DOI:
10.1039/C3NR03886F
(Paper)
Nanoscale, 2014,
6, 385-391
Resonance energy transfer between fluorescent BSA protected Au nanoclusters and organic fluorophores†
Received
26th July 2013
, Accepted 14th October 2013
First published on 17th October 2013
Abstract
Bovine serum albumin (BSA) protected nanoclusters (Au and Ag) represent a group of nanomaterials that holds great promise in biophysical applications due to their unique fluorescence properties and lack of toxicity. These metal nanoclusters have utility in a variety of disciplines including catalysis, biosensing, photonics, imaging and molecular electronics. However, they suffer from several disadvantages such as low fluorescence quantum efficiency (typically near 6%) and broad emission spectrum (540 nm to 800 nm). We describe an approach to enhance the apparent brightness of BSA Au clusters by linking them with a high extinction donor organic dye pacific blue (PB). In this conjugate PB acts as a donor to BSA Au clusters and enhances its brightness by resonance energy transfer (RET). We found that the emission of BSA Au clusters can be enhanced by a magnitude of two-fold by resonance energy transfer (RET) from the high extinction donor PB, and BSA Au clusters can act as an acceptor to nanosecond lifetime organic dyes. By pumping the BSA Au clusters using a high extinction donor, one can increase the effective brightness of less bright fluorophores like BSA Au clusters. Moreover, we prepared another conjugate of BSA Au clusters with the near infrared (NIR) dye Dylight 750 (Dy750), where BSA Au clusters act as a donor to Dy750. We observed that BSA Au clusters can function as a donor, showing 46% transfer efficiency to the NIR dye Dy750 with a long lifetime component in the acceptor decay through RET. Such RET-based probes can be used to prevent the problems of a broad emission spectrum associated with the BSA Au clusters. Moreover, transferring energy from BSA Au clusters to Dy750 will result in a RET probe with a narrow emission spectrum and long lifetime component which can be utilized in imaging applications.
Introduction
Considerable research efforts have been directed towards bioconjugated nanomaterials due to their possible applications in fluorescence tagging, imaging, diagnostics and multiplexed assays. The demand for novel materials has led to the synthesis of materials like quantum dots (QDs). Despite great potential in biomedical and optoelectronic applications, the use of QDs is limited by cytotoxicity and low biocompatibility in their unprotected form.1–3 This has further strengthened the pursuit for luminescent materials with biocompatible properties. Metal nanoclusters (Au, Ag and Cu) are one such group of nanomaterials which show immense potential for biophysical applications owing to their distinctive fluorescence properties and lack of toxicity.4–8 Unlike nanoparticles, clusters possess molecule-like absorption and luminescence properties due to their sub-nanometer size and discrete energy levels. Hence their electronic properties are different from those of plasmonic nanostructures.
The strong and tunable fluorescence of the protein-stabilized Au nanoclusters (Au NCs) in the visible spectrum has made them attractive targets for biochemical studies. Proteins with differing molecular weights and amino acid sequences have been used as a template to synthesize fluorescent metal nanoclusters, namely bovine serum albumin (BSA), human serum albumin (HSA), lysozyme, trypsin and the ferritin family of proteins.9–12 Among these, BSA protected clusters appear to be a well researched nanocluster preparation. Xie et al. used BSA when they introduced a facile protein directed synthesis, which was followed by many research groups for other protein–Au NC systems.13 These metal nanoclusters have vast applications in diverse fields – including catalysis, biosensing, photonics, imaging and molecular electronics.14–18 However, they have some drawbacks such as low quantum efficiency (6%) and a broad emission spectrum spread over 530 to 800 nm.
These disadvantages can be circumvented by creating resonance energy transfer (RET) probes of BSA Au clusters with organic dyes. In this study, we have prepared two different conjugates of BSA Au clusters with organic dyes. In the first one, pacific blue (PB) is conjugated to BSA Au clusters where PB acts as a donor to the BSA Au cluster. The choice of PB was based on properties such as good fluorescence quantum yield, extinction maximum, absorption suitable for 405 nm diode laser excitation and good spectral overlap with the BSA Au cluster absorption spectrum. The second one was made by conjugating the near infrared dye Dy750 to the BSA Au cluster where, the long lifetime BSA Au cluster acts as a donor to Dy750. Dy750 has several properties that make it a good acceptor. It does not significantly absorb at the chosen excitation wavelength, provides good spectral overlap with BSA Au cluster emission, the Dy750 emission peak is separated from BSA Au cluster emission maxima, and lastly, it is a good near infrared (NIR) probe with all the attributes of a NIR fluorophore such as emission around 800 nm where there is very less auto-fluorescence background.
