Silica–F127 nanohybrid-encapsulated manganese oxide nanoparticles for optimized T1 magnetic resonance relaxivity†
Benedict You
Wei Hsu
a,
Miao
Wang
b,
Yu
Zhang
c,
Vimalan
Vijayaragavan
d,
Siew Yee
Wong
e,
Alex
Yuang-Chi Chang
b,
Kishore Kumar
Bhakoo
d,
Xu
Li
*e and
John
Wang
*ac
aNUS Graduate School for Integrative Sciences and Engineering (NGS), National University of Singapore, 28 Medical Drive, 117456, Singapore. E-mail: msewangj@nus.edu.sg
bJohns Hopkins Singapore International Medical Center, 11 Jalan Tan Tock Seng, Singapore 308433
cDepartment of Materials Science & Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117576
dTranslational Molecular Imaging Group, Singapore Bioimaging Consortium, Agency for Science, Technology and Research (A*STAR), 11 Biopolis Way, 02-02 Helios, Singapore 138667
eInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 3 Research Link, Singapore 117602. E-mail: x-li@imre.a-star.edu.sg
First published on 9th October 2013
Abstract
To properly engineer MnO nanoparticles (MONPs) of high r1 relaxivity, a nanohybrid coating consisting of silica and F127 (PEO106PPO70PEO106) is designed to encapsulate MONPs. Achieved by an interfacial templating scheme, the nanohybrid encapsulating layer is highly permeable and hydrophilic to allow for an optimal access of water molecules to the encapsulated manganese oxide core. Hence, the efficacy of MONPs as MRI contrast agents is significantly improved, as demonstrated by an enhancement of the MR signal measured with a pre-clinical 7.0 T MRI scanner. The nanohybrid encapsulation strategy also confers high colloidal stability to the hydrophobic MONPs by the surface decoration of PEO chains and a small overall diameter (<100 nm) of the PEO–SiO2 nanohybrid-encapsulated MONPs (PEOMSNs). The PEOMSNs are not susceptible to Mn-ion leaching, and their biocompatibility is affirmed by a low toxicity profile. Moreover, these hybrid nanocapsules exhibit a nano-rattle structure, which would favor the facile loading of various therapeutic reagents for theranostic applications.
Introduction
Magnetic resonance imaging (MRI) is a non-invasive medical imaging modality that provides excellent soft tissue contrast for anatomical imaging with high spatial resolution. For T1-weighted MRI, gadolinium-based contrast agents (GBCAs) are commonly used. However, these GBCAs are contraindicated in patients with renal disease or liver transplant due to increased risk of nephrogenic systemic fibrosis (NSF).1 Hence, there has been great impetus for developing alternative MRI T1-contrast agents. In this regard, manganese (Mn) and its derivatives have appeared as promising replacements, owing to the reduced toxicity and comparable positive contrast when compared to Gd3+ ions.2 The seminal publication first describing the use of manganese oxide nanoparticles (MONPs) as T1 contrast agents for MRI was reported by Hyeon and co-workers.3 Compared to commercially available Mn2+ chelates (e.g. Teslascan®) for MRI, it was proposed that MnO nanoparticles would be preferred, as they offer a large payload of MR-active magnetic centers.4 This groundbreaking work has successfully demonstrated the application of MONPs as a T1 contrast agent in the mouse brain. Nevertheless, the maximum specific relaxivity (r1) is restricted by the use of a PEG-phospholipid coating to encapsulate the MONPs, which inevitably reduces the accessibility of water molecules to the MnO core.5,6 Therefore, there has been a strong ongoing emphasis to engineer MONPs and their encapsulation for higher r1 relaxivity.Modifying the particle size and morphology of MONPs has a profound effect on the r1 relaxivity. For example, Huang et al.2 observed that nanoplates showed the largest r1 relaxivity among the different nanostructured morphologies of MONPs. Hyeon et al.3 also showed that the r1 relaxation rate increased as the particle size of MONPs decreased. Shin et al.7 reported that hollow MONPs could exhibit a higher r1 relaxivity than those of their solid counterparts due to a higher surface-to-volume ratio. Nonetheless, it still remains a challenge to develop an ideal nanocoating shell for the encapsulation of these hydrophobic payloads. Ideally, the nanoshell should fulfill the following desirable functions for MRI: (i) render the MONPs biocompatible, water-dispersible and colloidally stable in physiological environment, (ii) be sufficiently thin so that the overall diameter remains between 5 and 100 nm; such a dimension ensures a prolonged blood circulation and reduced opsonization, (iii) allow the encapsulated manganese paramagnetic cores to interact readily with the surrounding water molecules for higher r1 relaxivity, and (iv) prevent Mn-ion leaching in order to avoid toxic accumulation of Mn2+ in tissues. However, to the best of our knowledge, none of the encapsulation strategies used at present has been demonstrated to satisfy all of these criteria.
