Discovery and development of first in class antifungal caspofungin (CANCIDAS®)—A case study
James M.
Balkovec
,
David L.
Hughes
,
Prakash S.
Masurekar
,
Carole A.
Sable
,
Robert E.
Schwartz
and
Sheo B.
Singh
*
Merck Research Laboratories, Rahway, NJ 07065, USA. E-mail: sheo.singh.215@gmail.com
First published on 22nd November 2013
Abstract
Covering: 1985 to 2001.
This paper describes a fifteen year journey from concept to clinical discovery and development of the first in class caspofungin acetate (CANCIDAS®) a parenteral antifungal agent. Caspofungin is a semisynthetic derivative of pneumocandin B0, a naturally occurring, lipophilic cyclic peptide isolated from the fungus, Glarea lozoyensis. While the echinocandins had been previously studied for antifungal activity by several organizations, the class was dropped for a variety of reasons. Merck subsequently initiated a research program leading to the discovery and development of caspofungin. The multitude of challenges that ensued during the discovery and development process and which were successfully resolved by multi-disciplinary teams constitute the content of this article. The article consists of five sections that describe the discovery and development of caspofungin in chronological order: (i) discovery of the natural product pneumocandin B0 from fungal fermentations, (ii) fermentation development to improve the titer of pneumocandin B0 to make it commercially viable, (iii) semisynthetic modification by medicinal chemistry to successfully improve the properties of pneumocandin B0 leading to the discovery of caspofungin, (iv) development of commercial semisynthesis and purification and formulation development to improve stability and (v) clinical development and approval of CANCIDAS® as an antifungal drug which subsequently saved thousands of lives.
 James M. Balkovec | James M. Balkovec received his PhD in 1985 in synthetic organic chemistry with Prof. Barry Trost at the University of Wisconsin. After postdoctoral studies with Prof. Gilbert Stork at Columbia University, he joined Merck in 1987. Jim held positions of increasing responsibility supporting many natural product leads in diverse therapeutic areas. Jim left Merck in 2011 as a Senior Scientific Director and is currently Principal and Owner of Chemtract LLC, a consulting business assisting companies in early stage drug development. He also serves on the Scientific Advisory Board of Sunnylife Pharma, Inc., an Indianapolis-based small molecule drug discovery company. |
 David L. Hughes | David Hughes received his PhD in physical organic chemistry with Prof. Bordwell at Northwestern University in 1981. After postdoctoral studies with Prof. Arnett at Duke University, Dave joined the Process Chemistry group at Merck and currently holds the position of Distinguished Scientist. He served on the Board of Editors of Organic Syntheses from 2008–2013. |
 Prakash S. Masurekar | Prakash S. Masurekar received his SM (1968) and PhD. (1973) in Biochemical Engineering from Massachusetts Institute of Technology under supervision of Prof. Arnold Demain. He joined Merck Microbiology and rose to rank of Director. He was involved in the development of processes for the production of antibiotics, antifungal agents, human therapeutic compounds and animal health products. His most notable contributions at Merck were the production processes for the anti-fungal cyclic lipopeptide, pneumocandin B0 and first approved cholesterol lowering agent, lovastatin. He served on the editorial board of Applied and Environmental Microbiology from 1980 to 1986. |
 Carole A. Sable | Carole Sable is an Infectious Disease Physician who trained at the University of Virginia. She joined Merck in 1995 and led the development of caspofungin, including worldwide approvals for multiple indications. In 2007, she became Chief Medical Officer of Novexel, SA. She established the US subsidiary and designed and conducted the first global clinical trials for Novexel. When Novexel was acquired by Astra Zeneca, she returned to Merck as VP and Franchise Integrator, Infectious Diseases. In 2011, Carole assumed the role of VP, Project Leadership and Management, Neurosciences, a position she currently holds. |
 Robert E. Schwartz | Robert completed his B.S. in Biology at Bradley University in 1975; followed by his Ph.D. in Chemistry at the University of Hawaii. In 1980, Robert became a National Institutes of Health Postdoctoral Fellow. He joined the Natural Products group at Merck in Rahway where he discovered the first potent HMGCoA Sythase inhibitor L-659,699, the antifungal natural products pneumocandin B0 (the starting material for Cancidas™), another antifungal agent called enfumafungin, and a potent tubulin synthesis inhibitor cryptophycin. In 2006 Robert shared the American Chemical Society's Heroes of Chemistry Award. From 2001–2008 he was in charge of the Compound Management Group at Merck; in 2008 Robert formed MyIslandBeach LLC to license software for Harmonized Tariff classification of chemicals. |
 Sheo B. Singh | Sheo B. Singh received his PhD in Natural Products Chemistry at CIMAP/Avadh University in India in 1981. After postdoctoral studies on synthesis and biosynthesis with Prof. Karl Overton at Glasgow University followed by various research appointments to study biologically active natural products with G. Robert Pettit at Arizona State University, Sheo joined the Natural Products Chemistry group at Merck in 1989 eventually leading the efforts of Natural Products as a department head. He currently holds the position of Senior Principal Scientist in the Discovery Chemistry. He currently serves on the Editorial Boards of Journal of Natural Products, Journal of Antibiotics and Natural Product Reports. |
1 Introduction
Opportunistic fungi of the genera Candida spp, Aspergillus spp, and Cryptococcus spp cause life-threatening infections, particularly in patient populations that are immunocompromised. In 1985, when Merck initiated this project there were two major treatment options for systemic fungal infections, amphotericin B and ketoconazole.1 Amphotericin B exerts its fungicidal action by binding to ergosterol and compromising the fungal membrane. Ketoconazole is a fungistatic agent which works by inhibiting sterol biosynthesis.Amphotericin B is an effective antifungal agent but has a very narrow therapeutic window. It was known alternately as “Amphoterrible” or “Shake and Bake” due to side effects related to its non-selective mode-of-action often leading to chills and high fevers, while ketoconazole's fungistatic mode-of-action had potential for developing a high rate of resistance. In addition, the azole class of drugs interact with cytochrome P450 enzymes leading to significant drug-drug interactions which can cause significant issues with seriously ill patients who often undergo polypharmacy. Clearly neither agent was optimal.
An ideal antifungal agent should be fungicidal and should have a fungal specific target. In other words it should target/interact with a protein that has no homologs in mammalian cells. Since mammalian cells do not have a cell wall the most obvious target is the fungal cell wall. Cell wall targets in bacteria have been incredibly successful in delivering highly useful and powerful antibacterial agents such as penicillins, cephalosporins, carbapenems and vancomycin.
1.1 Discovery of natural product cyclic lipopeptide lead pneumocandin B0
Echinocandin B (1a, Fig. 1) was one of the first cyclic lipopeptides of this class reported in 1974 by Sandoz. It was the lead for medicinal chemistry efforts at Sandoz (now Novartis) and subsequently at Eli Lilly. Merck's natural product screening efforts led to the discovery of the pneumocandin series which had two major structural differences compared to the echinocandins. The natural product pneumocandin B0 (2a, Fig. 1) eventually became the starting material for the synthesis of caspofungin acetate (CANCIDAS®). The discovery of pneumocandin B0 began in 1985 at CIBE, a subsidiary of Merck located in Madrid, Spain (Table 1) when Glarea lozoyensis (originally named as Zalerion arboricola)2 was isolated, fermented, and extracted by an organic solvent and determined to produce a potential cell-wall active antifungal agent.3,4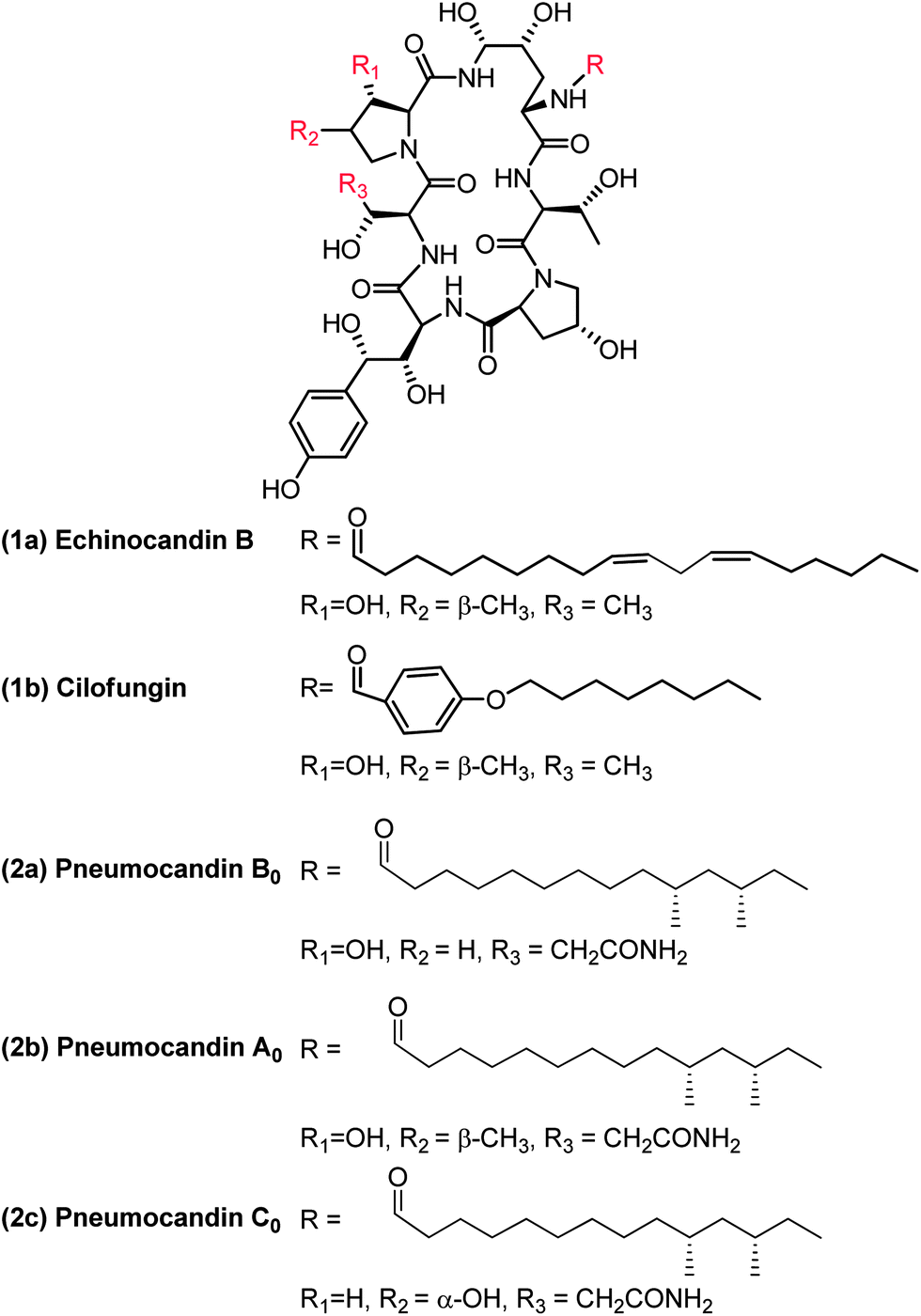 | ||
| Fig. 1 Structures of echinocandin B, cilofungin and pneumocandins A0, B0 and C0. | ||
| Date | Discovery Organization/Sponsor | Reference | |
|---|---|---|---|
| 1974 | Echinocandin B discovered | Sandoz (Novartis) | 8 |
| 1984 | Inhibition of 1,3-β-glucan synthase is the mode-of-action of echinocandins antifungal activity | Univ. Cambridge | 7 |
| 1985 | Glarea lozoyensis isolated by CIBE in Spain | Merck | |
| 1987 | Pneumocandin A0 isolated and structure determined; Pneumocandin A0 discontinued; Pneumocandin B0 discovered | Merck | 5 |
| 1988 | Cilofungin clinical trial halted | Eli Lilly | 24 |
| 1988 | PCP activity of pneumocandin A0 demonstrated | Merck | 13 |
| 1989 | Pneumocandin program reinvigorated based on PCP; Pneumocandin B0 chosen as natural product lead | Merck | |
| 1990 | Structure of pneumocandin B0 rigorously determined; First development candidate identified; | Merck | 14, 29 |
| 1991 | Potency and antifungal spectrum breakthroughs achieved | Merck | |
| 1992 | Pneumocandin B0 crystallized and absolute stereochemistry of the nucleus determined Silica gel chromatography developed | Merck | 15 |
| 1992 | L-743,872 (MK-0991, caspofungin) first synthesized | Merck | |
| 1993 | MK-0991 approved for development | Merck | |
| 1995 | SD/MD Phase I trial | Merck | |
| 1996 | Phase II trials in Candida esophagitis | Merck | |
| 1998 | Open label aspergillosis, Phase III Candida esophagitis and invasive candidiasis trials initiated | Merck | |
| 2000 | Invasive aspergillosis NDA filed, Empiric therapy trial initiated | Merck | |
| 2001 | Cancidas® approved for Invasive aspergillosis by US FDA | Merck | |
| 2002 | Approved for Esophageal candidiasis by US FDA | Merck | |
| 2003 | Approved for Invasive candidiasis by US FDA | Merck | |
| 2004 | Approved for empirical therapy by US FDA | Merck |
By March 1987 pneumocandin A0 (2b, Fig. 1) had been isolated, the structure determined and the biological characterization completed.5,6 It was shown to exert its antifungal activity by inhibiting synthesis of the fungal cell wall, probably via inhibition of 1,3-β-glucan synthase, the established target of the previously discovered, and structurally related echinocandins.7 In October of 1987 pneumocandin B0 was isolated and characterized as the desmethyl congener of pneumocandin A0. However, the actual structure had not been completed due to a lack of material and the fact that the pneumocandin A0 lead had just been discontinued owing to a lack of antifungal spectrum, poor oral bioavailability and water solubility, and structural similarity to echinocandin B (1a) and its semisynthetic analog, cilofungin (1b, Fig. 1).8
Two major events that were consequential to the project occurred in 1988. Firstly, the clinical trial for cilofungin was halted because of the toxicity of a co-solvent used for oral administration. Secondly, due to the DNA sequence of the ribosomal RNA gene9,10 and thymidylate synthase enzyme studies,11 it was suggested that Pneumocystis was a fungus rather than a protozoan (the infectious organisms in rats and humans are P. carinii and P. jiroveci, respectively). In addition, it was reported that the wall of the cyst form of P. carinii contained high levels of 1,3-β-glucan.12 With this information in hand, pneumocandin A0 was tested in an immunosuppressed rat model of Pneumocystis carinii pneumonia (PCP). It was shown to be superior to the existing therapy of trimethoprim/sulfamethoxazole.13 These events and data were sufficient to reinvigorate the program and pneumocandin B0 was chosen as the lead compound for a medicinal chemistry program to improve upon its properties to make a successful drug.