The interaction of organic fluorophores that results in fluorescence resonance energy transfer (FRET) has tremendous use in bioanalytical applications. The FRET technique is widely used in investigating the structural dynamics of the biomolecules such as single molecule protein folding, DNA hybridization and DNA cleavage.19,20 Analytical methods using FRET usually have higher sensitivity and are simpler to use in studying drug–receptor binding.21 These kinds of applications can be explored for BSA Au clusters, provided their ability to behave as an acceptor or a donor in the presence of suitable organic dyes. The work laid out in this manuscript will answer the following questions about BSA Au clusters: (1) can they be used as an acceptor from nanosecond lifetime probes and if we can, will it lead to an increase in the apparent brightness of BSA Au clusters? (2) Can they be used as donors to develop long lifetime, long wavelength RET probes?
Experimental section
Synthesis of BSA Au nanoclusters
The Au NCs used in this study were synthesized using an approach developed by Xie et al.13 Typically, 5 mL of 10 mM HAuCl4 was mixed with 5 mL of 50 mg mL−1 BSA followed by addition of 1 mL 1 M NaOH and kept at 37 °C overnight in the incubator. The light brown solution of clusters was further dialyzed (2000 MWCO membrane) against de-ionized water for a minimum of 12 h with periodic changes of water to remove any small impurities. Dialyzed cluster solution was filtered using a 0.02 μm syringe filter and used for subsequent measurements.
Synthesis of fluorophore–BSA Au cluster conjugates
Pacific blue–BSA Au cluster.
Typically, micro-molar solution of BSA Au clusters in 0.1 M bicarbonate buffer was reacted with 1.5 fold excess (in relative to BSA) of reactive pacific blue overnight at 4 °C. On the next day, the resulting labeled BSA Au clusters were purified using a pre-packed Sephadex G-25 column to remove free pacific blue. Also, pacific blue was conjugated to native BSA using the above protocol as a control, donor only sample.
BSA Au cluster–Dy750.
Typically, micro-molar solution of BSA Au clusters in 0.1 M bicarbonate buffer was reacted with 2 fold excess (in relative to BSA) of reactive Dy750 overnight at 4 °C. On the next day, the resulting labeled BSA Au clusters were purified using a pre-packed Sephadex G-25 column to remove free pacific blue. Dy750 was conjugated to native BSA using the above mentioned protocol as a control acceptor sample.
Spectroscopic measurements
UV-Vis absorption and fluorescence spectra were obtained using a Cary 50 bio UV-visible Spectrophotometer (Varian Inc.) and Cary Eclipse Spectrofluorometer (Varian Inc.) respectively. All the measurements were done in 1 cm × 0.5 cm cuvettes at room temperature. Fluorescence lifetime was measured on a FluoTime 200 fluorometer (PicoQuant, Inc.) using PicoQuant diode lasers 405 and 470 nm. The fluorometer is equipped with an ultrafast microchannel plate detector (MCP) from Hamamatsu, Inc. The fluorescence lifetimes were measured in the magic angle condition and data analyzed using FluoFit4 program from PicoQuant, Inc. (Germany) using a multi-exponential fitting model:| |  | (1) |
where αi is the amplitude of the decay of the ith component at time t and τi is the lifetime of the ith component. The intensity weighted average lifetime (τAvg) was calculated using the following equation:| |  | (2) |
Total photon counts were calculated using integration of fluorescence intensity decays obtained from time resolved fluorescence measurements with Origin graphing software version 8.0. Forster distances for each pair were calculated using the following equation given by Lackowicz, 1999.22
| | | R0 = 9.79 × (κ2n−4QDJ)1/6 | (3) |
where
κ2 is a factor describing the relative orientation in space of the transition dipoles of the donor and acceptor,
n is the refractive index of the medium,
QD is the fluorescence quantum yield of the donor in the absence of the acceptor, and
J is the overlap integral expressing the degree of spectral overlap between the donor emission and acceptor absorption (in M
−1 cm
3). The transfer efficiencies were calculated using steady state data with the following equation:
where
F is the net integral of the emission of the donor in the presence of the acceptor and
F0 is the integral in the absence of the acceptor. Moreover, time resolved data were also used to calculate the transfer efficiencies using the following equation:
where
τ is the fluorescence lifetime of the donor in the presence of the acceptor and
τ0 is the fluorescence lifetime of the donor in the absence of the acceptor.