Although the PEG-phospholipid coating has been the most widely used strategy thus far to functionalize MONPs, its efficiency for water exchange is the subject of much debate.5,6 Similarly, poly-lactic-co-glycolic acid (PLGA) has been used to encapsulate MONPs. However, the intact nanoparticles demonstrated a very low r1 relaxivity of 0.21 mM−1 s−1 under physiological pH.8 It has been argued that the use of a more hydrophilic encapsulation material can potentially increase the exchange rate of water molecules, thereby shortening the T1 relaxation time. As a proof-of-concept, Chen and co-workers5,6 encapsulated MONPs with either human serum albumin (HSA) or polyaspartic acid (PASP). Using a 7.0 T MRI scanner, both coating materials exhibited high r1 relaxivity values of 1.97 and 1.27 mM−1 s−1 respectively. It was however conceded that the HSA encapsulated MONPs lacked colloidal stability due to their tendency to precipitate upon prolonged storage.6 On the other hand, negatively charged PASP coated MONPs exhibit a low blood circulation half-life as the electrostatic interactions would favor the binding of opsonins to the nanoparticle surface.9 Another popular approach for the surface modification involves the use of polymeric ligands conjugated with dopamine moieties to yield highly water-soluble multifunctional MONPs.10,11 A major limitation of this strategy is the susceptibility of the MnO core to degrade in aqueous solution over time. A comparative study has suggested that silica encapsulated MONPs are a better alternative to prevent Mn-ion leaching.12 This approach is supported by Lee and co-workers,13 who demonstrated that among a number of encapsulating materials tested in vitro, silica encapsulation favors the slowest dissolution rate of the MnO core in a PBS buffer solution. Unfortunately, it has been shown that the formation of a silica nanoshell via the conventional reverse (water-in-oil) microemulsion technique often results in a dense silica framework that would limit the accessibility of water molecules to the MnO core.3,12,14 Therefore, mesoporous silica has been proposed as a viable solution to improve the r1 relaxivity.15,16 However, cetyltrimethylammonium bromide (CTAB), which is employed to generate the mesopores, is highly toxic. Therefore, any incomplete removal of this surfactant can impede their use for biomedical applications.
In order to address the various drawbacks of the encapsulation strategies outlined previously for MONPs, we have designed a novel silica–F127 nanohybrid. Firstly, block copolymer F127 (PEO106PPO70PEO106) is chosen due to its high hydrophilicity and proven biocompatibility. Secondly, the incorporation of a nano-silica framework not only serves to reinforce the F127 micellar coating, but also to protect the MnO core from ion leaching. Thirdly, the silica–F127 nanohybrid coating is fabricated by an interfacial templating technique, whereby the sol–gel synthesis of the silica nanoshell is performed at room temperature and without the use of an alkaline catalyst.17 This ensures the formation of a thin and highly permeable silica nanoshell that is perforated by the PEO chains of F127, thereby enhancing the accessibility of water molecules to the MnO core. Fourthly, unlike other designs of silica encapsulated MONPs, our bilayer encapsulated MONPs exhibit a nano-rattle structure, wherein the MnO core is not in direct contact with the peripheral silica nanoshell. Hence, the passage of water molecules to the core interior is not blocked, since the hydroxyl groups on the MnO surface are not involved in the sol–gel process.16 In this paper, we describe a detailed investigation into the encapsulation of MONPs with a novel silica–F127 nanohybrid coating and evaluate its efficacy as a T1 MRI contrast agent.