The relative stereochemistry was deduced by X-ray crystallography of crystalline pneumocandin B0, in which a large portion of the fatty side chain was disordered making the acquisition of the dataset not only extremely slow but also preventing the determination of the relative stereochemistry of the two methyl groups in the side chain (Fig. 2).
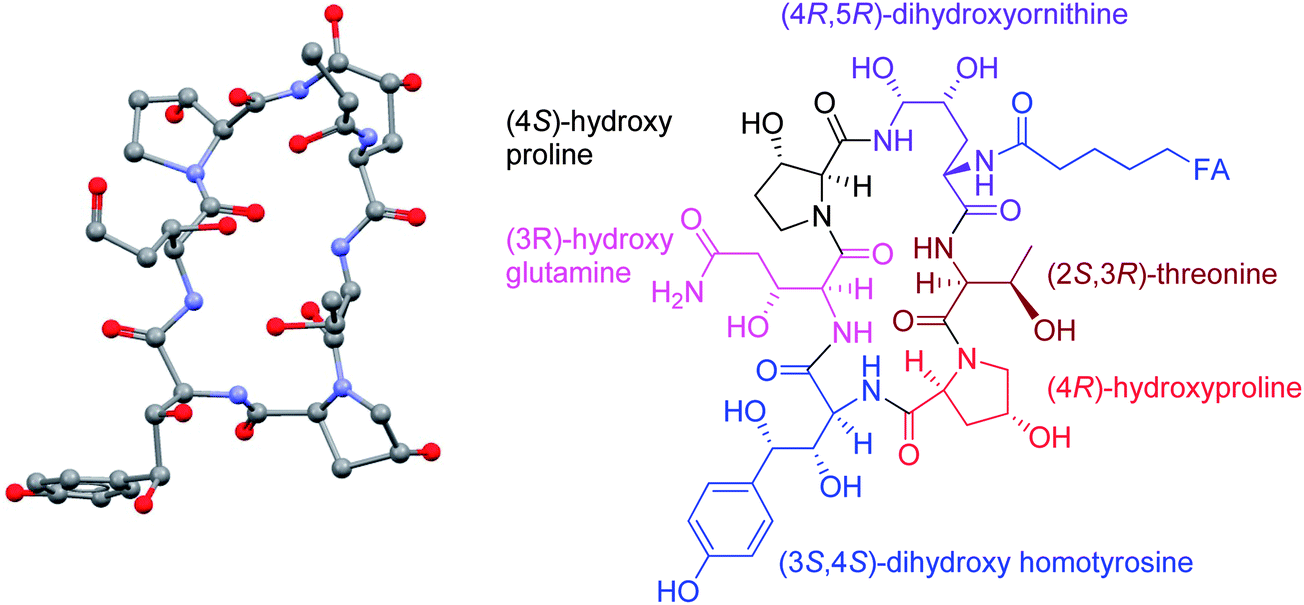 | ||
| Fig. 2 Single crystal X-ray structure analysis of pneumocandin B0 depicting stereo chemical assignment. Most of the side chain and hydrogen atoms are omitted in the ball and stick drawing on the left. | ||
The absolute stereochemistry of the threonine was determined from acid hydrolysis, followed by derivatization with α-methylbenzyl isothiocyanate, HPLC analysis and comparison to an authentic standard. When combined with the relative stereochemistry determined by X-ray, this established the absolute stereochemistry of the nucleus.15
The absolute configuration of the fatty acid side chain (10R, 12S-dimethyl myristic acid) was determined by comparison of the side chain fatty acid obtained from methanolysis of pneumocandin B0 with that of the acid synthesized by asymmetric synthesis.16
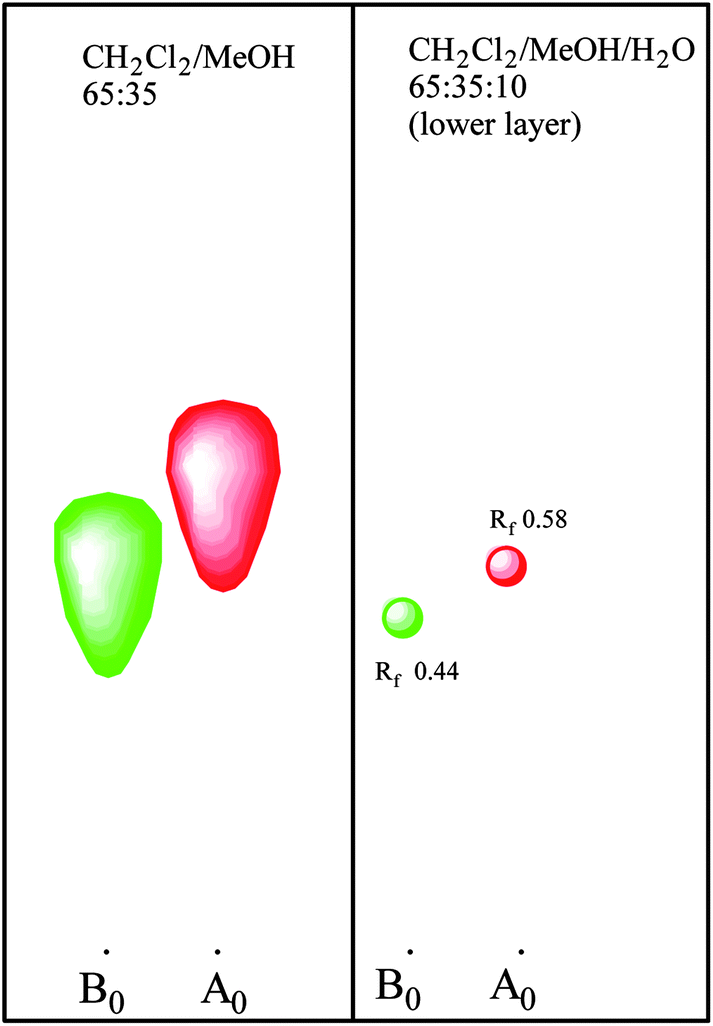 | ||
| Fig. 3 Thin layer chromatography and preparative chromatography of pneumocandin A0 and B0 on silica gel. | ||
1.2 Fermentation development of pneumocandin B0
Often, the levels of a bioactive natural product produced by a microorganism isolated from nature are miniscule and the purity is very low. The pneumocandin B0 story was no different. The levels of pneumocandins produced in the fermentation were of the order of few mg per liter and pneumocandin A0, the first pneumocandin discovered, was the dominant product. Structures of pneumocandin A0 and pneumocandin B0 are shown in Fig. 1. Both contain, starting at the 12 O'clock position, (4R, 5R)-dihydroxyornithine, (2S, 3R)-threonine, (4R)-hydroxy-proline, (3S, 4S)-dihydroxy-homotyrosine, (3R)-hydroxy-glutamine and another (3S)-hydroxy-proline. Pneumocandins also contain a (10R, 12S)-dimethyl myristic acid side chain. All these structural characteristics are shared by both compounds; however, they differ by the presence of a methyl group in the (3S)-hydroxy-proline at the ten O'clock position which is missing in pneumocandin B0.14–16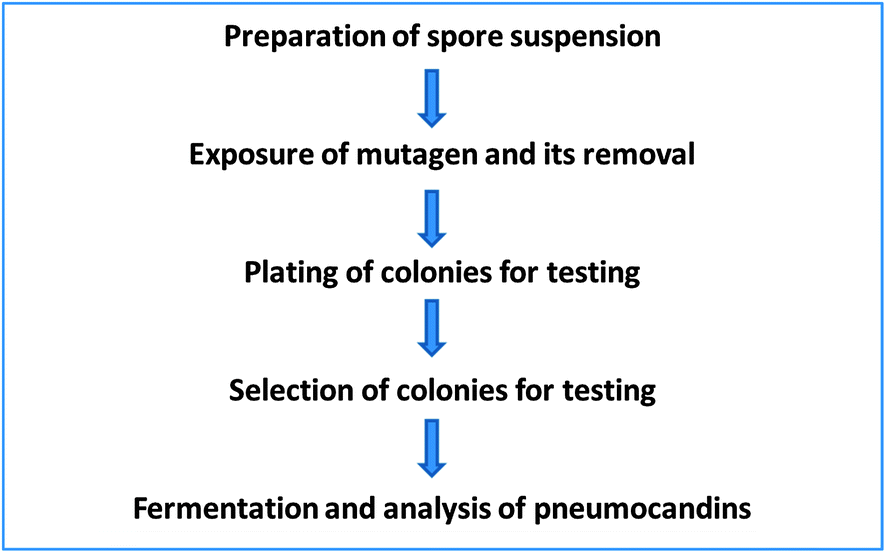 | ||
| Fig. 4 Flowchart of mutant selection and isolation process. | ||
Since at that time not much was known about the biosynthesis of these compounds, only an empirical approach was possible. We decided to tackle it by two approaches, environmental and genetic.19 The environment of a microbial process is defined by the operating conditions such as the medium composition, temperature, pH, oxygen transfer characteristics and carbon dioxide concentration. Although all these affect the outcome, more often the medium composition has a profound effect.
For successful mutagenesis it is important to have a single nucleus as a target. In the case of a filamentous fungus such as G. lozoyensis it is important to obtain single spores, which are propagules. That is, they develop into colonies. We were fortunate that this was the case for this fungus. The first step was to prepare spore suspensions. This was accomplished by growing the fungus on an agar medium long enough to allow the culture to sporulate and collect spores. Centrifugation or filtration was used to remove larger mycelial pieces and other debris. This spore suspension was then exposed to chemical mutagens at concentration, and under conditions, predetermined in preliminary experiments. After the treatment, the mutagen was removed by washing the treated spore suspension with either buffer or saline. The resulting cleaned spore suspension was diluted and plated for isolated colonies and allowed to grow. The colonies were then screened following fermentations and analysis of the produced products. This approach was used to isolate the desired mutants (Fig. 4).
As mentioned earlier, the main product of G. lozoyensis was pneumocandin A0 and the first goal was to isolate mutants which produced pneumocandin B0 as the main product. After several rounds of screening, the first mutant that produced B0 as the main product was isolated. The HPLC comparison showed that the mutant not only produced higher amounts of pneumocandin B0 it showed lower production of pneumocandin A0. Since the mutant still produced A0, which interfered with the purification of B0, further work continued.
After a few additional rounds of mutation and screening a mutant was identified, which met the requirements for facile isolation. ATCC 20957 was our first mutant which produced pneumocandin B0 as a major product. It produced 285 mg pneumocandin B0/liter with the ratio of pneumocandin B0/A0 of 10. Great progress had been already made with this first mutant where not only the overall titer of pneumocandin B0 had been improved but also the ratio of pneumocandin A0/B0 was reversed, particularly when compared with the original ratio of pneumocandin A0/B0 of 7 (a 70-fold improvement in ratio for pneumocandin B0 over A0). However, the second mutant was even better. Its yields of pneumocandin B0 were comparable to the first mutant but the ratio of pneumocandin B0/A0 was much better, i.e., 80.
At this point the program appeared viable and ready to deliver the required material to medicinal chemistry. However, just about this time, the Natural Product chemistry team discovered the regioisomeric pneumocandin C0 which contains a 4α-hydroxyproline instead of the 3α-hydroxyproline found in pneumocandin B0 (Fig. 1) at the ten O'clock position.
This discovery complicated the matter since the reversed phase HPLC system which was being used for mutant screening did not separate pneumocandin B0 and C0. As a matter of fact their retention times were identical. As described earlier, it was possible to separate them with the normal phase silica gel chromatography, so a low throughput normal phase TLC was substituted for the screening efforts. After additional rounds of mutagenesis and screening, a mutant was isolated which produced higher levels of pneumocandin B0 and lower levels of C0 (Fig. 5).