Results
Pacific blue–BSA Au clusters
Similar to metal–ligand complexes (MLC), BSA Au clusters possess a low extinction coefficient with a low fluorescence quantum yield. These shortcomings of the nanoclusters can be avoided using the BSA Au cluster as an acceptor to a high fluorescence quantum yield short wavelength donor. As shown in Scheme 1, we assume that even a small fraction of energy transferred from a high extinction donor will enhance the emission of long lifetime BSA Au clusters. Steady state fluorescence measurements were carried out on pacific blue conjugated BSA (PB–BSA), BSA Au clusters (BSA Au) and pacific blue conjugated BSA Au clusters (PB–BSA Au) to confirm if any energy transfer occurred. Fig. 1 shows the normalized spectral overlap of pacific blue emission and BSA Au clusters absorption indicative of possible energy transfer. Here, we have showed deconvoluted BSA Au absorption instead of its extinction spectrum. In our earlier report, we had explained the deconvolution procedure to get the real absorption of BSA Au clusters.23 The Forster distance, R0, using an n value equal to 1.33, donor fluorescence quantum yield of 0.5 and assuming K2 = 2/3 was calculated to be 32.3 Å from the acceptor molar extinction of 7000 M−1 cm−1. ESI Fig. 1† shows the absorption of PB–BSA, BSA Au clusters and PB–BSA Au clusters. Adequate care was taken to match the absorption of controls (PB–BSA and BSA Au clusters) with the absorption of PB–BSA Au clusters. The absorption spectrum of PB–BSA Au clusters closely approaches the sum of individual spectra of PB–BSA and BSA Au clusters. The steady state emission measurements were done using 405 nm excitation. In Fig. 2A, the magenta line shows the emission spectrum of BSA Au clusters (barely visible), the blue spectrum is an emission of PB–BSA and the green line shows the emission spectrum of PB–BSA Au clusters. Fig. 2B shows a magnified portion of Fig. 2A in the wavelength region between 580 nm and 800 nm. We see moderate quenching (12% change) of the donor (PB) and enhanced emission of clusters around 660 nm. There is increased emission intensity in the cluster emission region compared to the acceptor alone sample indicative of energy transferred from PB.
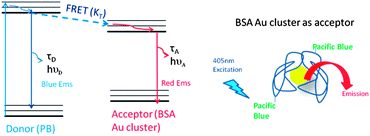 |
| | Scheme 1 Jablonski diagram (left) and cartoon for FRET occurring from pacific blue (donor) to the BSA Au cluster (acceptor). | |
 |
| | Fig. 1 Spectral overlap (marked by horizontal lines) between the donor pacific blue emission (dashed blue line) spectrum and acceptor BSA Au cluster absorption (solid magenta line). | |
 |
| | Fig. 2 (A) Steady state emission spectra of PB–BSA (blue), BSA Au clusters (magenta) and PB–BSA Au clusters (green). (B) Enlarged portion (580–800 nm) of plot A marked by a brown rectangle. | |
Furthermore, time resolved measurements were done in order to confirm the changes in steady state emission. Fig. 3 shows the fluorescence intensity decays of pacific blue on BSA as donor only and pacific blue conjugated to BSA Au clusters as a donor in the presence of the acceptor. During lifetime measurements, 405 nm was used as the excitation wavelength and emission was recorded at 450 nm. The lifetime change was moderate too. For donor only it was 2.4 ns and it changed to 2.3 ns in the presence of the acceptor. In addition, to confirm if this moderate quenching of the donor resulted in enhanced acceptor emission or not, we measured the number of photons collected in the acceptor region at 660 nm. The decays of the directly excited acceptors are short and emission from the directly excited acceptors will not be observed if the detection is off-gated for the first 30 ns after the excitation pulse. To our surprise, our hypothesis of energy transfer from pacific blue to BSA Au clusters proved right when we looked at the number of photons collected at 660 nm; Fig. 4 shows the fluorescence intensity decays and number of photons collected (inset graph) for pacific blue conjugated BSA Au cluster and the pacific blue on BSA sample. The D–A pair shows 2.3 times more photon or enhanced emission than the BSA Au clusters sample alone. The efficiency of transfer is 0.12 calculated by eqn (4). However, transfer efficiency calculated from time resolved data is 0.05 and differs from that of steady state data. Possible static quenching of PB in the presence of the BSA Au cluster could be a possible explanation for this discrepancy and is visible in Fig. 4, as the lower peak counts for the PB–BSA Au sample. The approximate distance between the donor and acceptor that we calculated is 52.4 Å in this case. We believe only a small population of pacific blue on BSA Au clusters is involved in energy transfer due to the heterogeneous nature of the system. The limitation of our study is that pacific blue labelling efficiency on the BSA Au cluster was low (1 PB per BSA molecule in the PB–BSA Au cluster sample) and can further be increased and optimized to enhance the effective brightness of BSA Au clusters manifold using resonance energy transfer (RET).