Experimental section
Materials
Block copolymer Pluronic® F127 (PEO106PPO70PEO106), TMOS (98%), THF, 1-octadecene (technical grade, 90%), manganese(II) tetrahydrate (99%), oleic acid and pyrene were purchased from Sigma Aldrich. CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay MTS was procured from Promega. Fetal bovine serum (FBS) was obtained from Hyclone. Dulbecco's Modified Eagle's Medium (DMEM) containing glucose, sodium pyruvate, L-glutamin, penicillin, and streptomycin were purchased from PAA Laboratories GmbH, Austria. 10× phosphate buffered saline (PBS) was obtained from 1st base and 1× PBS was prepared by diluting the 10× PBS with Milli-Q deionized water. All chemicals were used as received without any further purification.Synthesis of MnO nanoparticles
The preparation procedures for manganese oleate precursor and MnO nanoparticles (MONPs) were described in previously published reports.18 Briefly, MONPs were synthesized by thermal decomposition of a manganese oleate precursor dissolved in 1-octadecene at elevated temperatures. The as-synthesized MONPs were dispersed in THF to produce a stock solution of 20 mg mL−1.Synthesis of PEO–SiO2-encapsulated MONPs (PEOMSNs)
Firstly, 15 mg of F127 was dissolved in 450 μL of THF. 450 μL of the MnO stock solution and 20 μL of TMOS were then added and stirred for 2 h at room temperature to obtain a homogeneous mixture. Next, the mixture solution was slowly injected into 10 g of deionized water while being stirred rapidly. Stirring was continued for an additional 4 days to ensure complete hydrolysis of TMOS. The resulting aqueous dispersion was dialyzed against deionized water to remove any unreacted silica precursors. Empty silica nanocapsules were then removed by centrifuging at 6000 rpm, discarding the supernatant and re-dispersing the nanoparticles in deionized water. The final concentration of purified PEOMSNs was determined to be 61.8 μg Mn mL−1 by inductively coupled plasma atomic emission spectroscopy (ICP-AES).Characterization
Transmission electron microscopy (TEM, JEM-2010F, 200 kV) was employed to study the size and morphology of nanoparticles. Dynamic light scattering (DLS, Malvern Zetasizer Nanoseries) was performed using a HeNe laser (633 nm) to measure the particle size and distribution in solution. X-ray diffraction (XRD, Bruker AXS D8 Advance) was conducted to identify the crystalline phase of MONPs. Phase analyses were carried out under Cu Kα radiation (λ = 1.5406 Å) at a scanning rate of 2θ = 2° min−1 over a range of 2θ = 15–90°. Fourier transform infrared spectroscopy (FT-IR, Varian 3100 Excalibur) and X-ray photoelectron spectroscopy (XPS, ESCALAB 220I-XL Thermo Scientific) were employed to determine the chemical composition of PEOMSNs. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was used to determine the manganese concentration in the as-synthesized PEOMSNs. UV-vis-NIR absorption spectroscopy (Shimadzu UV-3101 PC spectrometer) was utilized to measure the controlled release kinetics of pyrene-encapsulated PEOMSNs.Antifouling study
PEOMSNs were incubated in PBS containing 10% (v/v) FBS at 37 °C in a water bath. Their hydrodynamic sizes were measured by DLS at regular time intervals during the 24 h incubation in order to determine the degree of protein adsorption onto the surface of PEOMSNs.19 For the control, the hydrodynamic size of silica-coated MONPs incubated in a PBS/FBS solution was monitored using DLS. The silica coated MONPs were synthesized by repeated extraction of PEOMSNs with ethanol at 60 °C to remove the F127.Mn2+ ion-leaching experiments
An aqueous PEOMSN solution was first divided into several equal aliquots, each of which was then allowed to incubate at room temperature. At each 5 day interval, one aliquot was selected for ultracentrifugation and the supernatant was analyzed by ICP-AES for Mn content. The likelihood of Mn2+ ion-leaching from the MONPs was quantified by monitoring the variation of Mn concentrations in the supernatants over an extended period of time.3Controlled release experiments