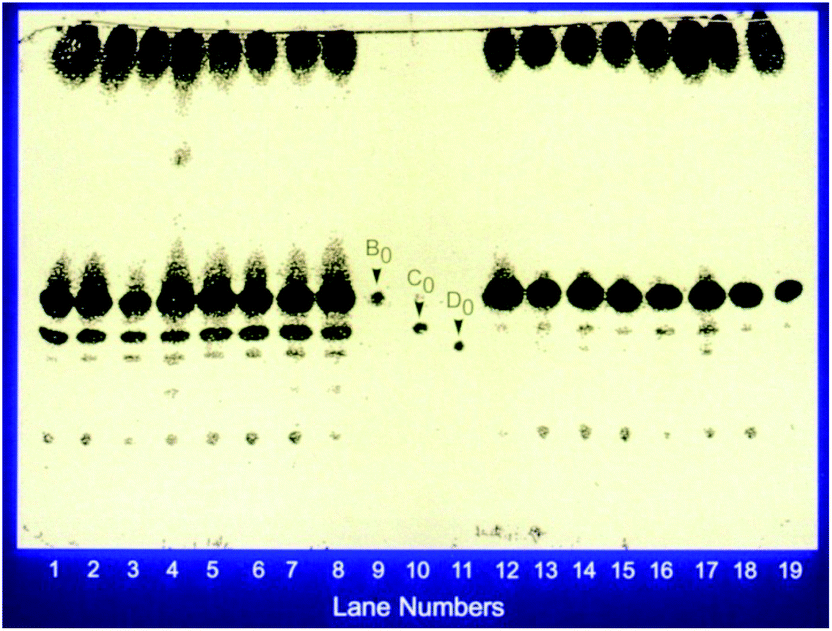 | ||
| Fig. 5 Production of pneumocandin by the parent and the new mutant. Exemplified by a silica gel TLC plate showing the separation of pneumocandins. The plate was developed in methyelene chloride/methanol/water: 65/35/10. Lanes 1–8 represent clones of the parent and lanes 12–19 represent clones of the new mutant. Lanes 9–11 represent standards. This dramatically showed the reduction in the production of C0. | ||
| L-Proline (g L−1) | Pneumocandins B0 + A0 (mg liter−1) | Pneumocandin C0 (%) |
|---|---|---|
| 0 | 145 | 9 |
| 2 | 199 | 8 |
| 4 | 219 | 7 |
| 8 | 224 | 3 |
Our efforts continued until we had developed the manufacturing process. In this process, pneumocandin A0 is undetectable and pneumocandin C0 is less than 5%. The production of pneumocandin B0 has been improved to the g liter−1 range from initial low mg liter−1 range. Thus, the goals of the project of delivering pneumocandin B0 of high purity and in good yields had been realized which made the project commercially viable.
1.3 Semisynthetic optimization of pneumocandin B0 and selection of the preclinical candidate
In the previous sections, the discovery, isolation and fermentation optimization of the pneumocandins were detailed. While pneumocandin B0 possessed potent fungicidal activity, the physicochemical and pharmacological properties were less than desirable and thus required optimization. Several points needed to be addressed: solubility, stability, antifungal spectrum and pharmacokinetic properties. This section of the article describes the medicinal chemical optimization of the lipopeptide natural product, pneumocandin B0, leading to the selection of caspofungin as the preclinical candidate.The medicinal chemistry program around the pneumocandins began at the Merck Research Laboratories in Rahway, NJ, in early 1989 and consisted of a small exploratory effort initially focused on improving the water solubility and/or stability of the lipopeptide. Several related natural products and derivatives had been described in the literature in the early 1970's.8,20–24 Echinocandin B was the subject of considerable investigation at several laboratories but it was Eli Lilly researchers who derived the first clinical agent cilofungin (LY-121019).25 Unlike echinocandin B, cilofungin proved non-lytic to red blood cells (RBCs) at microbiologically relevant concentrations. Lilly scientists achieved this by enzymatic deacylation of the linoleoyl side chain and reacylation of the echinocandin B nucleus with a 4-octyloxybenzoyl group. Little improvement in the solubility of the compound was achieved and the I.V. formulation required 26% polyethylene glycol as a co-solvent. The co-solvent led to metabolic acidosis and renal insufficiency and thus halting development of the candidate. A comprehensive effort by the Lilly team over several years led to a firm understanding of the side chain structure–activity requirements26 and a successful clinical candidate (LY303366) which ultimately became known as anidulafungin.27
The key structural differences between the pneumocandins and the echinocandins are the presence of a 3R-hydroxyglutamine in place of a threonine and a 10R,12S-dimethylmyristoyl side chain versus linoleoyl, respectively (Fig. 1). These differences render the pneumocandins non-lytic to RBCs at concentrations well above their MICs.28 Due to this inherent advantage and the competition around side chain derivatives, Merck medicinal chemistry efforts focused on modifications of the cyclopeptide core of the natural product.
Other considerations that guided the work were the following. The echinocandins and pneumocandins possess an acid and base labile N-acyl hemiaminal group. While the group had sufficient chemical stability, the metabolic stability was unknown. The natural products and cilofungin displayed potent activity against relatively few clinically important Candida spp. and lacked useful activity against Aspergillus spp.29,30 Finally, cilofungin required several daily infusions to achieve the desired plasma levels31–34 and displayed non-linear kinetics at high doses.35
A key foundation of the work described below was the availability of robust biological assays and rodent efficacy models. In addition to the 1,3-β-D-glucan synthase inhibition and standardized minimum inhibitory/fungicidal concentration (MIC/MFC) assays,36–38 mouse disseminated candidiasis (TOKA for Target Organ Kidney Assay) and disseminated aspergillosis (ASP) models were developed.39 The former measured fungal burden in kidneys (and other tissues) after inoculation of mice with C. albicans and twice daily dosing over four days with a test compound. The latter model measured 28 day survival after infection with A. fumigatus and five days of twice daily dosing of test compound. Both models had considerable throughput allowing the evaluation of at least ten compounds per week with low mass requirements for compounds. Importantly, SAR was being driven by the performance of compounds in the in vivo models. Additionally, a rat pneumocystis pneumonia (PCP) model was available for evaluation of compounds13 against P. carinii but will not be discussed in this section. Despite the fact that the discovery of PCP activity in addition to C. albicans activity was what had ignited the initial program, the P. carinii activity played only a secondary role in the selection of potential development candidates.
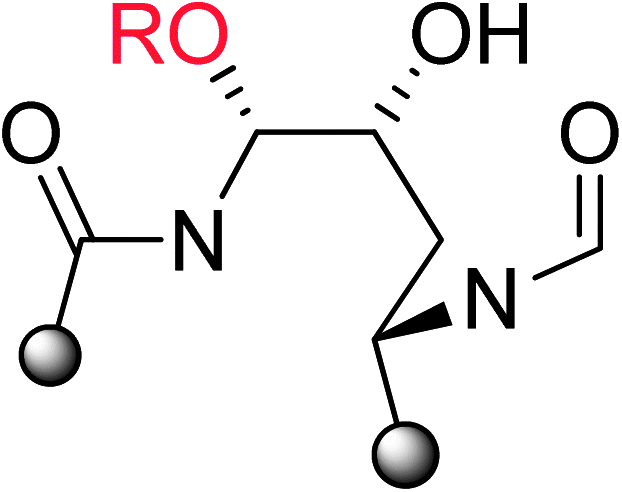