 |
| | Fig. 3 Fluorescence intensity decay of PB–BSA (blue) as donor only and PB–BSA Au clusters (green) as a donor in the presence of the acceptor. The excitation used was 405 nm and the observation wavelength was 450 nm. | |
 |
| | Fig. 4 Fluorescence intensity decay of BSA Au clusters (magenta) and PB–BSA Au clusters (green) when excited using 405 nm and observed at 700 nm with 30 ns delay to remove direct PB contribution. The inset shows the comparison of the number of photons emitted suggesting increased photon output at a longer wavelength in the presence of the high extinction donor. | |
BSA Au cluster–Dy750
In order to test our hypothesis that BSA Au clusters can be used as the donor despite low extinction, long wavelength Dy750 was conjugated to BSA Au clusters. Scheme 2 shows the Jablonski diagram showing FRET between the BSA Au cluster and Dy750 along with an illustration of BSA Au cluster conjugated Dy750. Fig. 5 shows the spectral overlap between the BSA Au cluster emission and Dy750 absorption spectrum. Donor emission significantly overlaps with the acceptor absorption and shows high possibility of energy transfer between them. Next, we measured the absorption of Dy750 labelled native BSA (red), BSA Au cluster (magenta) and Dy750 conjugated BSA Au cluster (green). Consistent with the previous case, absorption of the D–A pair was matched with that of control samples (ESI Fig. 2†). The absorption spectrum of Dy750–BSA Au clusters closely approaches the sum of individual spectra of Dy750–BSA and BSA Au clusters. For resonance energy transfer studies, we obtained the Forster distance, R0, by means of the spectral overlap based on eqn (3). Using n equal to 1.33, donor fluorescence quantum yield of 0.06 and assuming K2 = 2/3, R0 was calculated to be 51.3 Å from the acceptor molar extinction of 220![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. To record the steady state emission, samples were excited at 470 nm. Fig. 6 shows all the emission spectra collected. The red line shows the emission for Dy750 labelled native BSA (A only), the magenta line shows the emission spectrum of the BSA Au cluster (D only) and the green line shows the Dy750 conjugated BSA Au clusters (D–A pair). The presence of energy transfer shows increased acceptor emission intensity compared to the acceptor only sample and decreased donor emission intensity (∼50% change).
000 M−1 cm−1. To record the steady state emission, samples were excited at 470 nm. Fig. 6 shows all the emission spectra collected. The red line shows the emission for Dy750 labelled native BSA (A only), the magenta line shows the emission spectrum of the BSA Au cluster (D only) and the green line shows the Dy750 conjugated BSA Au clusters (D–A pair). The presence of energy transfer shows increased acceptor emission intensity compared to the acceptor only sample and decreased donor emission intensity (∼50% change).
 |
| | Scheme 2 Jablonski diagram (left) and cartoon for FRET occurring from the BSA Au cluster (donor) to NIR dye Dy750 (acceptor). | |
 |
| | Fig. 5 Spectral overlap (marked by horizontal lines) between the donor BSA Au cluster emission (dashed magenta line) spectrum and Dy750 absorption (solid red line) spectrum. | |
 |
| | Fig. 6 Steady state emission spectrum of Dy750–BSA (red), BSA Au clusters (magenta) and Dy750–BSA Au clusters (green) when excited at 470 nm. A shoulder around 710 nm is due to Wood's effect. Left and right arrows show decrease in donor emission and enhanced acceptor emission respectively in the D–A pair. | |
Time resolved measurements further confirmed the steady state changes. All the samples were excited at 470 nm and donor emission was observed at 660 nm. Fig. 7 shows the fluorescence intensity decay of the BSA Au cluster (donor) and Dy750 conjugated BSA Au cluster (donor + acceptor). The donor only sample had an average lifetime of 1.4 μs. However, in the presence of the acceptor, it decreased to 0.8 μs confirming the steady state emission data. The decay of the directly excited Dy750 is short and even little emission from the directly excited acceptors will not be observed if the detection is off-gated for the first 10 ns after the excitation pulse. So when we compared the photons in the acceptor channel, the D–A pair showed a moderately higher number of photons than the donor alone sample (data not shown here). It is clear from the examination of the D–A decay in Fig. 8 that the energy transfer took place on a very short time scale. The efficiency of the transfer is 0.46 as calculated by using steady state data. Moreover, the efficiency calculated using time resolved data is 0.43 and closely matches with steady state data. The approximate distance between the donor and acceptor that we calculated is 52.5 Å for this pair. Lastly, the presence of a long lifetime component in the acceptor decay is an important finding from an imaging point of view, as it can be used to off-gate the short-lived auto-fluorescence background.