The pyrene-encapsulated PEOMSNs were prepared by the interfacial templating scheme as described above. Briefly, 2 mg of pyrene is dissolved in the homogeneous mixture of MONPs–F127–TMOS–THF prior to injection into the deionized water. The resulting mixture was kept under stirring for four days at room temperature. Excess pyrene molecules were removed by centrifugation at 6000 rpm for 10 min. The as-prepared pyrene-encapsulated PEOMSNs in the supernatant were then transferred to a dialysis tube and immersed in an aqueous media at 37 °C to monitor the release profile of pyrene. At regular time intervals, small aliquots of the aqueous media are subsequently removed for UV-vis measurements and replaced with fresh deionized water.Cell culture and cytotoxicity assay
A MTS assay was performed to assess the cytotoxicity of PEOMSNs. Briefly, MDA-MB-231 cells obtained from American Type Culture Collection (ATCC) were seeded into a 96-well plate at a density of 8000 cells per well in the presence of a growth medium. The culture medium contains completed DMEM with glucose (4.5 g g−1), sodium pyruvate and L-glutamin, 10% FBS, 100 units penicillin and 100 mg ml−1 of streptomycin. Subsequently, various concentrations of PEOMSNs were added to the cells and incubated at 37 °C in 5% CO2. After 24 h, the growth medium was discarded and replaced by a MTS/phenazine methosulfate solution. The plate was further incubated for another 3 h. Cell viability was quantified by using a Benchmark Plus microplate spectrophotometer (Bio-RAD) to measure the optical absorbance of each well at absorbance wavelengths of 490 nm. All samples were assayed in triplicate.MRI relaxivity measurement
MRI relaxivities of the PEOMSNs were evaluated by using a 7-Tesla Bruker Clinscan MRI system. T1 relaxation times were determined by an inversion recovery experiment with a number of TIs (inversion times) (10 TIs; TI: 100–7000 ms; echo times (TE): 12 ms). T2 relaxation times were determined from a multi-echo spin-echo sequence (repetition time (TR): 3000 ms; TE: 6.7–214.4 ms). The longitudinal (r1) and transverse (r2) relaxivities were obtained from the slope of 1/T1 or 1/T2versus molar [Mn] concentration plots.In vitro MRI study
MDA-MB-231 cells were grown in an 8-well chamber at a density of 15000 cells per well by using the same cell culture conditions mentioned above. After 24 h of incubation, the culture medium was replaced by fresh DMEM containing PEOMSNs at various concentrations. After another 24 h of incubation in the presence of the nanoparticles, each well was washed with PBS three times. The cells were then transferred into a 0.2 mL Eppendorf tube and fixed with a 4% paraformaldehyde solution. MR imaging of the MDA-MB-231 cells was then performed by using a 7.0 Tesla MR scanner (Bruker Clinscan system). Prior to imaging, a phantom was prepared by centrifuging the cells in the Eppendorf tube at 400 G for 5 min. Each tube was then placed inside a 50.0 mL Falcon centrifuge tube, which was subsequently filled with a 1% agarose solution. A spin echo sequence was optimized to generate T1-weighted images of the MDA-MB-231 cells, the relevant acquisition parameters being TR/TE = 400/12 ms, number of averages (NA) = 4.Results and discussion
An outline for the synthesis of PEOMSNs is illustrated in Scheme 1. Hydrophobic MONPs with an average particle size of 16.6 nm (σ = 8.6%) were selected for investigation. The phase purity and high crystallinity of MONPs was confirmed by XRD (Fig. S1†), while FT-IR revealed the presence of oleyl surface ligands, which enable the MONPs to remain stable against aggregation and oxidation (Fig. S2†). | ||
| Scheme 1 Schematic illustration of: (a) the synthesis of PEO–SiO2-coated MONPs and (b) 3D-structure of a single MONP encapsulated within the nanohybrid coating. | ||
A TEM image (Fig. 1b) shows that the majority of as-synthesized PEOMSNs are composed of individual MONPs encapsulated within a PEO–SiO2 nano-rattle structure. This nanohybrid coating is carefully engineered by confining the silica (SiO2) shell formation to the core–corona interface of F127 micelles. Hence, the free PEO chains perforate through the silica shell to form an outer periphery, while rendering the MONPs water-dispersible and biocompatible. Although the PEO chains cannot be observed in the conventional TEM image due to their low electron density, their presence on the surface of PEOMSNs is confirmed with DLS, which indicates a hydrodynamic size of 76.4 nm (Fig. S3†).