At pHs outside a range of approximately 5 to 8, pneumocandin B0 undergoes accelerated ionization or ring opening at the hemiaminal (Fig. 6).40 The resultant electrophile, either an N-acylimine (acidic pH) or aldehyde (alkaline pH), undergoes addition of the amide nitrogen of the side chain to produce a five-membered ring product (Fig. 6). Under sufficiently acidic conditions, both the hemiaminal and benzylic hydroxyl groups are ionized and delivery of hydride (i.e., reduction) provided an analog with a similar microbiological profile but with greatly improved acid and base stability.36,41 This chemistry was key to the later discovery of analogs substituted by nitrogen at the C5-ornithine position that were the basis for the discovery of caspofungin.
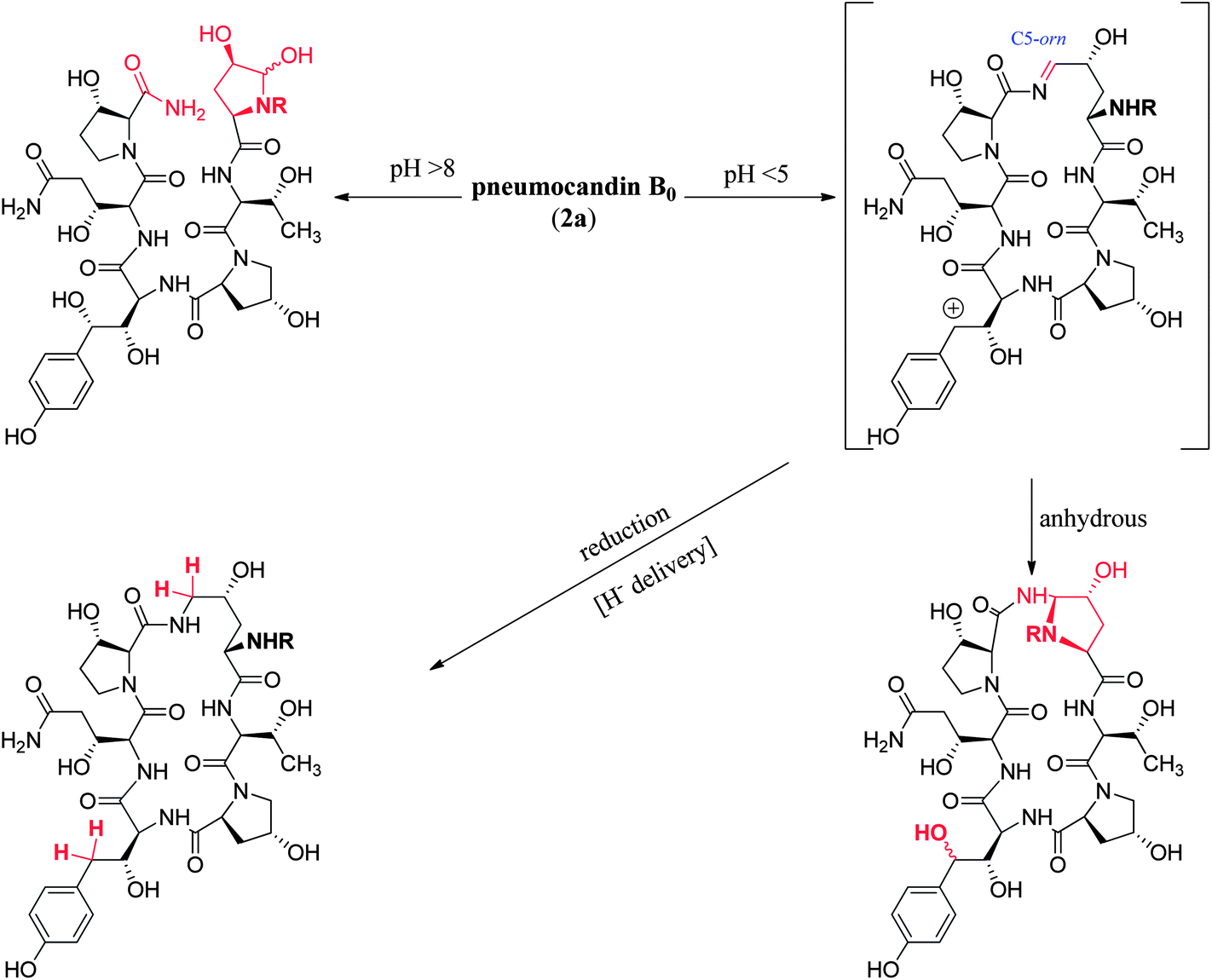 | ||
| Fig. 6 Acid and base degradation pathways of pneumocandin B0. | ||
|
|
C. albicans GS (IC50, nM) | C. albicans MFC (μg mL−1) | TOKA (ED99, mg kg−1) | ASP (ED50, mg kg−1) |
|---|---|---|---|---|
| OH | 70 | 0.25 | 3 | >20 |
| OCH3 | 100 | 2 | >6 | 1.8 |
| OCH2CH3 | 800 | 4 | — | — |
| OCH2CH2OH | 400 | 2 | — | — |
| OCH2CH2NH2 (3) | 10 | 0.125 | 0.0.3 | 0.06 |
|
|
C. albicans GS (IC50, nM) | C. albicans MFC (μg mL−1) | TOKA (ED99, mg kg−1) |
|---|---|---|---|
| CONH2 | 70 | 0.25 | 3 |
| CH2NH2 (4) | 10 | <0.06 | 0.2 |
| CH2N(CH3)3+ | 9 | 0.5 | 0.2 |
| CH2NHCOCH2NH2 | 15 | 0.5 | 0.3 |
| CH2NH-ornithine | 3 | <0.06 | 0.2 |
| CH2NHCOCH3 | 300 | 4 | 10 |
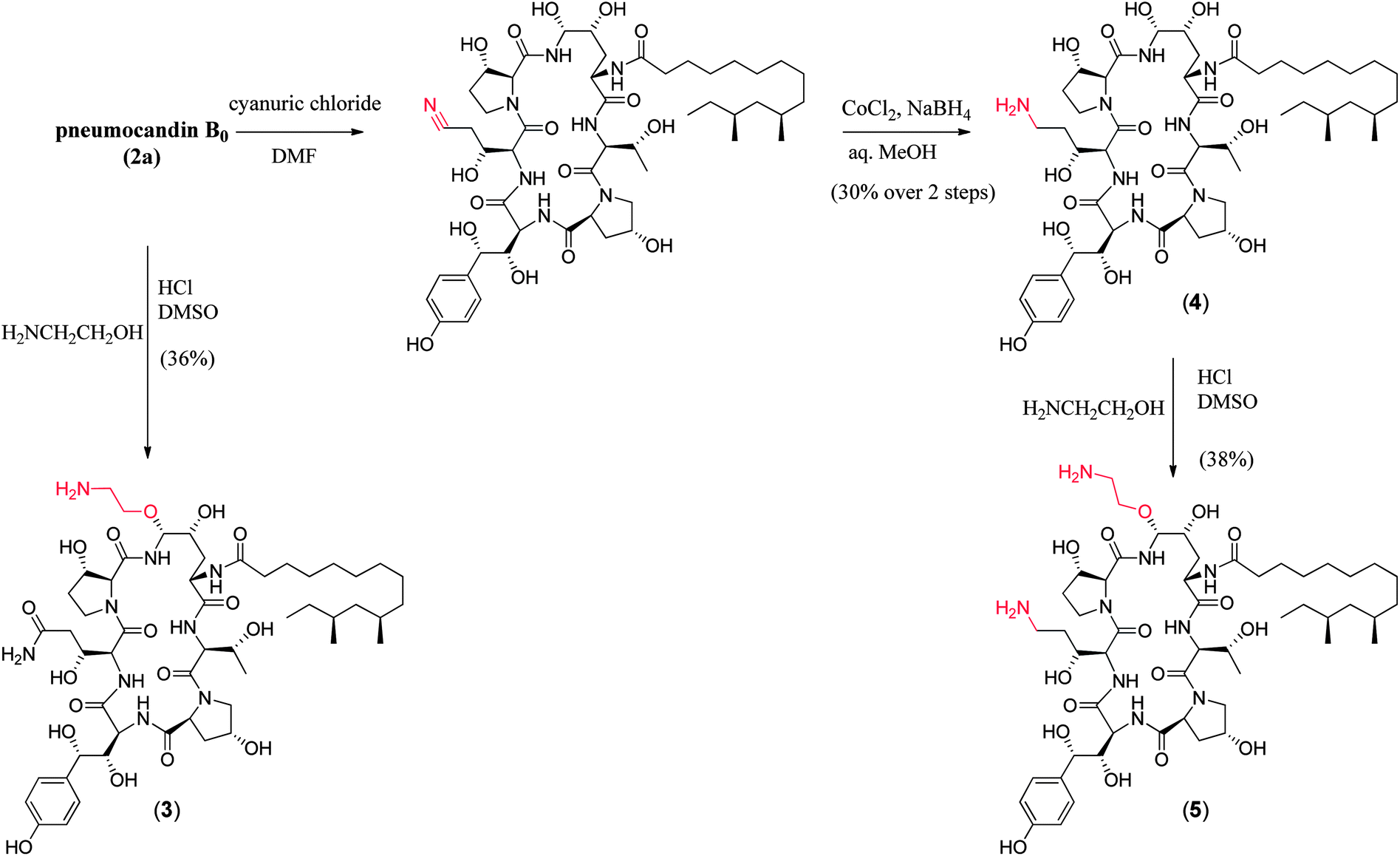 | ||
| Fig. 7 Synthesis of key semisynthetic pneumocandin derivatives (3–5). | ||
| Cmpd # | C. albicans GS (IC50, nM) | C. albicans MFC (μg mL−1) | C. parapsilosis MFC (μg mL−1) | A. fumigatus MEC (μg mL−1) | TOKA ED99.9, (mg kg−1, % MB cure) | ASP (ED50, mg kg−1) |
|---|---|---|---|---|---|---|
| 2a | 70 | 0.25 | 4 | 1 | 6 (20%) | >20 |
| 3 | 10 | 0.125 | 1 | 0.015 | 0.75 (80%) | 0.06 |
| 4 | 10 | 0.125 | 0.125 | 2 | 0.375 (70%) | >20 |
| 5 | 1 | <0.06 | 0.125 | 0.015 | 0.09 (80%) | 0.03 |
Compared to pneumocandin B0, diamino compound 5 had a 70-fold improvement in its glucan synthase IC50, an improvement in MFCs against Candida spp. including activity against the intrinsically more resistant C. parapsilosis, and a lower MEC against A. fumigatus (Table 5).46 An in vivo dose as low as 0.09 mg kg−1 gave over a four log10 reduction in CFUs per gram of kidney with microbiological (MB) cure in 80% of mice. In the aspergillosis model, the ED50 was 0.03 mg kg−1 and 90–100% survival could be obtained at higher doses.39 A pharmacodynamic analysis of the TOKA results suggested that full efficacy could be achieved by sustaining plasma levels of 5 above the MFC for the pathogen for at least two-thirds of the dosing interval. When considering plasma levels in the mouse, rat and rhesus monkey, this suggested that a 1 mg kg−1 I.V. daily dose would cover most Candida pathogens.47 Initial data suggested that 5 was non-toxic at clinically relevant doses and so a final evaluation was planned to qualify the compound as a pre-clinical candidate. Chimpanzee pharmacokinetics was considered a good predictor of human PK and the compound was administered to a single ape at 1 mg kg−1. The plasma levels obtained from this experiment were disappointingly low due to rapid early phase clearance (Fig. 8). Although the half-life was similar to that obtained in rodents and monkeys, the rapid distribution phase would necessitate a much higher or more frequent dosing regimen. In addition, it was noted that the chimpanzee was lethargic with a poor appetite and serum biochemistry showed that ALT and AST were significantly elevated suggesting hepatic injury.
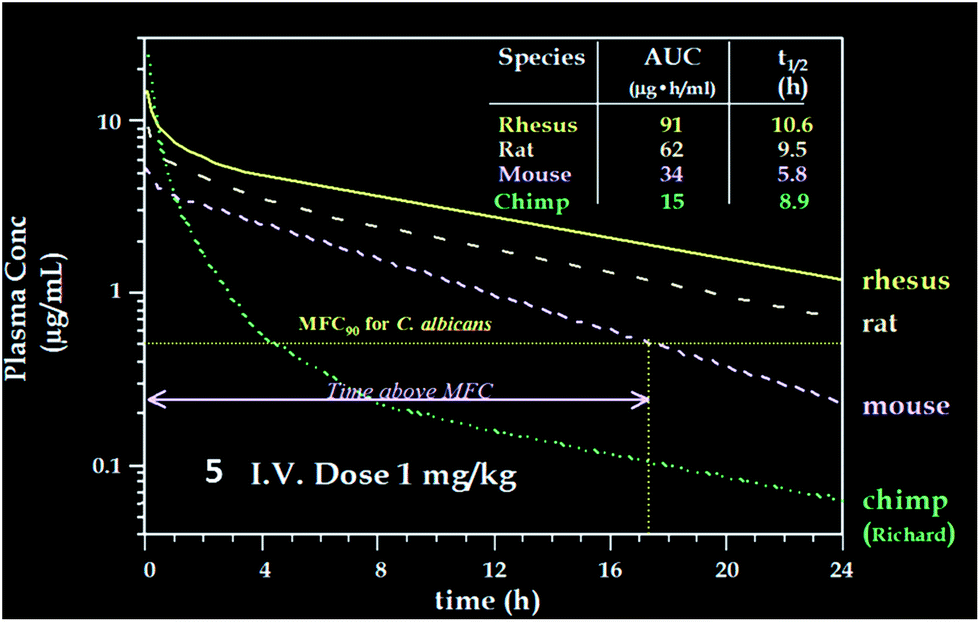 | ||
| Fig. 8 Pharmacokinetics of 5 in four species. | ||
These results were devastating to the program. However, after several months while trying to establish in vitro and in vivo rodent models of hepatic dysfunction, an examination of the chimpanzee that had received 5 was undertaken. Ketamine was the anesthetic used during the PK experiments. There were some reports in the literature that ketamine could cause abnormal liver function tests in humans48 thus ketamine alone was dosed to the chimpanzee that received compound 5 in the first experiment. The ALT levels effectively matched the levels from the first experiment and it was concluded that ketamine was responsible for the liver abnormalities. This result was a significant breakthrough for the program but the utility of 5 as a viable clinical agent remained in question. Identification of a new candidate with similar potency and properties and an acceptable toxicological profile was therefore necessary.
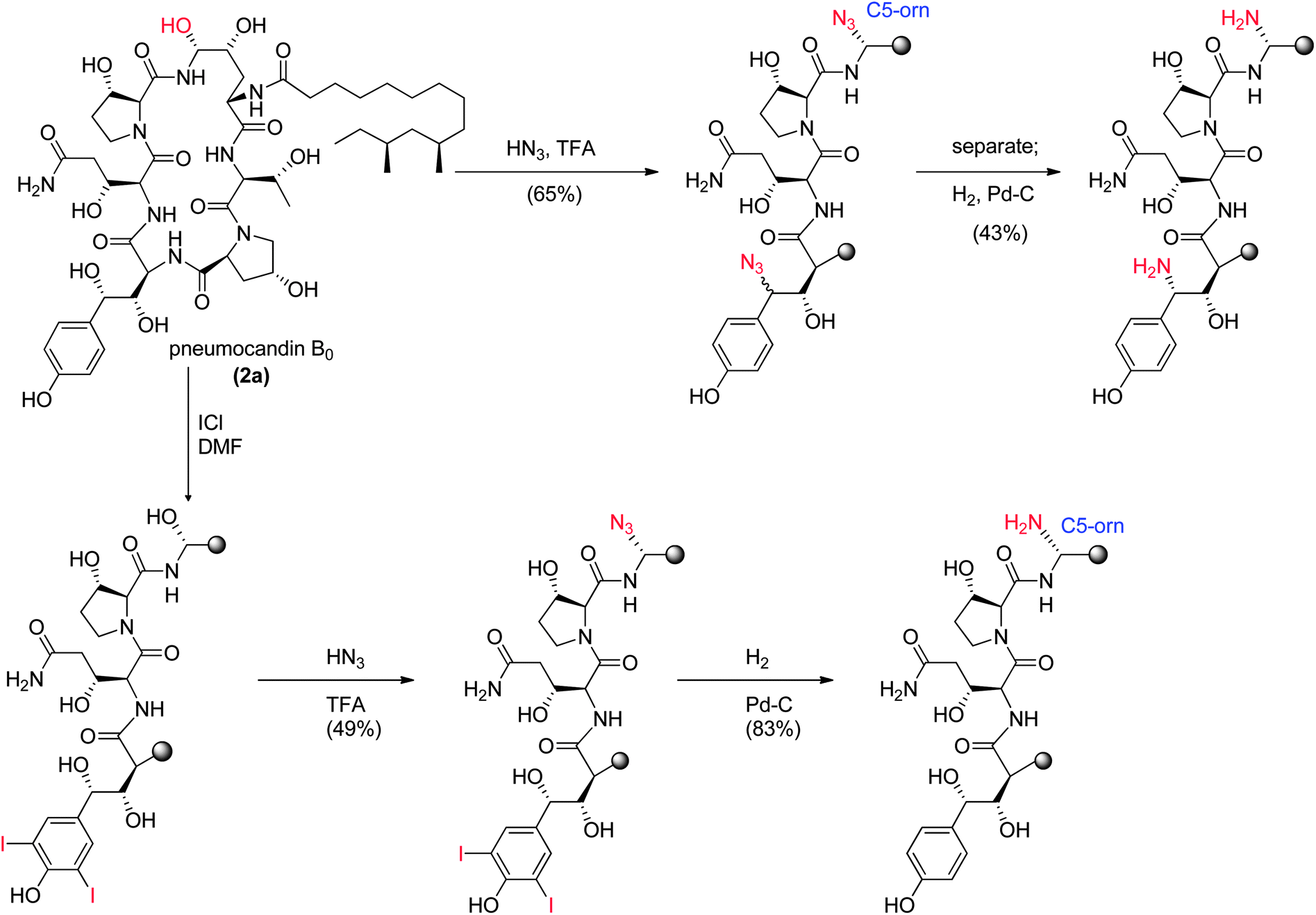 | ||
| Fig. 9 Nitrogen substituted analogs of pneumocandin B0. | ||
To append an ethylenediamine moiety onto 4, the chemistry shown in Fig. 10 was developed. The aminoethyl-thioether derivative was prepared by acid catalyzed exchange of the hemiaminal with cysteamine. Oxidation to the sulfone produced an analog that could easily be displaced at the C5-ornithine position by nucleophiles under neutral conditions. Treatment with ethylenediamine effected rapid displacement of the sulfone group. Interestingly, when the β-sulfone was used, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of α:β aminoethyl amines was obtained while use of the α-sulfone gave a 9:1 mixture of the desired α- to undesired β-isomer. This suggests that substitution of the α- and β-sulfones occur by a mixed mechanism or through different intermediates which could include direct substitution, geometric isomers of the N-acylimine or neighboring group participation of the 4-hydroxyl group of the ring ornithine residue.
:1 mixture of α:β aminoethyl amines was obtained while use of the α-sulfone gave a 9:1 mixture of the desired α- to undesired β-isomer. This suggests that substitution of the α- and β-sulfones occur by a mixed mechanism or through different intermediates which could include direct substitution, geometric isomers of the N-acylimine or neighboring group participation of the 4-hydroxyl group of the ring ornithine residue.