 |
| | Fig. 7 Fluorescence intensity decay of BSA Au clusters (magenta) as donor only and Dy750–BSA Au clusters (green) as the donor in the presence of the acceptor. The excitation used was 470 nm and the observation wavelength was 660 nm with 10 ns delay to avoid direct excited contribution from Dy750. | |
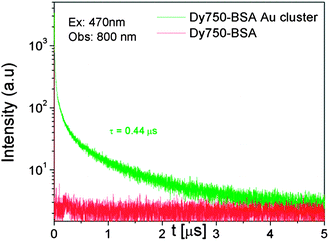 |
| | Fig. 8 Fluorescence intensity decay of Dy750–BSA Au clusters (green) and Dy750–BSA (red) when excited using 470 nm and observed at 800 nm with 10 ns delay. Dy750 in the presence of the BSA Au cluster shows a long decay time. | |
Discussion
Several studies have investigated the interaction of gold nanoparticles with BSA, HSA and other organic fluorophores.24–26 The ability of gold to quench the intrinsic tryptophan in proteins and other organic fluorophores has also been well documented.26–28 However, the interaction of BSA Au clusters with organic dyes is yet unexplored. While Liang et al. have reported the quenching of CdTe quantum dots of different diameters by BSA gold clusters,29 Liu et al. conjugated the oligomer-substituted polyhedral oligomeric silsesquioxane (POSSFF) as a donor to BSA Au clusters for use in intracellular mercury ion sensing.30 The results of our experiments showed that BSA gold nanoclusters can interact with organic fluorophores to behave as resonance energy donor and acceptor as well.
Although metal nanoclusters are useful fluorophores, they suffer from several disadvantages such as low quantum efficiency typically near 6% to 10% and a broad emission spectrum. The broad emission spectrum results in considerable overlap of emission spectra of different fluorophores and renders them incapable of being used at multiple wavelengths to resolve multiple species. Furthermore, the broad emission spectrum decreases sensitivity owing to the autofluorescence over the wide range of wavelengths which contributes to the measured intensity. RET probes can be used to circumvent these problems. RET probes with BSA Au clusters as an acceptor to high extinction donors such as pacific blue can enhance the brightness of an acceptor by several fold. This approach can make a low fluorescence quantum yield probe a bright fluorophore which can then be applied to imaging and microscopy.
Numerous reports have suggested the use of fluorescent metal–ligand complexes as long lifetime donors to long wavelength organic fluorophores.31–33
Similarly, we have attempted to create a RET probe using long lifetime BSA Au clusters and NIR probe Dy750. The advantage of such a RET probe is that the emission spectra of red and NIR fluorophores are typically narrow on the wavelength scale, whereas the emission spectra of nanoclusters are broad. Since the autofluorescence from biological samples is distributed broadly on the wavelength scale, the concentration of the emission into the narrow spectral range by the acceptor should improve detection in terms of an increased signal to noise ratio. These RET probes have potential applications in DNA hybridization, fluorescence in situ hybridization, or as molecular beacons. Following our approach, most of the species labelled with a donor or acceptor alone will display little emission. In contrast, D–A pairs due to macromolecular association will be brightly fluorescent. Additionally, the NIR acceptor emission will be long lived and using time-gated detection of a brightly fluorescent spot can become apparent against an autofluorescence background or short decay time. This technique can also be used to create D–A pairs that act as a single luminophore or to distinguish interactions in samples containing species labelled with a donor or acceptor. Thus, it seems likely that long wavelength long lifetime probes will find wide-ranging applications in biochemical and biomedical research.
Conclusion
In summary, we demonstrated that BSA Au clusters can be employed in FRET-based studies. BSA Au clusters can act as an acceptor and donor to short lifetime organic fluorophores to create RET probes. These RET probes can circumvent the intrinsic disadvantages of these metal nanoclusters such as low quantum efficiency and broad emission spectrum. Such RET probes have potential applications in chemical and biological sensing, catalysis, microscopy and imaging.