 | ||
| Fig. 1 TEM images of: (a) ‘naked’ and (b) PEO–SiO2 coated MnO nanoparticles. (c) FT-IR spectra, and (d) XRD patterns of ‘naked’ and PEO–SiO2 encapsulated MnO nanoparticles. | ||
This is larger than the particle size derived from TEM (45 ± 4 nm), because the light scattering measurement includes the contribution of the PEO chains extended in solution.17 In addition, the nano-rattle structure of PEOMSNs was re-confirmed by XPS, which indicated the presence of Si2s, Si2p and the comparatively weaker Mn2p peaks (Fig. S4†). Since XPS is a surface-sensitive technique, it suggests that the MONPs are well-encapsulated within the core interior, thereby resulting in a limited ability of XPS to detect these structures. It also confirms the chemical stability of MONPs because the high resolution XPS scans of the Mn2p region correspond closely to the standard spectra of bulk MnO. Hence, this shows that Mn2+ is not readily oxidised to the higher valence states despite being stored for several days. Formation of the nano-rattle structure is achieved by employing the interfacial templating technique, wherein the silica precursor (TMOS), hydrophobic MONPs and the block copolymer F127 are simultaneously introduced into the aqueous solution. During the micellization process, TMOS and MONPs are first sequestered into the hydrophobic core of F127 micelles. The subsequent hydrolysis and condensation of TMOS in the interfacial region between the PPO core and PEO corona will thus prevent the MnO core from being in direct contact with the peripheral silica nanoshell. Moreover, FT-IR analysis indicates that the vibration band intensities of the oleyl surface ligands decrease significantly upon encapsulation (Fig. 1c). Instead, a strong and broad absorption band appears in the region of 1000–1200 cm−1. This is attributed to the superposition of multiple peaks belonging to both silica and F127: asymmetric O–Si–O stretching vibrations (1097 cm−1) and the typical triplet of intense overlapped bands in the C–O–C stretching region of F127.20 Hence, a successful exchange of the hydrophobic oleate ligands by the nanohybrid F127/SiO2 coating layer in PEOMSNs has taken place. The presence of MONPs in PEOMSNs is also reflected by the absorption bands of the Mn–O bond (623 cm−1, 511 cm−1). This is further verified by the XRD trace of PEOMSNs (Fig. 1d), which shows the evaporation-induced crystalline peaks of F127,20 as well as the (111), (200) and (220) peaks of MnO.
Biocompatibility and biostability are two essential prerequisites for biomedical applications of MRI contrast agents. To evaluate the colloidal stability of PEOMSNs upon systemic administration, we investigated the nanoparticles for their behavior in non-specific protein adsorption and mechanical stability under extreme dilution conditions. The former is demonstrated by incubating PEOMSNs in a phosphate buffered saline (PBS) solution containing 10% fetal bovine serum (FBS) at 37 °C. DLS was then used to monitor the changes in the hydrodynamic diameter of the nanoparticles. Ideally, the PEO segment of our nanohybrid coating layer should enhance water solubility, while reducing non-specific protein adsorption. This is evidenced in Fig. 2a, where it can be observed that there is no significant change in the hydrodynamic diameter of PEOMSNs upon incubation in PBS containing 10% FBS. It also suggests that particle agglomeration is avoided, as the FBS proteins do not readily adhere to the nanoparticles. In contrast, when the PEO corona is removed by ethanol, there is a marked increase in the DLS size after 12 hours of incubation. Therefore, these experimental results demonstrate that the free PEO chains trail on the surface of PEOMSNs, and confer antifouling properties by preventing particle agglomeration due to the non-specific binding to protein. On the other hand, F127 micelles alone did not provide a sufficiently stable dispersion due to their susceptibility to disruption once below the critical micelle concentration (CMC). This is observed in Fig. 2b, which shows a marked increase in the hydrodynamic diameter of F127-encapsulated MONPs when the dilution factor is 2. However, the onset of particle agglomeration for PEOMSNs is significantly reduced until extreme dilution conditions (200 times). This suggests that silica plays an important role in reinforcing the F127 micellar structure and our selection of a silica–F127 nanohybrid coating would favor a synergistic improvement in the colloidal stability of MONPs in physiological environment.