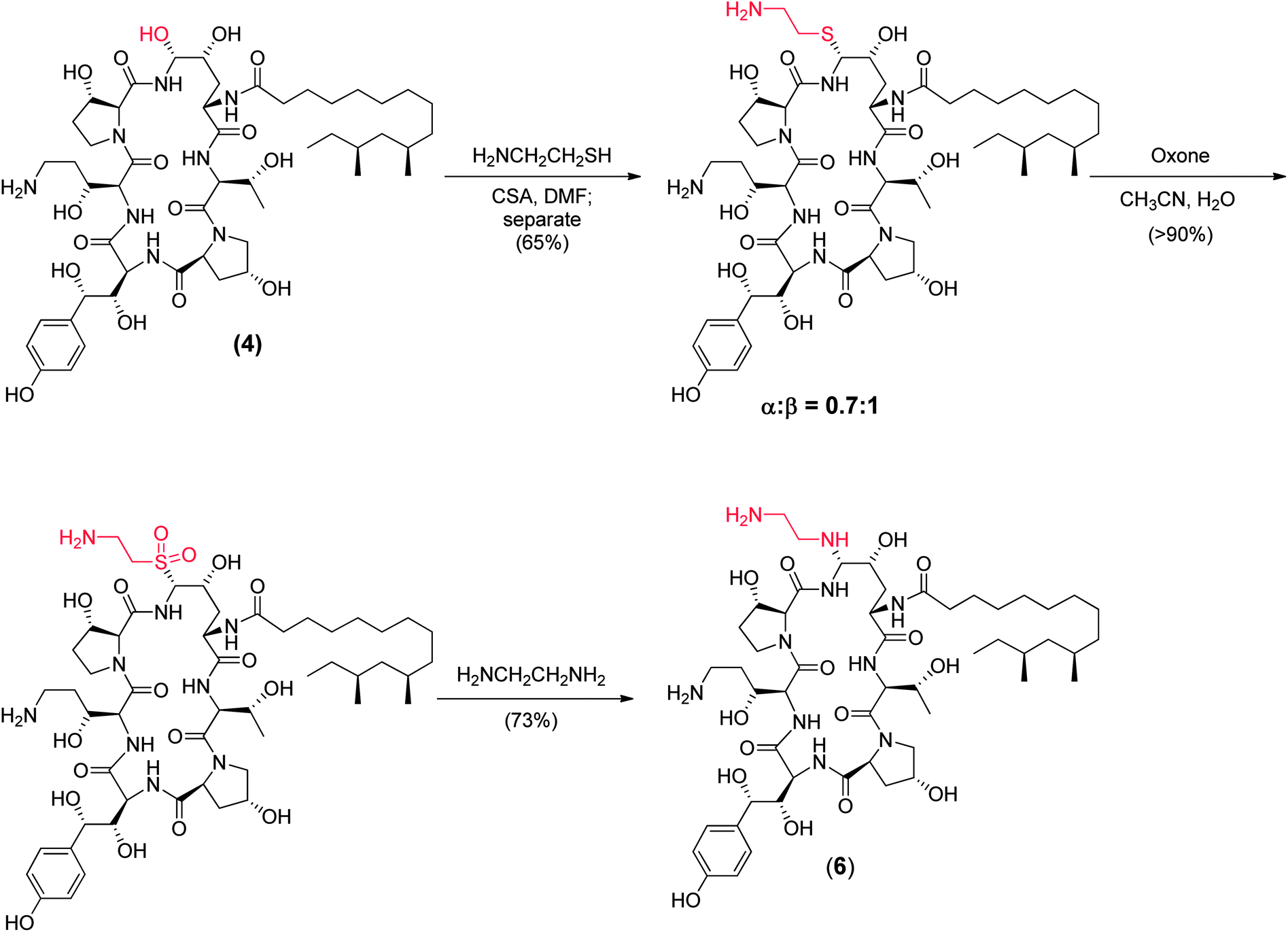 | ||
| Fig. 10 Medicinal chemistry synthesis of caspofungin (6). | ||
The potency of aza analog 6, was similar to oxa analog 5 (Table 6). The glucan synthase IC50 was 0.6 nM with potent Candida MFCs and Aspergillus MECs.50 Compound 6 proved approximately twice as potent in the TOKA and about half as potent in the ASP models compared to 5.51 The acute toxicity in mice was 50 mg kg−1, well above the efficacious dose with a far more favorable toxicity profile compared to amphotericin B. Importantly, compound 6 had consistent pharmacokinetics with sustained plasma levels in four species (Fig. 11)52 and supported a once daily I.V. dosing regimen of 0.5 to 1 mg kg−1 to cover 90% of Candida spp. and Aspergillus spp. Disseminated candidiasis and aspergillosis infection models in rodents rendered pancytopenic with cyclophosphamide showing that 6 had potency similar to amphotericin B.53 Additional microbiology, short term toxicology, pharmacology and drug metabolism studies supported 6 (later named MK-0991 and subsequently caspofungin) as a viable pre-clinical candidate. It was approved for development in December 1993 approximately one year after its initial synthesis and four years after the medicinal chemistry program had begun.
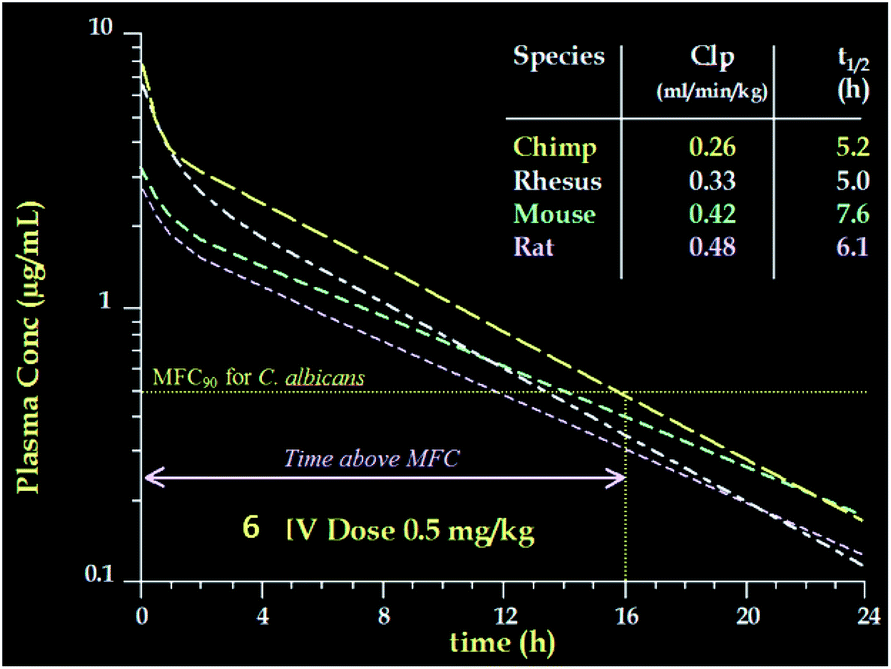 | ||
| Fig. 11 Pharmacokinetics of 6 in four species. | ||
| Cmpd # | C. albicans GS (IC50, nM) | C. albicans MFC (μg mL−1) | A. fumigatus MEC (μg mL−1) | TOKA (ED99, mg kg−1) | ASP (ED50, mg kg−1) | Acute tox (mg kg−1) |
|---|---|---|---|---|---|---|
| 5 | 1 | <0.06 | 0.015 | 0.045 | 0.03 | 30 |
| 6 | 0.60 | 0.125 | 0.008 | 0.027 | 0.05 | 50 |
| Amb | NA | 0.25 | 1 | 0.013 | 0.06 | 4 |
1.4 Development formulation and manufacturing process for caspofungin acetate (CANCIDAS®)
Previous sections have outlined the discovery of pneumocandin B0 as a lead antifungal candidate, the optimization of its fermentation, and the medicinal chemistry that culminated in the discovery of the drug candidate, caspofungin acetate. This section covers the development of the manufacturing process from the isolation of pneumocandin B0 from the fermentation broth, through the preparation of the API, to the development of a sterile IV formulation.During development, the bioprocess engineers worked closely with the process chemists and engineers to identify and control impurity levels in pneumocandin B0, especially those the chemists had determined to be difficult to remove during the subsequent synthetic steps and isolations. Thus, the HPLC purification was continually modified during development to control critical impurities while maximizing yield. As might be imagined, the analytical chemistry effort associated with the development of analytical assays to separate analogs was particularly challenging and was critical for development of the entire process, but is outside the scope of this article.
The isolation began with a series of novel extractions of the fermentation broth that separated the pneumocandins from most non-analog materials (Fig. 12).54 First, iso-butanol was added to the fermentation broth, resulting in a two-phase mixture with product partitioning into the alcohol layer. After concentration to reduce volume, heptane and methanol were added, resulting in a two-phase system in which the desired product now partitioned into the aqueous phase, removing non-polar impurities such as fatty acids which partitioned into the organic phase. The aqueous phase is then concentrated and additional iso-butanol was added, forming a two-phase system with the product now moved in the organic phase. The aqueous phase was discarded, removing polar impurities such as amino acids. The iso-butanol layer was concentrated, resulting in partial precipitation, which was improved after addition of acetonitrile to provide crude solid product with 63% purity.54
 | ||
| Fig. 12 Isolation of pneumocandin B0. | ||
The purification of this crude material involved a silica gel based chromatography to remove analogs, with a mobile phase consisting of ethyl acetate, MeOH, and water (Fig. 12).17,18 The purification productivity was originally low due to the poor solubility of pneumocandin B0 in the mobile phase and the limited separation of several pneumocandin analogs. The observation that some runs were providing improved separation led to a detailed investigation that revealed that low levels of amino acids, present at varying levels in the HPLC feed, could greatly improve separation of several analogs. This discovery led to greatly improved performance by the deliberate addition of amino acids such as L-proline to the eluent.55,56 The rich cuts from the HPLC were concentrated, resulting in precipitation of the product with about 90% purity. This material became the starting point for the synthetic steps described below.
As outlined in the previous section, the medicinal chemistry effort focused on modifying the peptide core to find an analog with improved potency, water solubility, and half-life, understandably without much concern for the route of preparation. Once the lead candidate was identified, the process chemistry group focused on the development of a practical and scalable process with the ultimate goal of defining a route that was robust, green, and economically viable. The starting point for the synthesis as the compound moved into development was a 5-step route with a 6–8% overall yield with poor α:β selectivity at the hemiaminal center.
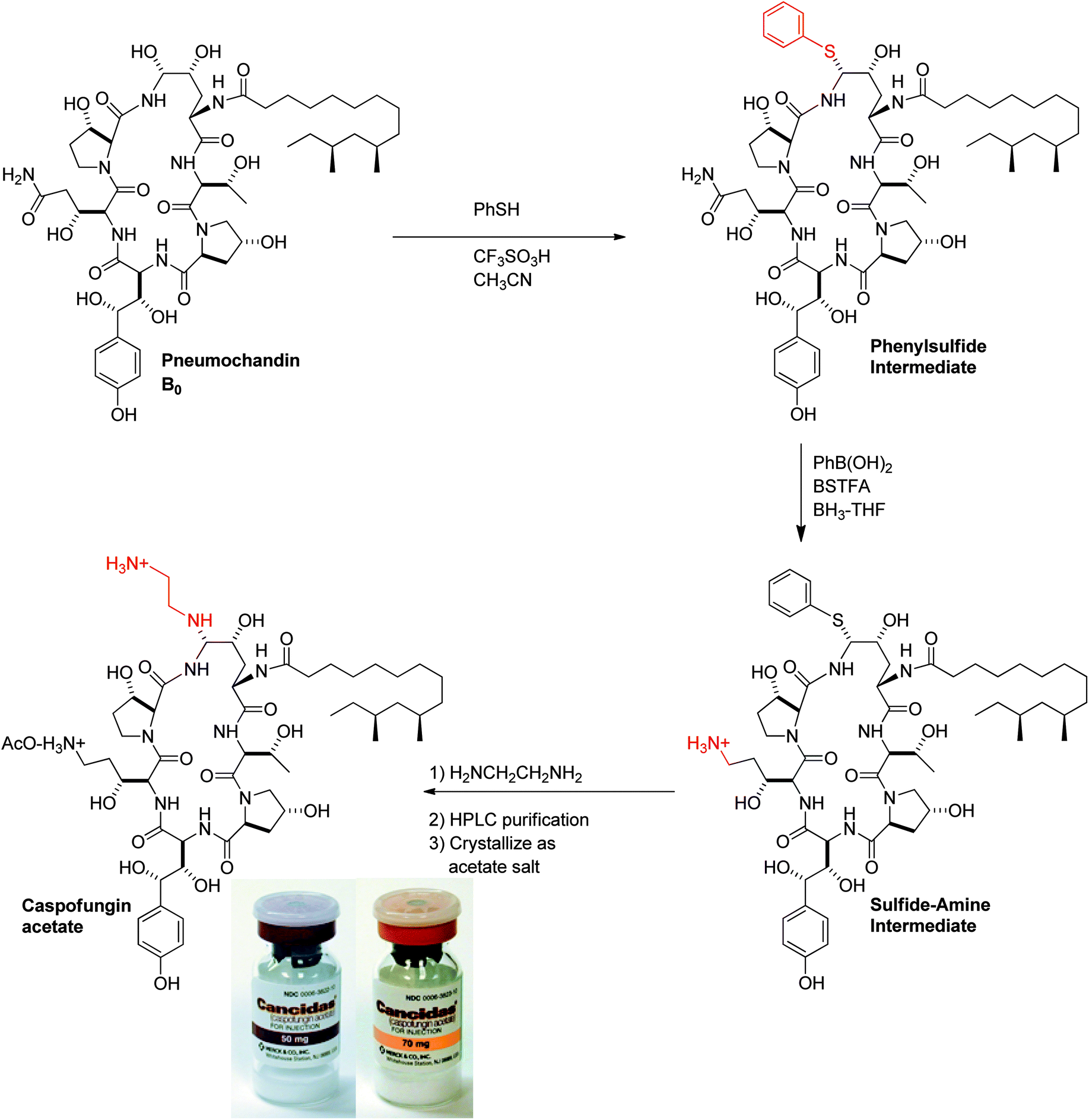
The highlights of the 3-step manufacturing synthesis, providing CANCIDAS® in 45% overall yield, are outlined below (Fig. 13).57
 | ||
| Fig. 13 Manufacturing route to caspofungin acetate (CANCIDAS®). | ||
:7 ratio of α:β isomers. The ratio could not be further improved by changes in solvent or temperature. The reaction is reverse-quenched into aqueous acetic acid, keeping the pH between 5.0 and 5.5 to minimize decomposition. The reaction is then extracted with heptane to remove thiophenol.