Acknowledgements
This work was supported by the NIH grant R01EB12003 (Z.G.), NSF grant CBET-1264608 (I.G.) and National Science Centre – N202 112340 (R.L.). We would like to thank Dr Badri Maliwal for preparing the BSA Au cluster conjugates with organic fluorophores and for valuable suggestions in writing this manuscript. We also would like to thank Deepanwita Pal for proofreading the manuscript.
Notes and references
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- A. M. Derfus, W. C. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. Chan, Small, 2010, 6, 138–144 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- H. Xu and K. S. Suslick, Adv. Mater., 2010, 22, 1078–1082 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 CrossRef CAS PubMed.
- L. Hu, S. Han, S. Parveen, Y. Yuan, L. Zhang and G. Xu, Biosens. Bioelectron., 2012, 32, 297–299 CrossRef CAS PubMed.
- X. Le Guevel, N. Daum and M. Schneider, Nanotechnology, 2011, 22, 275103 CrossRef PubMed.
- T. Zhou, Y. Huang, W. Li, Z. Cai, F. Luo, C. J. Yang and X. Chen, Nanoscale, 2012, 4, 5312–5315 RSC.
- T. H. Chen and W. L. Tseng, Small, 2012, 8, 1912–1919 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- X. X. Wang, Q. Wu, Z. Shan and Q. M. Huang, Biosens. Bioelectron., 2011, 26, 3614–3619 CrossRef CAS PubMed.
- Y. M. Fang, J. Song, J. Li, Y. W. Wang, H. H. Yang, J. J. Sun and G. N. Chen, Chem. Commun., 2011, 47, 2369–2371 RSC.
- C. V. Durgadas, C. P. Sharma and K. Sreenivasan, Analyst, 2011, 136, 933–940 RSC.
- C. V. Durgadas, C. P. Sharma and K. Sreenivasan, Nanoscale, 2011, 3, 4780–4787 RSC.
- G. Ma, A. Binder, M. Chi, C. Liu, R. Jin, D. E. Jiang, J. Fan and S. Dai, Chem. Commun., 2012, 48, 11413–11415 RSC.
- B. Schuler and W. A. Eaton, Curr. Opin. Struct. Biol., 2008, 18, 16–26 CrossRef CAS PubMed.
- V. V. Didenko, BioTechniques, 2001, 31, 1106 CAS.
- N. Boute, R. Jockers and T. Issad, Trends Pharmacol. Sci., 2002, 23, 351–354 CrossRef CAS.
-
Topics in Fluorescence Spectroscopy, ed. J. Lackowicz, Plenum Press, New York, 1997 Search PubMed.
- S. Raut, R. Chib, R. M. Rich, D. Shumilov, Z. Gryczynski and I. Gryzcynski, Nanoscale, 2013, 5, 3441–3446 RSC.
- T. Sen, K. K. Haldar and A. Patra, J. Phys. Chem. C, 2008, 112, 17945–17951 CAS.
- T. Sen, S. Mandal, S. Haldar, K. Chattopadhyay and A. Patra, J. Phys. Chem. C, 2011, 115, 24037–24044 CAS.
- S. H. De Paoli Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2009, 4, 365–379 CrossRef PubMed.
- G. Schneider, G. Decher, N. Nerambourg, R. Praho, M. H. Werts and M. Blanchard-Desce, Nano Lett., 2006, 6, 530–536 CrossRef CAS PubMed.
- S. Chowdhury, Z. Wu, A. Jaquins-Gerstl, S. Liu, A. Dembska, B. A. Armitage, R. Jin and L. A. Peteanu, J. Phys. Chem. C, 2011, 115, 20105–20112 CAS.
- H. Wang, C. Zheng, T. Dong, K. Liu, H. Han and J. Liang, J. Phys. Chem. C, 2013, 117, 3011–3018 CAS.
- K. Pu, Z. Luo, K. Li, J. Xie and B. Liu, J. Phys. Chem. C, 2011, 115, 13069–13075 CAS.
- L. Li, I. Gryczynski and J. R. Lakowicz, Chem. Phys. Lipids, 1999, 101, 243–253 CrossRef CAS.
- B. P. Maliwal, Z. Gryczynski and J. R. Lakowicz, Anal. Chem., 2001, 73, 4277–4285 CrossRef CAS.
- J. S. Kang, G. Piszczek and J. R. Lakowicz, J. Fluoresc., 2002, 12, 97–103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr03886f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 