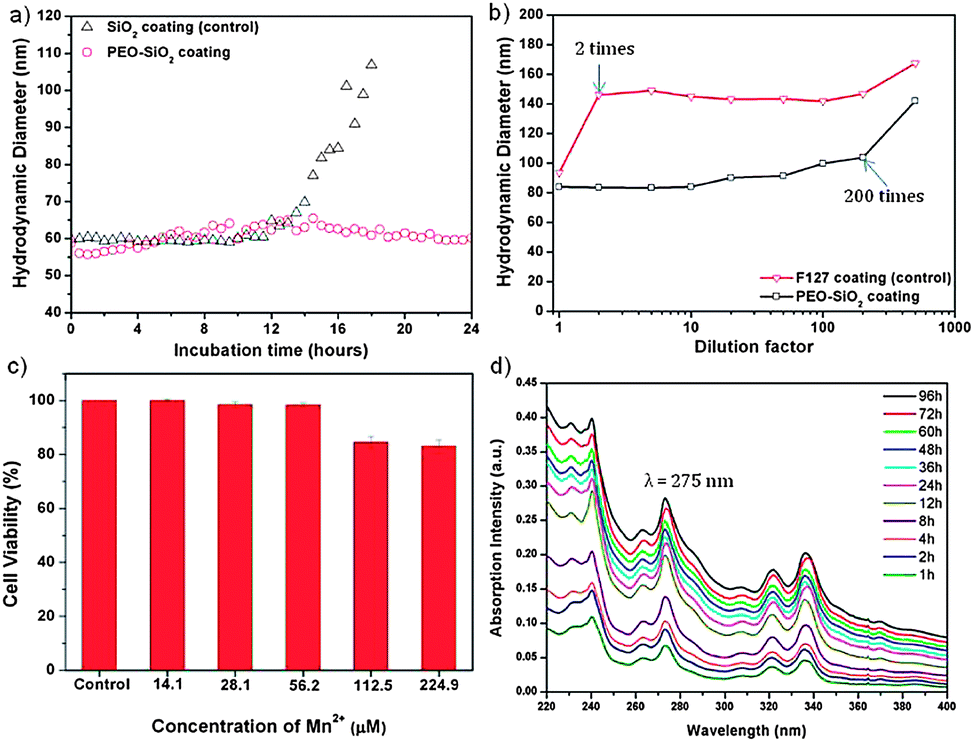 | ||
| Fig. 2 (a) Antifouling behavior. Change in hydrodynamic diameter of nanoparticles measured as a function of time upon incubation in PBS containing 10% FBS at 37 °C. (b) Particle stability under dilution. DLS particle size change as a function of dilution factor. (c) MTS assay of MDA-MB-231 cells. Cells were incubated with different concentrations of PEOMSNs for 24 hours. (d) UV-vis absorption spectra at different times of the aqueous media in which pyrene was released from the pyrene-encapsulated PEOMSNs. | ||
In addition, it is also of great importance to minimize the toxicity of nanoparticles. In this regard, PEOMSNs must not only be non-toxic to cells, the MnO core inside the silica shells should also be stable against Mn-ion leaching. Otherwise, a toxic accumulation of Mn2+ in tissues can result in hepatic failure and cardiac toxicity.4 Hence, in the present study two tests were carried out to evaluate the cytotoxicity of PEOMSNs. Firstly, a MTS assay was performed to evaluate the viability of MDA-MB-231 cells, which were incubated with varying concentrations of PEOMSNs for 24 hours. Fig. 2c shows that PEOMSNs are relatively non-cytotoxic as a high cell viability (83%) can be maintained even at a concentration of 224.9 μM Mn. Secondly, the likelihood of Mn-ion leaching is quantified by monitoring the variation in Mn concentrations of the supernatants after centrifugation at regular 5 day intervals (Fig. S5†). As ICP-AES indicates no substantial increase in the Mn content from the supernatants despite an incubation period of 30 days, it suggests that the highly stable MnO core is not susceptible to appreciable leaching-out over an extended period of time.
A high r1 relaxivity is favored with improved accessibility of water molecules to the paramagnetic MnO core. In this regard, MONPs are able to exert a more efficient water exchange if the silica framework is highly permeable, thin and hydrophilic. Yet, the conventional means to form the silica nanoshell would typically result in a dense silica framework.12,14 In order to demonstrate the highly permeable nature of our nanohybrid encapsulating layer, PEOMSNs are dispersed in an acetate buffer solution (pH 5.0) and stirred for 16 hours to etch away the MnO core. Fig. S6† shows the formation of an interior void in the encapsulated MONPs when subjected to such acidic conditions. This suggests that the acidic solution can penetrate readily through the PEO–SiO2 hybrid coating to bring about a gradual dissolution of MONPs. A hollow interior is subsequently formed due to the preferential dissolution of the MnO core over the peripheral Mn3O4.7 More importantly, it verifies the porosity of the silica shell, which is successfully engineered in this manner via the interfacial templating scheme, whereby the hydrolysis and condensation of silicon alkoxides are confined to the interfacial region between the core and corona of the F127 micelles. As a result, the silica nanoshell formed is perforated with PEO chains, which can act as hydrophilic nanochannels for the rapid penetration of water molecules to the MnO core. Moreover, the interfacial templating scheme is specifically carried out at room temperature and without the use of an alkaline catalyst (e.g. ammonia). This favors the formation of a low-density silica framework that can still remain intact despite a 12 hour acidic immersion. Therefore, MnO payloads encapsulated in the nanocore will not be prematurely released into the surroundings because the PEO–SiO2 nanohybrid coating is chemically stable to moderate pH changes. Pyrene was also selected as a model drug to evaluate the controlled release behavior of PEOMSNs. This was achieved by the simultaneous encapsulation of pyrene and MONPs within the silica–F127 hybrid nanocapsules, which is likely facilitated by the nano-rattle structure of PEOMSNs. The controlled release of pyrene by PEOMSNs was monitored by UV-vis absorption analysis (Fig. 2d). The increasing absorption intensity of the peak at ca. 275 nm indicates that the pyrene content is gradually released into the surrounding aqueous media with time. Therefore, it provides further verification of the highly permeable nature of the silica–F127 nanohybrid coating.
In order to illustrate the enhancement in T1 contrast as afforded by the porosified PEO–SiO2 nanocoating, the relaxation times of an aqueous PEOMSN solution were measured on a pre-clinical 7.0 T MRI scanner. Fig. 3a shows the T1- and T2-weighted phantom MR images at different Mn concentrations. Clearly, its potential to function as a MRI contrast agent is confirmed as the image contrast is significantly enhanced with a minute change in concentration. The specific relaxivities (r1 and r2), derived from the plots of 1/T1 and 1/T2versus Mn concentration (Fig. 3b), were determined to be 1.17 and 30.73 mM−1 s−1, respectively. Compared to other commonly used surface coatings of MONPs, there is indeed an increase in the r1 relaxivity of MONPs if all other factors remain unchanged. For example, r1 values for non-hollow MONPs of 15 nm size are reported to be 0.11, 0.08 and 0.65 mM−1 s−1, when encapsulated with PEG-phospholipid, dense silica and mesoporous silica coatings, respectively.15 For the same concentration range, PEOMSNs developed in the present work are able to exhibit substantial contrast in the T2-weighted images, and thus can function as a suitable T1–T2 dual mode MRI contrast agent. This has apparent advantages in clinical settings, as the T1-weighted images can be used to highlight the anatomy, while the T2-weighted images are more suited for the identification of pathology.21Fig. 3c confirms the effectiveness of PEOMSNs to function as a MRI probe for cell labeling. When the MDA-MB-231 cells were incubated with PEOMSNs, the nanoparticles could be successfully internalized by the cancer cells to give a concentration-dependent signal enhancement in the corresponding T1-weighted images.
 | ||
| Fig. 3 (a) T1- and T2-weighted MR images of PEOMSNs at various Mn concentrations in water. (b) Plot of 1/T1 and 1/T2versus Mn concentration. The slope indicates the corresponding specific relaxivities r1 and r2. (c) T1-weighted MRI images of MDA-MB-231 cells incubated with PEOMSNs at various concentrations for 24 h. | ||
Conclusions
We have designed and successfully developed a novel silica–F127 nanohybrid coating layer to encapsulate MONPs. Following an interfacial templating scheme, the nanohybrid coating layer thus developed consists of a thin and porosified silica nanoshell, which is perforated by the PEO chains of F127 on the surface. Besides a synergistic improvement in the colloidal stability of MONPs, the bilayer, nanohybrid-encapsulated MONPs demonstrate a relatively high T1 relaxivity due to the improved accessibility of water molecules to the MnO nanoparticle core. The biocompatibility of these PEOMSNs is confirmed by a MTS assay, while ion-leaching experiments suggest that they are not liable to Mn-ion leaching. The unique nano-rattle structure of PEOMSNs not only improves the passage of water molecules into the MnO core, it would also allow for the facile loading of therapeutic reagents for theranostic applications, which is demonstrated by the controlled release of encapsulated pyrene as a model drug.Acknowledgements
This work is supported by the Agency for Science, Technology and Research (A-Star, Singapore), BMRC–SERC Joint Grant (Grant number 112 1480002), and NMRC, Ministry of Health, Singapore (Grant: EDG10may036). All the biological experiments were carried out in the research lab of Urology Department and Department of Clinical Research of Singapore General Hospital.Notes and references
- D. Pan, S. D. Caruthers, A. Senpan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 162–173 CrossRef CAS PubMed.
- C. C. Huang, N. H. Khu and C. S. Yeh, Biomaterials, 2010, 31, 4073–4078 CrossRef CAS PubMed.
- H. B. Na, J. H. Lee, K. An, Y. I. Park, M. Park, I. S. Lee, D. H. Nam, S. T. Kim, S. H. Kim, S. W. Kim, K. H. Lim, K. S. Kim, S. O. Kim and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 5397–5401 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- J. Huang, J. Xie, K. Chen, L. Bu, S. Lee, Z. Cheng, X. Li and X. Chen, Chem. Commun., 2010, 46, 6684–6686 RSC.
- R. Xing, F. Zhang, J. Xie, M. Aronova, G. Zhang, N. Guo, X. Huang, X. Sun, G. Liu, L. H. Bryant, A. Bhirde, A. Liang, Y. Hou, R. D. Leapman, S. Sun and X. Chen, Nanoscale, 2011, 3, 4943–4945 RSC.
- J. Shin, R. M. Anisur, M. K. Ko, G. H. Im, J. H. Lee and I. S. Lee, Angew. Chem., Int. Ed., 2009, 48, 321–324 CrossRef CAS PubMed.
- M. F. Bennewitz, T. L. Lobo, M. K. Nkansah, G. Ulas, G. W. Brudvig and E. M. Shapiro, ACS Nano, 2011, 5, 3438–3446 CrossRef CAS PubMed.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- M. I. Shukoor, F. Natalio, M. N. Tahir, M. Wiens, M. Tarantola, H. A. Therese, M. Barz, S. Weber, M. Terekhov, H. C. Schröder, W. E. G. Müller, A. Janshoff, P. Theato, R. Zentel, L. M. Schreiber and W. Tremel, Adv. Funct. Mater., 2009, 19, 3717–3725 CrossRef CAS.
- T. D. Schladt, K. Schneider, M. I. Shukoor, F. Natalio, H. Bauer, M. N. Tahir, S. Weber, L. M. Schreiber, W. E. G. Müller and W. Tremel, J. Mater. Chem., 2010, 20, 8297–8304 RSC.
- T. D. Schladt, K. Koll, S. Prüfer, H. Bauer, F. Natalio, O. Dumele, R. Raidoo, S. Weber, U. Wolfum, L. M. Schreiber, M. P. Radsak, H. Schild and W. Tremel, J. Mater. Chem., 2012, 22, 9253–9262 RSC.
- Y. C. Lee, D. Y. Chen, S. J. Dodd, N. Bouraoud, A. P. Koretsky and K. M. Krishnan, Biomaterials, 2012, 33, 3560–3567 CrossRef CAS PubMed.
- H. Yang, Y. Zhuang, H. Hu, X. Du, C. Zhang, X. Shi, H. Wu and S. Yang, Adv. Funct. Mater., 2010, 20, 1733–1741 CrossRef CAS.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Q. Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955–2961 CrossRef CAS PubMed.
- Y. K. Peng, C. W. Lai, C. L. Liu, H. C. Chen, Y. H. Hsiao, W. L. Liu, K. C. Tang, Y. Chi, J. K. Hsiao, K. E. Lim, H. E. Liao, J. J. Shyue and P. T. Chou, ACS Nano, 2011, 5, 4177–4187 CrossRef CAS PubMed.
- H. Tan, N. S. Liu, B. He, S. Y. Wong, Z. K. Chen, X. Li and J. Wang, Chem. Commun., 2009, 6240–6242 RSC.
- T. D. Schladt, T. Graf and W. Tremel, Chem. Mater., 2009, 21, 3183–3190 CrossRef CAS.
- H. Tan, J. M. Xue, B. Shuter, X. Li and J. Wang, Adv. Funct. Mater., 2010, 20, 722–731 CrossRef CAS.
- P. Innocenzi, L. Malfatti, M. Piccinini and A. Marcelli, J. Phys. Chem. A, 2010, 114, 304–308 CrossRef CAS PubMed.
- K. Armoogum, Magnetic Resonance Imaging IPEM Training Portfolio Chapter 6-Clinical Applications, http://www.medicalphysicist.co.uk/mriportfolio.htm, accessed 06/06/13.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c3nr04378a |
| This journal is © The Royal Society of Chemistry 2014 |