A critical breakthrough early in development was the crystallization of the bis-acetate salt of caspofungin. This crystallization only provided modest purity upgrade, but more importantly, provided much improved stability versus the amorphous solid. The final crystallization process was developed as crystallization from ethanol/ethyl acetate/water. The rich cuts containing acetonitrile/aqueous acetic acid were diafiltered to 9:1 ethanol/water and ethyl acetate was added to crystallize the diacetate salt of caspofungin. Overall yield across the final chemical step and purification is typically 70%.
Three formulation options were initially pursued: (1) sterile solution, (2) sterile crystalline powder for reconstitution, and (3) lyophilized powder for reconstitution. The sterile solution was quickly ruled out since caspofungin acetate does not have adequate solution stability at any pH at refrigerated conditions. The sterile crystalline diacetate salt was also ruled out since this material must be stored at −70 °C for adequate stability over 2 years. This left the lyophilized powder as the only viable route for commercialization.
Early work on the lyophilized powder indicated improved stability vs. the crystalline diacetate salt, with several options such as buffers and excipients available to improve stability further. After significant screening and optimization efforts, a formulation was developed that had 2-year stability at 2–8 °C. This formulation consisted of the bulking agent's mannitol and sucrose which 1) serve to sequester water and reduce hydrolysis, 2) act as a diluent to minimize dimerization, and 3) form an elegant cake upon lyophilization. Finally, the vials (Fig. 13) were sealed under argon to avoid any oxidation during storage.58
1.5 Clinical development of caspofungin acetate (CANCIDAS®, MK-0991, L-743, 872)
It took close to a decade between the start of the discovery program and the entrance of caspofungin into clinical development in 1995, when HIV was pandemic. Opportunistic infections were prevalent with significant associated morbidity and mortality. Protease inhibitors were just becoming available and resulted in significant changes in the approach to anti-retroviral therapy. The new combination treatments were accompanied by concerns about drug–drug interactions among anti-retroviral therapies and also with the other treatments required for concurrent infections and conditions. In parallel, the field of oncology was advancing rapidly with increasingly more aggressive chemotherapy, and bone marrow and stem cell transplantations performed for a wider range of diseases. These advances in cancer care were associated with an increase in the number of immunocompromised patients and a parallel rise in the frequency of opportunistic fungal infections.59–61Candida infections were the most common invasive fungal infection (IFI), responsible for ∼85% of all infections, followed by invasive aspergillosis causing 6% of IFI.59 Systemic antifungal therapeutic options were limited; amphotericin B formulations had a broad spectrum of activity but were available only intravenously (IV) and were associated with often dose limiting nephrotoxicity and infusion related reactions. Fluconazole had been introduced in the early 1990's and had the advantage of an oral and IV formulation and a more favorable safety and tolerability profile, but had a more limited spectrum of activity, specifically Candida spp. and some other fungi but no clinically relevant activity against Aspergillus spp.Initial evaluation of caspofungin in animal models and in healthy subjects in phase 1 study demonstrated that caspofungin acetate had poor oral bioavailability so could therefore be administered only by IV infusion. The plasma half-life of 9 to 11 h in phase 1 studies supported once daily dosing. Caspofungin is highly protein bound (∼97%) and plasma pharmacokinetics is controlled primarily by distribution. Tissue uptake is thought to be mediated through active transport. Caspofungin undergoes non-oxidative metabolism and the inactive metabolites formed are products of chemical degradation, hydrolysis, and N-acetylation. Approximately half the metabolites are excreted in urine and the remainder excreted via the biliary system. As caspofungin is not a substrate for, nor an inhibitor of the cytochrome P-450 system, few clinically significant drug–drug interactions are expected.
The caspofungin development program was designed to demonstrate the safety, tolerability and efficacy of caspofungin in well-documented fungal infections due to Candida and Aspergillus spp. in comparison to standard of care. Ideally, caspofungin would be at least as effective as amphotericin B and fluconazole in the treatment of patients with Candida infections and have a favorable safety and tolerability profile with few drug related adverse events and few important drug interactions. A number of important factors had to be taken into consideration in the initial efficacy evaluation of caspofungin. Diagnosis of infection and evaluation of outcomes is difficult, often requiring biopsy and culture or histopathology of infected organs. Caspofungin is active against fungal pathogens that cause invasive fungal infections (IFI). IFIs typically occur in immunocompromised patients such as those with HIV infection or malignancies and are associated with considerable morbidity and mortality.59–61 These invasive procedures pose increased risk in immunocompromised patients and the challenge of making a definitive diagnosis results in infections being classified as “probable” or “possible” in addition to “proven”.62 Although in clinical practice treatment is often empirical, in clinical trials it is necessary to identify the specific causative pathogen and antifungal susceptibility pattern to provide compelling evidence that a new agent is an appropriate treatment for a proposed infection. An additional challenge was that there was no definitive information about the pharmacokinetic/pharmacodynamics relationship for pneumocandins, making dose selection less certain. The first clinical efficacy study had to balance the severity of the infection being studied, including the risk of morbidity and mortality if the drug was ineffective, the need to conduct dose ranging, and the need to study an infection where intravenous treatment was appropriate. The ability to confirm the diagnosis and assess the adequacy of treatment was also critical.
While in some cases a phase 1b study can provide some efficacy information, the first study of caspofungin for the treatment of a fungal infection was a Phase 2 study in esophageal candidiasis.63 In 1996 when the study was initiated, esophageal candidiasis was among the most common opportunistic infections in patients with advanced HIV infection64 and was responsible for significant morbidity. Infections typically responded to treatment, but tended to recur as long as the underlying immune compromise persisted.65 Amphotericin B, despite its numerous toxicities, was generally regarded as the treatment of choice for symptomatic patients requiring IV therapy or having failed treatment with azoles such as fluconazole.66 The presence of esophageal candidiasis could be readily diagnosed and outcome assessed by endoscopy with culture and/or biopsy. A number of variables that could potentially influence the outcome of treatment had to be considered in the design and analysis plans for the study, including: Candida spp., underlying disease, degree of immunosuppression, severity of infection, in vitro susceptibility to caspofungin and amphotericin B, the duration of treatment, and the definition of response.
The study was conducted at seven sites in Latin America. Patients were randomized to receive caspofungin at 50 or 70 mg day−1 or amphotericin B at 0.5 mg kg−1 daily for 14 days. The doses of caspofungin were selected based on a conservative target of maintaining plasma concentrations above ≥1 μg mL−1 throughout the 24 h dosing interval. In vitro susceptibility data demonstrated that the MIC90 for Candida spp. was ≤1 μg ml−1 and multiple daily doses of 50 mg resulted in a C24 h ≥ 1 μg mL−1 in 95% of patients, indicating 50 mg was a reasonable target dose. Patients were prospectively stratified at entry based on the severity of esophageal lesions on endoscopy based on a four grade scales (0 normal to 4 most severe). The primary outcome measure for efficacy was pre-specified as the combined clinical and endoscopic response assessed 14 days after completion of treatment. Patient responses were classified as “favorable” only if they had both complete resolution of all esophageal symptoms and either total clearing of esophageal lesions (grade 0) or a reduction in endoscopy score by at least 2 grade levels. A favorable microbiological response was a secondary endpoint and required eradication of the pathogen(s) as supported by negative post-treatment stains and/or cultures for Candida. Strict definitions of response were considered important to better delineate dose response and ensure that adequate dose(s) of caspofungin were selected for study of other, more serious infections.
The study included 128 adult patients of whom 103 had HIV infections. Baseline characteristics were comparable across treatment groups. Candida spp. were recovered from 120 patients; 101 specimens (84%) yielded pure cultures of Candida albicans, with caspofungin MICs ranging from 0.06 to 2 μg mL−1 as determined by the standard National Committee for Clinical Laboratory Standards protocol (M27-A). The remaining infections were caused by a variety of other Candida pathogens or were mixed infections with multiple Candida spp. isolated.
All 3 treatment regimens were highly effective in achieving favorable combined symptom and endoscopy responses (Table 7); numerically the response rate was highest for those receiving caspofungin at 70 mg and lowest for the amphotericin B group at both time points. The mean differences in response rates for caspofungin versus amphotericin B were 11% (95% CI, −9% to 32%) and 26% (95% CI, 4%–50%) for those receiving 50 and 70 mg, respectively, at the primary end point 2 weeks after completion of therapy. More than half the patients in each treatment arm had resolution of all symptoms by day 4 of therapy. Symptoms persisted in 3 of 46 (7%), 0 of 28 (0%), and 7 of 54 (13%) patients at the end of therapy in the groups receiving caspofungin at 50 mg, caspofungin at 70 mg, and amphotericin B, respectively. The proportion of patients with endoscopic improvement was slightly higher in the caspofungin than amphotericin B groups. At the 14-day follow-up time point, 34 of 46 (74%) patients receiving caspofungin at 50 mg, 25 of 28 (89%) patients receiving caspofungin at 70 mg, and 34 of 54 (63%) patients receiving amphotericin B had a marked reduction in endoscopic grade. Illustrative examples of patients with a favorable response to caspofungin therapy are in Fig. 14.
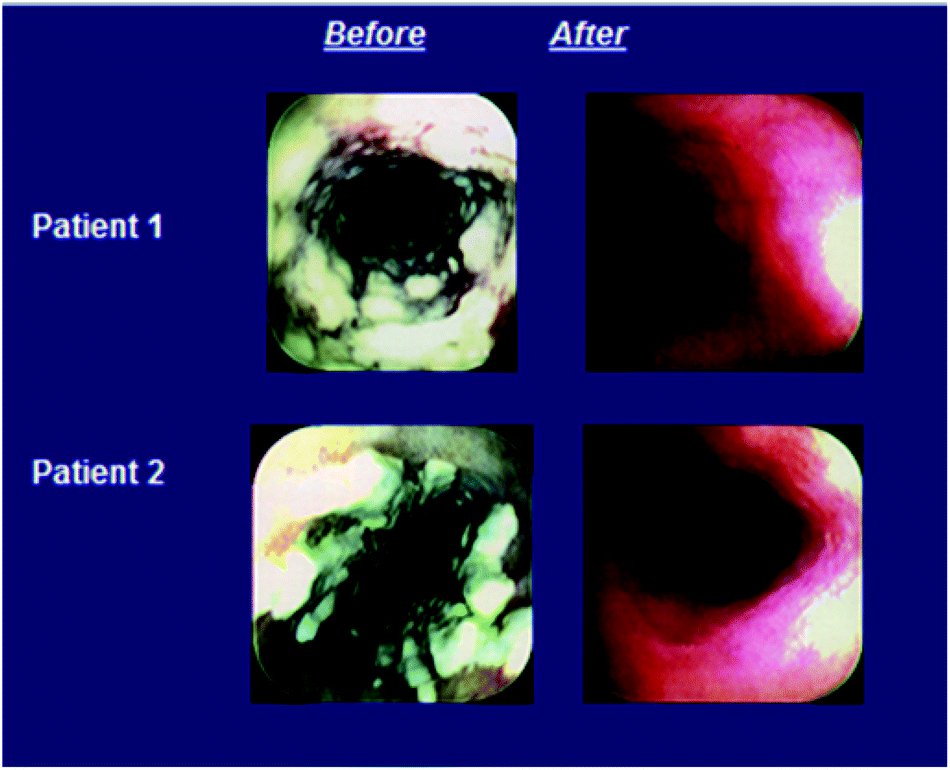 | ||
| Fig. 14 Examples of patients with a favorable response to caspofungin treatment for esophageal candidiasis. | ||
| Caspofungin 50 mg | Caspofungin 70 mg | Amphotericin B | |
|---|---|---|---|
| No. (%) 95% CI | No. (%) 95% CI | No. (%) 95% CI | |
| 14-day post-therapy follow-up, both strata | 34 (74) 59–86 | 25 (89) 72–98 | 34 (63) 49–76 |
| Stratum I | 22 (76) 57–90 | 11 (85) 55–98 | 19 (66) 46–82 |
| Stratum II | 12 (71) 44–90 | 14 (93) 68–100 | 15 (60) 39–79 |
Patients receiving amphotericin B had a significantly higher incidence of drug-related clinical adverse events (93%) than did patients receiving caspofungin at 50 mg (61%) or 70 mg (68%) (P < .01 for each caspofungin arm vs. the amphotericin B group). The most frequently reported drug related clinical adverse events in the caspofungin groups were fever, phlebitis, headache, and rash (Table 8). Significantly fewer patients in the study arm receiving caspofungin at 50 mg compared with the amphotericin B group experienced drug-related fever (P < .01), chills (P < .01), or nausea (P < .05); significantly fewer patients in the study arm receiving caspofungin at 70 mg compared with the amphotericin B group developed fever (P < .05) or chills (P < .01). Serum creatinine concentrations increased in 16 patients (30%) in the amphotericin B group but in only one caspofungin patient (P < .01 for comparisons vs. amphotericin B).
| Caspofungin 50mg N = 46 (% of patient) | Caspofungin 70mg N = 28 (% of patient) | Amphotericin B 0.5mg kg−1 N = 54 (% of patient) | |
|---|---|---|---|
| Chills | 2 | 0 | 72 |
| Fever | 28 | 39 | 69 |
| Phlebitis | 15 | 25 | 28 |
| Nausea | 2 | 4 | 15 |
| Vomiting | 2 | 4 | 7 |
| Anemia | 7 | 0 | 13 |
| ALT increased | 13 | 14 | 22 |
| AST increased | 15 | 11 | 20 |
| BUN increased | 0 | 0 | 15 |
| Creatinine increased | 0 | 4 | 30 |
| Potassium decreased | 4 | 11 | 28 |
| Hemoglobin decreased | 17 | 0 | 43 |
In this first study evaluating the pneumocandin/echinocandin antifungal in the treatment of human fungal infections, caspofungin at 50 mg or 70 mg day−1 appeared to be at least as effective as amphotericin B at 0.5 mg kg−1 day−1 in the treatment of Candida esophagitis, as assessed by the response of symptoms and/or esophageal lesions. Caspofungin was generally well tolerated, and the side effects typically associated with amphotericin B occurred significantly less often with caspofungin. This study demonstrated proof of concept for the class of pneumocandins/echinocandins and provided the first characterization of the clinical profile of caspofungin.
After the successful completion of this study, caspofungin was evaluated in other comparative studies in esophageal candidiasis as well as in a Phase 3 trial of invasive candidiasis.67,68 A study in invasive aspergillosis (IA) was initiated as data accumulated to indicate that caspofungin was effective in treating Candida infections and that it had a favorable safety profile. There was a high unmet need for new therapies for invasive aspergillosis as mortality in the most severely immunocompromised patients approached 90% despite treatment.69,70 Amphotericin B and the lipid formulations of amphotericin were the only reliable drugs available in the late 1990's for the treatment of IA. Studies indicated that the response rate to treatment with amphotericin B was <40% and as low as 10%–15% among patients undergoing allogeneic stem cell transplantation for cancer.71,72 The lipid formulations of amphotericin B were associated with less toxicity at higher dosages; however, their overall efficacy at therapeutic doses appeared similar to that of standard amphotericin B in the primary treatment of IA. At the time, oral itraconazole was indicated for IA, but its use was limited by the fact that it was fungistatic not fungicidal, lacked an IV formulation, and had unreliable absorption. It was also associated with drug-related hepatotoxicity and significant drug interactions. The available in vitro and animal model data indicated that caspofungin had activity against Aspergillus spp., but was not fungicidal. With no clinical data to support the utility of caspofungin in aspergillosis, this initial trial of an echinocandin/pneumocandin in IA focused on only those patients that were either refractory to or intolerant of standard therapy. The multi-center study was non-comparative and open-label.73 Patients who met the entry criteria of having documented, proven or probable pulmonary invasive aspergillosis and who were refractory to or intolerant of standard therapy, received caspofungin at 50 mg daily (following 70 mg loading dose on Day 1).
In this study, “refractory” was defined as of progression of disease or failure to improve clinically despite receiving at least 7 days of standard therapy. Intolerance referred to the development of nephrotoxicity, pre-existing renal impairment, or any other significant intolerance to prior therapy. Duration of therapy was based on the severity of underlying disease, recovery from immunosuppression, and clinical response. In general, patients were treated for a minimum of 28 days and for at least 7 days after resolution of symptoms. Patients with neutropenia were treated for at least 14 days after resolution of neutropenia. Favorable response was defined as Complete or Partial Response. “Complete response” required resolution of all signs and symptoms attributable to IA and complete resolution of radiographic or bronchoscopic abnormalities. The term “partial response” was defined as clinically meaningful improvement of signs and symptoms and at least 50% improvement of radiographic or bronchoscopic abnormalities. Patients who did not meet the criteria above were considered to have an unfavorable response.
Because of the complexities involved in establishing a diagnosis of IA and assessing outcome after treatment, an independent panel of 3 experts in medical mycology was established to independently evaluate the diagnosis of IA, whether patients were refractory to or intolerant of standard therapy, and the response to caspofungin therapy. The expert assessment was the basis for efficacy analysis. The primary efficacy analysis was the modified intention to treat (MITT) analysis and included all patients who had protocol defined IA and had received at least 1 dose of caspofungin and had sufficient data on which to base an assessment. A second analysis was performed for patients who received at least 7 days of caspofungin therapy (evaluable patients analysis) and corroborated the results in the primary analysis.
Fifty eight patients were reviewed by the Expert Panel of whom fifty four were included in the primary efficacy assessment. As expected, most (71.4%) patients had pulmonary aspergillosis while 18.2% had disseminated disease. Eighty two percent were refractory to standard therapy, including 33% who had received multiple prior therapies. The overall response rate in the MITT population was 40.7% with favorable responses seen in patients who were refractory to prior therapy (34.1%) as well as those intolerant to standard treatment (70%) (Table 9). Illustrative examples of some patients who responded to caspofungin therapy are shown in Fig. 15. Due to the compelling results seen in this salvage invasive aspergillosis study in patients with limited therapeutic options, after discussions with the US FDA, a New Drug Application (NDA) was submitted to the FDA in July 2000. Caspofungin was approved for the treatment of invasive aspergillosis in patients who were refractory to or intolerant of standard antifungal therapies in January 2001.
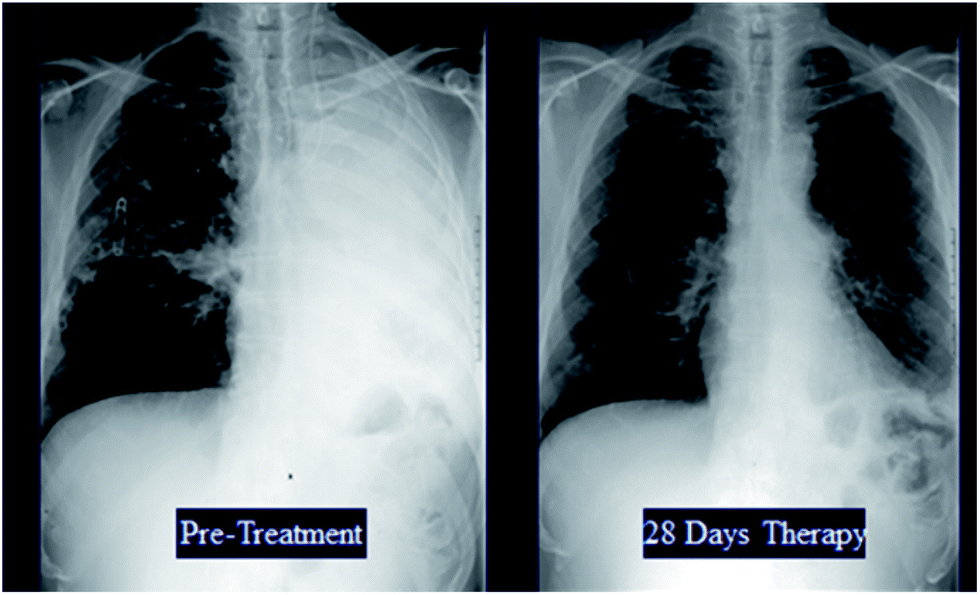 | ||
| Fig. 15 Illustrative examples of patients with invasive aspergillosis who responded to caspofungin therapy. | ||
| Caspofungin 70 mg X1, then 50 mg day−1 n/m (%) | |
|---|---|
| MITT analysis | 22/54 (40.7%) |
| Evaluable patients analysis (>7days caspofungin) | 22/45 (48.9%) |
| Subsets of MITT analysis | |
| Refractory to prior therapy | 15/44 (34.1%) |
| Intolerant of prior therapy | 7/10 (70%) |
Although the initial results of the clinical studies of caspofungin were promising, clinical development continued to better characterize the profile of caspofungin and define its utility across a broad range of indications. Subsequently caspofungin was approved in over 80 countries making it the first pneumocandin/echinocandin to be marketed worldwide.
It has been approved in the US for the following indications:
–Invasive aspergillosis in patients who are refractory to or intolerant of standard antifungal therapies (January 2001)
–Esophageal Candidiasis (April 2002)
–Candidemia and other Candida infections: peritonitis, intra-abdominal abscess, and pleural space infections (January 2003)
–Empirical therapy for presumed fungal infections in febrile, neutropenic patients (September 2004)
Drug discovery and development is an arduous and complex process starting with selection of disease area to study, biological target selection, lead finding, and lead optimization and drug development. Each of the drug discovery and development areas requires the commitment of numerous highly talented scientists with variety of expertise, patience, perseverance, and problem solving skills. Each drug also has one or more champions who are willing to tackle the numerous road blocks that arise during the complex development process. The financial commitment for discovery, development and successful launch of a drug is substantial. However, in the uncommon circumstance of success, it is a very rewarding experience for all involved as exemplified by the successful approval of CANCIDAS®. By 2006, CANCIDAS® had become the number one intravenous antifungal drug worldwide and has saved the lives of tens of thousands of patients. It is this type of success that makes the years of trial and failure in the pharmaceutical industry worthwhile.
2 Acknowledgements
JMB wishes to acknowledge the invaluable contributions of Regina M. Black, F. Aileen Bouffard, James F. Dropinski, Milton L. Hammond, James V. Heck, Catherine James and Robert A. Zambias to the design, synthesis and medicinal chemical aspects of this work.3 References
- T. J. Walsh and A. Pizzo, Eur. J. Clin. Microbiol. Infect. Dis., 1988, 7, 460–475 CrossRef CAS.
- G. F. Bills, G. Platas, F. Pelaez and P. Masurekar, Mycological Res., 1999, 103, 179–192 CrossRef CAS.
- R. E. Schwartz, R. A. Giacobbe, J. A. Bland and R. L. Monaghan, J. Antibiot., 1989, 42, 163–167 CrossRef CAS.
- M. B. Kurtz and J. H. Rex, Adv. Prot. Chem., 2001, 56, 423–475 CrossRef CAS.
- C. F. Wichmann, J. M. Liesch and R. E. Schwartz, J. Antibiot., 1989, 42, 168–173 CrossRef CAS.
- R. A. Fromtling and G. K. Abruzzo, J. Antibiot., 1989, 42, 174–178 CrossRef CAS.
- E. T. Sawistowska-Schroder, D. Kerridge and H. Perry, FEBS Lett., 1984, 173, 134–138 CrossRef CAS.
- F. Benz, F. Knüsel, J. Nüesch, H. Treichler, W. Voser, R. Nyfeler and W. Keller-Schierlein, Helv. Chim. Acta, 1974, 57, 2459–2477 CrossRef CAS PubMed.
- J. C. Edman, J. A. Kovacs, H. Masur, D. V. Santi, H. J. Elwood and M. L. Sogin, Nature, 1988, 334, 519–522 CrossRef CAS PubMed.
- S. L. Stringer, J. R. Stringer, M. A. Blase, P. D. Walzer and M. T. Cushion, Exp. Parasitol., 1989, 68, 450–461 CrossRef CAS.
- U. Edman, J. C. Edman, B. Lundgren and D. V. Santi, Proc. Natl. Acad. Sci. U S A, 1989, 86, 6503–6507 CrossRef CAS.
- Y. Matsumoto, S. Matsuda and T. Tegoshi, J. Protozool., 1989, 36, 21S–22S CAS.
- D. M. Schmatz, M. A. Romancheck, L. A. Pittarelli, R. E. Schwartz, R. A. Fromtling, K. H. Nollstadt, F. L. Vanmiddlesworth, K. E. Wilson and M. J. Turner, Proc. Natl. Acad. Sci. U S A, 1990, 87, 5950–5954 CrossRef CAS.
- O. D. Hensens, J. M. Liesch, D. L. Zink, J. L. Smith, C. F. Wichmann and R. E. Schwartz, J. Antibiot., 1992, 45, 1875–1885 CrossRef CAS.
- R. E. Schwartz, P. S. Masurekar, and R. F. White, In Cutaneous Antifungal Agents, J. W. Rippon, and R. A. Fromtling, ed., Marcel Dekker, Inc.: New York, 1993, pp 375–393 Search PubMed.
- W. R. Leonard, Jr, K. M. Belyk, D. R. Bender, D. A. Conlon, D. L. Hughes and P. J. Reider, Org. Lett., 2002, 4, 4201–4204 CrossRef PubMed.
- A. E. Osawa, R. Sitrin and S. S. Lee, J. Chromatography A, 1999, 831, 217–225 CrossRef.
- D. J. Roush, F. D. Antia and K. E. Goklen, J.Chromatography A, 1998, 827, 373–389 CrossRef CAS.
- P. S. Masurekar, J. M. Fountoulakis, T. C. Hallada, M. S. Sosa and L. Kaplan, J. Antibiot., 1992, 45, 1867–1874 CrossRef CAS.
- K. Mizuno, A. Yagi, S. Satoi, M. Takada and M. Hayashi, J. Antibiot., 1977, 30, 297–302 CrossRef CAS.
- S. Satoi, A. Yagi, K. Asano, K. Mizuno and T. Watanabe, J. Antibiot., 1977, 30, 303–307 CrossRef CAS.
- J. Mizoguchi, T. Saito, K. Mizuno and K. Hayano, J. Antibiot., 1977, 30, 308–313 CrossRef CAS.
- K. Roy, T. Mukhopadhyay, G. C. S. Reddy, K. R. Desikan and B. N. Ganguli, J. Antibiot., 1987, 40, 275–280 CrossRef CAS.
- M. Debono, B. J. Abbott, J. R. Turner, L. C. Howard, R. S. Gordee, A. S. Hunt, M. Barnhart, R. M. Molloy, K. E. Willard and D. Fukuda, et al. , Ann. N Y Acad. Sci., 1988, 544, 152–167 CrossRef CAS.
- R. S. Gordee, D. J. Zeckner, L. F. Ellis, A. L. Thakkar and L. C. Howard, J. Antibiot., 1984, 37, 1054–1065 CrossRef CAS.
- M. Debono, W. W. Turner, L. LaGrandeur, F. J. Burkhardt, J. S. Nissen, K. K. Nichols, M. J. Rodriguez, M. J. Zweifel, D. J. Zeckner and R. S. Gordee, et al. , J. Med. Chem., 1995, 38, 3271–3281 CrossRef CAS.
- M. J. Rüping, J. J. Vehreschild, F. Farowski and O. A. Cornely, Expert Rev. Clin. Pharmacol., 2008, 1, 207–216 CrossRef.
- J. M. Balkovec, R. M. Black, M. L. Hammond, J. V. Heck, R. A. Zambias, G. Abruzzo, K. Bartizal, H. Kropp, C. Trainor, R. E. Schwartz, D. C. McFadden, K. H. Nollstadt, L. Pittarelli, M. A. Powles and D. Schmatz, J. Med. Chem., 1992, 35, 194–198 CrossRef CAS.
- M. L. Hammond, In Cutaneous antifungal agents, J. W. Rippon, R. A. Fromtling, ed., Marcel Dekker, Inc.: New York, 1993, p 395–420 Search PubMed.
- D. W. Denning and D. A. Stevens, Antimicrob. Agents Chemother., 1991, 35, 1329–1333 CrossRef CAS.
- H. R. Black, G. L. Brier, J. D. Wolny, and S. H. Dorrbecker In 29th Interscience Conference on Antimicrobial Agents and ChemotherapyHouston, 1989, p Abstract 1557 Search PubMed.
- C. R. Copley-Merriman, N. Ransburg, L. R. Crane, T. M. Kerkering, P. G. Pappas, J. C. Pottage, and D. L. Hyslop In 30th Interscience Conference on Antimicrobial Agents and ChemotherapyAtlanta, 1990, p Abstract 581 Search PubMed.
- C. R. Copley-Merriman, H. Gallis, J. R. Graybill, B. N. Doebbeling, and D. L. Hyslop In 30th Interscience Conference on Antimicrobial Agents and ChemotherapyAtlanta, 1990, p Abstract 582 Search PubMed.
- B. N. Doebbeling, B. D. Fine, M. A. Pfaller, C. T. Sheetz, J. B. Stokes, and R. P. Wenzel In 30th Interscience Conference on Antimicrobial Agents and ChemotherapyAtlanta, 1990, p Abstract 583 Search PubMed.
- J. W. Lee, P. Kelly, J. Lecciones, D. Coleman, R. Gordee, P. A. Pizzo and T. J. Walsh, Antimicrob. Agents Chemother., 1990, 34, 2240–2245 CrossRef CAS.
- D. M. Schmatz, G. Abruzzo, M. A. Powles, D. C. McFadden, J. M. Balkovec, R. M. Black, K. Nollstadt and K. Bartizal, J. Antibiot., 1992, 45, 1886–1891 CrossRef CAS.
- K. Bartizal, G. Abruzzo, C. Trainor, D. Krupa, K. Nollstadt, D. Schmatz, R. Schwartz, M. Hammond, J. Balkovec and F. Vanmiddlesworth, Antimicrob. Agents Chemother., 1992, 36, 1648–1657 CrossRef CAS.
- M. B. Kurtz, I. B. Heath, J. Marrinan, S. Dreikorn, J. Onishi and C. Douglas, Antimicrob. Agents Chemother., 1994, 38, 1480–1489 CrossRef CAS.
- G. K. Abruzzo, A. M. Flattery, C. J. Gill, L. Kong, J. G. Smith, D. Krupa, V. B. Pikounis, H. Kropp and K. Bartizal, Antimicrob. Agents Chemother., 1995, 39, 1077–1081 CrossRef CAS.
- F. A. Bouffard, M. L. Hammond and B. H. Arison, Tetrahedron Lett., 1995, 36, 1405–1408 CrossRef CAS.
- J. M. Balkovec and R. M. Black, Tetrahedron Lett., 1992, 33, 4529–4532 CrossRef.
- F. A. Bouffard, R. A. Zambias, J. F. Dropinski, J. M. Balkovec, M. L. Hammond, G. K. Abruzzo, K. F. Bartizal, J. A. Marrinan, M. B. Kurtz, D. C. McFadden, K. H. Nollstadt, M. A. Powles and D. M. Schmatz, J. Med. Chem., 1994, 37, 222–225 CrossRef CAS.
- R. A. Zambias, C. James, G. K. Abruzzo, K. F. Bartizal, R. Hajdu, R. Thompson, K. H. Nollstadt, J. Marrinan and J. M. Balkovec, Bioorg. Med. Chem. Lett., 1997, 7, 2021–2026 CrossRef CAS.
- R. A. Zambias, C. James, M. L. Hammond, G. K. Abruzzo, K. F. Bartizal, K. H. Nollstadt, C. Douglas, J. Marrinan and J. M. Balkovec, Bioorg. Med. Chem. Lett., 1995, 5, 2357–2362 CrossRef CAS.
- T. Satoh, S. Suzuki, Y. Suzuki, Y. Miyaji and Z. Imai, Tetrahedron Lett., 1969, 10, 4555–4558 CrossRef.
- M. B. Kurtz, C. Douglas, J. Marrinan, K. Nollstadt, J. Onishi, S. Dreikorn, J. Milligan, S. Mandala, J. Thompson and J. M. Balkovec, et al. , Antimicrob. Agents Chemother., 1994, 38, 2750–2757 CrossRef CAS.
- R. Hajdu, R. Thompson, K. White, B. Stark-Murphy, and H. Kropp In Interscience Conference on Antimicrobial Agents and ChemotherapyNew Orleans, 1993, p Abstract 357 Search PubMed.
- J. W. Dundee, J. P. Fee, J. Moore, P. D. McIlroy and D. B. Wilson, Anaesthesia, 1980, 35, 12–16 CrossRef CAS.
- J. M. Balkovec, R. M. Black, F. A. Bouffard, J. F. Dropinski, and M. L. Hammond In XIVth International Symposium on Medicinal ChemistryF. Awouters, Ed.; Elsevier Science B. V.: 1997 Search PubMed.
- K. Bartizal, C. J. Gill, G. K. Abruzzo, A. M. Flattery, L. Kong, P. M. Scott, J. G. Smith, C. E. Leighton, A. Bouffard, J. F. Dropinski and J. Balkovec, Antimicrob. Agents Chemother., 1997, 41, 2326–2332 CAS.
- G. K. Abruzzo, A. M. Flattery, C. J. Gill, L. Kong, J. G. Smith, V. B. Pikounis, J. M. Balkovec, A. F. Bouffard, J. F. Dropinski, H. Rosen, H. Kropp and K. Bartizal, Antimicrob. Agents Chemother., 1997, 41, 2333–2338 CAS.
- R. Hajdu, R. Thompson, J. G. Sundelof, B. A. Pelak, F. A. Bouffard, J. F. Dropinski and H. Kropp, Antimicrob. Agents Chemother., 1997, 41, 2339–2344 CAS.
- G. K. Abruzzo, C. J. Gill, A. M. Flattery, L. Kong, C. Leighton, J. G. Smith, V. B. Pikounis, K. Bartizal and H. Rosen, Antimicrob. Agents Chemother., 2000, 44, 2310–2318 CrossRef CAS.
- M. A. Chandler, K. E. Goklen, S. S. Lee, and D. J. Roush, US 6610822 ( 2003).
- J. Nti-Gyabaah, F. D. Antia, M. E. Dahlgren and K. E. Göklen, Biotechnol. Prog., 2006, 22, 538–546 CrossRef CAS PubMed.
- D. J. Roush, L. Y. Hwang and F. D. Antia, J. Chromatography A, 2005, 1098, 55–65 CrossRef CAS PubMed.
- W. R. Leonard, Jr, K. M. Belyk, D. A. Conlon, D. R. Bender, L. M. DiMichele, J. Liu and D. L. Hughes, J. Org. Chem., 2007, 72, 2335–2343 CrossRef PubMed.
- M. J. Nerurkar, W. A. Hunke, and M. J. Kaufman, US 5952300 ( 1993).
- N. Singh, Clin Infect Dis, 2001, 33, 1692–1696 CrossRef CAS PubMed.
- G. Bodey, B. Bueltmann, W. Duguid, D. Gibbs, H. Hanak, M. Hotchi, G. Mall, P. Martino, F. Meunier, S. Milliken, S. Naoe, M. Okudaira, S. Scevola and J. van'tWout, Eur. J. Clin. Microbiol. Infect. Dis., 1992, 11, 99–109 CrossRef CAS.
- M. A. Pfaller, P. G. Pappas and J. R. Wingard, Clin. Infect. Dis., 2006, 43, S3–S14 CrossRef CAS.
- S. Ascioglu, J. H. Rex, B. de Pauw, J. E. Bennett, J. Bille, F. Crokaert, D. W. Denning, J. P. Donnelly, J. E. Edwards, Z. Erjavec, D. Fiere, O. Lortholary, J. Maertens, J. F. Meis, T. F. Patterson, J. Ritter, D. Selleslag, P. M. Shah, D. A. Stevens, T. J. Walsh and C. Invasive Fungal Infections, Clin. Infect. Dis., 2002, 34, 7–14 CrossRef CAS PubMed.
- A. Villanueva, E. G. Arathoon, E. Gotuzzo, R. S. Berman, M. J. DiNubile and C. A. Sable, Clin. Infect. Dis., 2001, 33, 1529–1535 CrossRef CAS PubMed.
- J. E. Kaplan, D. Hanson, M. S. Dworkin, T. Frederick, J. Bertolli, M. L. Lindegren, S. Holmberg and J. L. Jones, Clin. Infect. Dis., 2000, 30, S5–S14 CrossRef PubMed.
- J. A. Sangeorzan, S. F. Bradley, X. G. He, L. T. Zarins, G. L. Ridenour, R. N. Tiballi and C. A. Kauffman, Am. J. Med., 1994, 97, 339–346 CrossRef CAS.
- J. H. Rex, T. J. Walsh, J. D. Sobel, S. G. Filler, P. G. Pappas, W. E. Dismukes and J. E. Edwards, Clin. Infect. Dis., 2000, 30, 662–678 CrossRef CAS PubMed.
- E. G. Arathoon, E. Gotuzzo, L. M. Noriega, R. S. Berman, M. J. DiNubile and C. A. Sable, Antimicrob. Agents Chemother., 2002, 46, 451–457 CrossRef CAS.
- J. Mora-Duarte, R. Betts, C. Rotstein, A. L. Colombo, L. Thompson-Moya, J. Smietana, R. Lupinacci, C. Sable, N. Kartsonis, J. Perfect and S. Caspofungin Invasive Candidiasis, New Eng. J. Med., 2002, 347, 2020–2029 CrossRef CAS PubMed.
- T. F. Patterson, W. R. Kirkpatrick, M. White, J. W. Hiemenz, J. R. Wingard, B. Dupont, M. G. Rinaldi, D. A. Stevens, J. R. Graybill and I. A. S. Grp, Medicine, 2000, 79, 250–260 CrossRef CAS PubMed.
- K. A. Marr, R. A. Carter, F. Crippa, A. Wald and L. Corey, Clin. Infect. Dis., 2002, 34, 909–917 CrossRef PubMed.
- D. L. Paterson and N. Singh, Medicine, 1999, 78, 123–138 CrossRef CAS PubMed.
- S. J. Lin, J. Schranz and S. M. Teutsch, Clin. Infect. Dis., 2001, 32, 358–366 CrossRef CAS PubMed.
- J. Maertens, I. Raad, G. Petrikkos, M. Boogaerts, D. Selleslag, F. B. Petersen, C. A. Sable, N. A. Kartsonis, A. Ngai, A. Taylor, T. F. Patterson, D. W. Denning and T. J. Walsh, Clin. Infect. Dis., 2004, 39, 1563–1571 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |