. Oncol., 2012, 14, 83–93 CrossRef CAS PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3–44 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- H. S. Choi and J. V. Frangioni, Mol. Imaging, 2010, 9, 291–310 CAS.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS PubMed.
- V. Mirshafiee, M. Mahmoudi, K. Lou, J. Cheng and M. L. Kraft, Chem. Commun., 2013, 49, 2557–2559 RSC.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. Radomski, K. A. Dawson and C. Aberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- S. R. Hubbard and W. T. Miller, Curr. Opin. Cell Biol., 2007, 19, 117–123 CrossRef CAS PubMed.
- J. Schlessinger, Cell, 2000, 103, 211–225 CrossRef CAS.
- A. Gschwind, O. M. Fischer and A. Ullrich, Nat. Rev. Cancer, 2004, 4, 361–370 CrossRef CAS PubMed.
- T. Holbro, G. Civenni and N. E. Hynes, Exp. Cell Res., 2003, 284, 99–110 CrossRef CAS.
- N. E. Hynes and G. MacDonald, Curr. Opin. Cell Biol., 2009, 21, 177–184 CrossRef CAS PubMed.
- M. Baumann and M. Krause, Radiother. Oncol., 2004, 72, 257–266 CrossRef CAS PubMed.
- M. Baumann, M. Krause, E. Dikomey, K. Dittmann, W. Dorr, U. Kasten-Pisula and H. P. Rodemann, Radiother. Oncol., 2007, 83, 238–248 CrossRef CAS PubMed.
- M. Krause, K. Gurtner, Y. Deuse and M. Baumann, Int. J. Radiat. Biol., 2009, 85, 943–954 CrossRef CAS PubMed.
- D. Zips, M. Krause, A. Yaromina, A. Dorfler, W. Eicheler, C. Schutze, K. Gurtner and M. Baumann, J. Pharm. Pharmacol., 2008, 60, 1019–1028 CrossRef CAS PubMed.
- A. Elbakri, P. N. Nelson and R. O. Abu Odeh, Hum. Immunol., 2010, 71, 1243–1250 CrossRef CAS PubMed.
- C. Van de Wiele, H. Revets and N. Mertens, Q. J. Nucl. Med. Mol. Imag., 2004, 48, 317–325 CAS.
- L. O. Gainkam, L. Huang, V. Caveliers, M. Keyaerts, S. Hernot, I. Vaneycken, C. Vanhove, H. Revets, P. De Baetselier and T. Lahoutte, J. Nucl. Med., 2008, 49, 788–795 CrossRef CAS PubMed.
- L. O. Gainkam, M. Keyaerts, V. Caveliers, N. Devoogdt, C. Vanhove, L. Van Grunsven, S. Muyldermans and T. Lahoutte, Mol. Imag. Biol., 2011, 13, 940–948 CrossRef PubMed.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978–1985 CrossRef CAS PubMed.
- S. Song, D. Liu, J. Peng, Y. Sun, Z. Li, J. R. Gu and Y. Xu, Int. J. Pharm., 2008, 363, 155–161 CrossRef CAS PubMed.
- F. M. Mickler, L. Mockl, N. Ruthardt, M. Ogris, E. Wagner and C. Brauchle, Nano Lett., 2012, 12, 3417–3423 CrossRef CAS PubMed.
- B. G. Ongarora, K. R. Fontenot, X. Hu, I. Sehgal, S. D. Satyanarayana-Jois and M. G. Vicente, J. Med. Chem., 2012, 55, 3725–3738 CrossRef CAS PubMed.
- S. Song, D. Liu, J. Peng, H. Deng, Y. Guo, L. X. Xu, A. D. Miller and Y. Xu, FASEB J., 2009, 23, 1396–1404 CrossRef CAS PubMed.
- K. Zarschler, S. Witecy, F. Kapplusch, C. Foerster and H. Stephan, Microb. Cell Fact., 2013, 12, 97 CrossRef PubMed.
- S. R. Rangan, Cancer, 1972, 29, 117–121 CrossRef CAS.
- I. King and A. C. Sartorelli, Cancer Res., 1989, 49, 5677–5681 CAS.
- J. Y. Song, S. W. Lee, J. P. Hong, S. E. Chang, H. Choe and J. Choi, Cancer Lett., 2009, 283, 135–142 CrossRef CAS PubMed.
- R. Cailleau, M. Olive and Q. V. Cruciger, In Vitro, 1978, 14, 911–915 CrossRef CAS.
- D. J. Giard, S. A. Aaronson, G. J. Todaro, P. Arnstein, J. H. Kersey, H. Dosik and W. P. Parks, J. Natl. Cancer Inst., 1973, 51, 1417–1423 CAS.
- Z. Novy, P. Barta, J. Mandikova, M. Laznicek and F. Trejtnar, Nucl. Med. Biol., 2012, 39, 893–896 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- A. S. Pitek, D. O'Connell, E. Mahon, M. P. Monopoli, F. Baldelli Bombelli and K. A. Dawson, PLoS One, 2012, 7, e40685 CAS.
- K. Pombo-García, K. Zarschler, J. A. Barreto, J. Hesse, L. Spiccia, B. Graham and H. Stephan, RSC Adv., 2013, 3, 22443 RSC.
- A. T. Baron, J. M. Lafky, D. C. Connolly, J. Peoples, D. J. O'Kane, V. J. Suman, C. H. Boardman, K. C. Podratz and N. J. Maihle, J. Immunol. Methods, 1998, 219, 23–43 CrossRef CAS.
- J. M. Lafky, J. A. Wilken, A. T. Baron and N. J. Maihle, Biochim. Biophys. Acta, 2008, 1785, 232–265 CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- V. Maheshwari, J. L. J. Dearling, S. T. Treves and A. B. Packard, Inorg. Chim. Acta, 2012, 393, 318–323 CrossRef CAS PubMed.
- J. Brzezinski and A. Lewinski, Eur. J. Endocrinol., 1998, 138, 388–393 CrossRef CAS PubMed.
- A. Pietrzak, R. Miturski, D. Krasowska, K. Postawski and B. Lecewicz-Torun, J. Eur. Acad. Dermatol. Venereol., 1999, 12, 1–5 CrossRef CAS.
- S. Thieme, M. Walther, H. J. Pietzsch, J. Henniger, S. Preusche, P. Mading and J. Steinbach, Appl. Radiat. Isot., 2012, 70, 602–608 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr00595c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |