DOI:
10.1039/C4NJ01363H
(Paper)
New J. Chem., 2015,
39, 508-519
Spin crossover and high spin electroneutral mononuclear iron(III) Schiff base complexes involving terminal pseudohalido ligands†
Received
(in Montpellier, France)
12th August 2014
, Accepted 28th October 2014
First published on 28th October 2014
Abstract
Investigations into a series of six novel mononuclear iron(III) Schiff base complexes with the general formula [Fe(L)X] (where H2L is a pentadentate Schiff-base ligand, X = pseudohalido ligand) are reported. Several different aromatic 2-hydroxyaldehyde derivatives were used in combination with N,N′-bis(2-aminoethyl)-1,3-propanediamine (compounds 1–5) and 2,2′-diaminodiethylamine (for 6) to synthesize the H2L Schiff base ligands. The consecutive reactions with iron(III) chloride resulted in the preparation of the [Fe(L)Cl] precursor complexes which were left to react with pseudohalido ligands (NCS− for 1, 2, 3, 4, 6; N3− for 4). Structural investigations revealed a usual coordination of the pentadentate Schiff base ligands via N3O2 donor atoms and the sixth coordination place is occupied by the N donor of the corresponding pseudohalido ligand. The spin crossover was observed in two cases with transition temperatures: Tc = 83 K (hysteresis width of ΔT = 2 K) for 1 and Tc = 174 K for 2. Magnetic investigations of compounds 3–6 revealed high spin behaviour. The magnetic data of all compounds were analysed using the spin Hamiltonian formalism including the zero-field splitting term and the molecular field effect. In the case of the spin crossover complexes 1 and 2, the Ising-like model was applied.
Introduction
Current projection of spin crossover (abbr. SCO) material research is focused on the goal-directed development of coordination compounds with application in memory devices,1 in displays2 or sensors3 and more recently in hybrid electronics4 and optoelectronics.5 Besides the well-known technology requirements for the abrupt, room temperature and hysteretic shape of the transition,1b,6 new trends place importance on the eventuality to utilize SCO nanoparticles,7 thin films or monolayers8 and on the manufacturing of functionalized surfaces in a logical pattern.9 Thus, it is still a great challenge to design and build SCO switchable systems with molecular structures capable of attaching on the surface by various lithography9 or sublimation10 techniques.
Among the possible candidates for manufacturing of SCO bistable nanoparticles or surfaces, the family of iron(III) complexes with pentadentate Schiff base N3O2 donor ligands (H2L) has an outstanding position. The simplicity of the ligand synthesis and high stability provided by the chelating effects signify advantages of the pentadentate Schiff base ligands. The corresponding iron(III) complexes of the general formula [Fe(L)Cl] offer the possibility of replacing the chlorido ligands with other N-donor ligands and in this sense, to vary structural, physical and chemical parameters of the {Fe(L)}+ paramagnetic moiety. For instance, the employment of various bridging polycyanometalate anions [M(CN)x]y− resulted in a wide pallet of low dimensional polynuclear compounds with a large ground spin state11 or with gradual spin crossover behaviours.12 In contrast, more abrupt,13 two step14 or room temperature spin transitions15 were observed when the interconnection of the iron(III)–N3O2 paramagnetic units was mediated via the pyridine, pyrazine or imidazole type of bridging ligands. From the application point of view, special attention is paid to mononuclear complexes with terminal pyridine or imidazole photoactive ligands (i.e. containing stilben, azapyridines etc.) which are able to create ligand driven light induced SCO.16 The introduction of pseudohalido ligand anions (X) into the coordination sphere of the iron(III) central atoms leads to the formation of mononuclear complexes [Fe(L)X]. Besides the low-spin (LS) cyanido complexes, the presence of anions providing weaker ligand field strengths i.e. X = NCS−, NCSe−, NCO−, or N3− often induces the SCO behaviour. Beneficially, the sulfur atom involved in pseudohalides and/or functional groups of the Schiff base ligand opens up the possibility of anchoring this type of SCO material on the gold surfaces or nanoparticles.17 Moreover, the neutral character of the complexes predetermines this class of SCO materials for the surface deposition by sublimation.
The design of the already reported [Fe(L)X] SCO complexes has been deliberately oriented toward the use of Schiff base ligands with asymmetric aliphatic parts derived from propyl-ethyl-triamine.18 The cooperativity between the potential SCO units was energized by systematic introduction of alkoxy groups, halogen atoms, aromatic moieties and/or by variation of terminal pseudohalido ligands. However, since the observed spin state transitions possess rather gradual character with a very rare occurrence of a thermal hysteresis loop, it is still necessary to explore and understand more structural, chemical and physical effects within the corresponding family of neutral SCO compounds.
Herein we report on the new contribution to the series of iron(III) Schiff base complexes with terminal pseudohalido ligands. Six new compounds were prepared by a bottom-up synthetic approach from the corresponding pentadentate ligand (H2L) and chlorido precursor complexes [Fe(L)Cl]. Magnetic investigations revealed the presence of spin transition bistability in two cases; the other four compounds stay in the high-spin (HS) state over the whole temperature range. All the complexes were structurally characterized; the crystal structures of spin crossover complexes were investigated for both LS and HS states.
Results and discussion
Synthesis and spectral properties of the reported compounds
The pentadentate Schiff base ligands H2L1–H2L6 (Scheme 1) were prepared in almost quantitative yields by Schiff condensation between the derivatives of 2-hydroxybenzaldehyde or 2-hydroxynaphthaldehyde and corresponding aliphatic triamine. In the consecutive complexation with iron(III) salt, the mononuclear complexes of the general formula [Fe(L)Cl] were synthesized and used in the third step, where the chlorido ligand was replaced by pseudohalide anions, which resulted in the formation of complexes 1–6 with the general formula [Fe(L)X] (X = NCS− for 1, 2, 3, 5, 6; X = N3− for 4). Single crystals suitable for diffraction analysis were obtained in several days after controlled evaporation of acetone or methanol solutions.
 |
| | Scheme 1 Schematic representations of three ligand structural types: (a) R1 = –O–CH2–CH3, R2 = H, H2L1 = N,N′-bis(3-ethoxy-2-hydroxybenzylidene)-1,6-diamino-3-azahexane; R1 = H, R2 = –NO2, H2L3 = N,N′-bis(2-hydroxy-5-nitro-benzylidene)-1,6-diamino-3-azahexane; R1 = –C–(CH3)3, R2 = –C–(CH3)3, H2L5 = N,N′-bis(3,5-di-tertbutyl 2-hydroxybenzylidene)-1,6-diamino-3-azahexane; (b) H2L2 = N,N′-bis(-2-hydroxynaphtylidene)-1,6-diamino-3-azahexane; (c) H2L6 = N,N′-bis(3-ethoxy-2-hydroxybenzylidene)-1,5-diamino-3-azapentane. | |
Infrared spectroscopy revealed many common vibrational bands of 1–6 and the corresponding mononuclear [Fe(L)Cl] complexes, especially in the fingerprint region. The major difference lies in the presence of the strong vibration around 2050 cm−1 related to the pseudohalido ligand stretching vibrations. A significant red shift was found in the azido vibration of complex 4 (2056 cm−1), whereas the vibration of the uncoordinated azido anion in NaN3 can be found at 2128 cm−1. On the other hand, the coordinated NCS− anions of compounds 1–6 vary in the range of 2044–2070 cm−1 which is typical for isothiocyanato ligands and show only tiny differences in comparison to the vibrations of uncoordinated KNCS (2049 cm−1). The vibrational bands of the secondary amino group in the family of [Fe(L)X] complexes usually adopt values from the range of 3260–3220 cm−1. The red shift of valence vibration frequencies below 3220 cm−1 indicates the presence of hydrogen bonded interconnections. Indeed, in the case of the compounds 1, 2, 3 and 6 rather strong hydrogen bonds between the secondary amino nitrogen atom and either oxygen or sulfur atoms (vide infra) were observed and therefore their N–H vibrations are placed bellow 3220 cm−1. In particular, the lowest N–H vibration was observed at 3158 cm−1 in the family of [Fe(L)X] compounds for the compound 6, where the length of N⋯O hydrogen bonds is 2.878(2) Å at 100 K.
In general, the electronic spectra of iron(III) complexes are not a very sensitive probe of the structure. The d–d transitions of HS iron(III) with the ground state 6A1 are both spin and Laporte forbidden and are usually obscured by strong ligand-to-ligand or metal-to-ligand charge transfer bands.19 UV-VIS absorption spectra of the herein reported iron(III) complexes were recorded in the acetonitrile solutions (Fig. S1, ESI†). The absorption bands located in the visible light region (524–561 nm) are typical to the ligand-to-metal charge transfer of phenolato and pseudohalido ligands to the iron(III) central atom.20 For the bands in the region 305–355 nm the assignment pπ → dσ* charge transfer is suggested. The high intensity of the charge transfer bands is likely caused by mixing of the low-lying phenyl and pseudohalido π → π* transition.
Structural properties
Single crystal X-ray diffraction studies revealed very similar molecular structures for the herein reported mononuclear complexes. The iron(III) central atom is coordinated by the corresponding pentadentate Schiff base ligand through N3O2 donor atoms (Fig. 1). Two imino and one amino nitrogen donors of the Schiff base ligand are placed in the facial conformation only in the structure of 6, whereby the meridian arrangement was observed in all other cases of the reported complexes. All the compounds contain two phenolato oxygen atoms arranged in the cis position. The sixth coordination site is occupied by one N-coordinated pseudohalido ligand (NCS− for 1, 2, 3, 5, 6; N3− for 4), thus all six complexes are neutral compounds and do not contain lattice solvent molecules in their crystal structures.
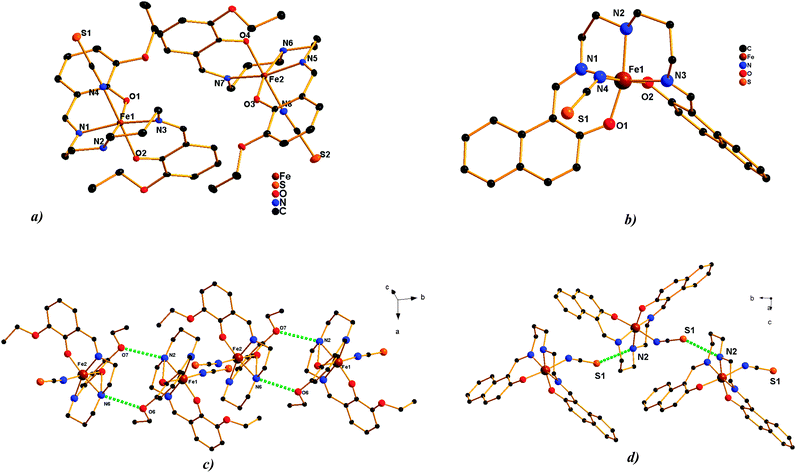 |
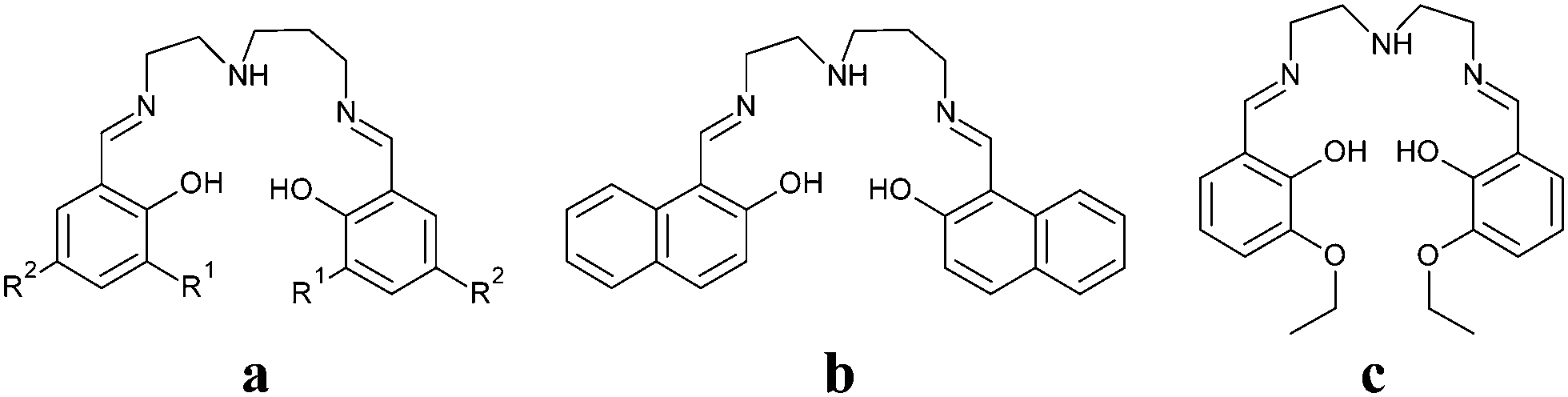
| | Fig. 1 Asymmetric units of complex 1 (a) and complex 2 (b) a perspective view on N(am)⋯O(ethoxy) hydrogen bonding (green dashed line) in 1 (c). Corresponding bond distances: d(N6⋯O6) = 3.179(4) Å, (293 K); 3.129(2), (100 K); 3.095(2), (15 K) and d(N2⋯O7) = 3.191(4) Å, (293 K); 3.140(2), (100 K); 3.156(2), (15 K). A perspective view on N(am)⋯S hydrogen bonding (green dashed line) in 2 (d). The corresponding bond distances: d(N2⋯S1) = 3.442(2), Å (298 K); 3.279(2), (100 K). | |
In order to detect the structural changes induced by spin transition (vide infra), single crystal X-ray diffraction analysis of compound 1 was performed at three different temperatures: 15 K, 100 K and 298 K. All three measurements revealed the monoclinic P21/c space group with the unit cell parameters listed in Table 1. A noticeable increase of the c lattice constant (about 5%) as well as growing of unit cell volume by about 107 Å3 (2.3%) are associated with both SCO and the increase in temperature from 15 K to 100 K. The asymmetric unit consists of two molecules of 1 with the general formula [Fe(L1)(NCS)]. At 15 K, the Fe–N(im) and Fe–N(ps) bond distances are indicative of the LS iron(III) compounds, being 1.943 Å in average, whereas the Fe–N(am) bond lengths acquire slightly higher values (2.016 Å). Upon going through the spin transition, there is a very marked increase in nitrogen bond distances. The Fe–N(im) bonds increase by about 0.16 Å (Fe1–N1, Fe2–N5) or 0.12 Å (Fe1–N3, Fe2–N7), Fe–N(ps) by about 0.14 Å and the most significant prolongation was observed in the case of the Fe–N(am) bond (Δd = 0.18 Å). As was already observed previously, the Fe–O bond distances do not reflect the changes upon the spin transition noticeably and the tiny difference observed between the LS and HS structures is most probably related to thermal variation only.18a During the SCO action the coordination polyhedron becomes more distorted from the ideal octahedral geometry, as demonstrated by an increase in the spin-state dependent Σ parameter,21Table 2. The equatorial angles created between the imino nitrogen, iron(III) centre and oxygen atom located on the other side of the Schiff base ligand (O1–Fe1–N3(im) and O3–Fe2–N7(im), Fig. 1) undergo the most significant deformations upon the spin transition (Table 2). Their HS and LS values differ by about 15 degrees and therefore they present the most relevant contribution to the variation of the Σ parameter. Investigation of intermolecular contacts in 1 revealed the presence of hydrogen bonding interconnections (Fig. 1) of two neighbouring molecules between the methoxy oxygen (O6, O7) and amino nitrogen atoms (N2, N6). The N⋯O distances get slightly shorter upon cooling (Fig. 1).
Table 1 Crystallographic data for the reported compounds
| |
Compound 1 |
Compound 2 |
 . .
|
| Formula |
C24H29FeN4O4S |
C24H29FeN4O4S |
C24H29FeN4O4S |
C28H25FeN4O2S |
C28H25FeN4O2S |
| Formula weight per g mol−1 |
525.42 |
525.42 |
525.42 |
537.43 |
537.43 |
| Crystal colour |
Black |
Black |
Black |
Black |
Black |
| Temperature/K |
298(2) |
100(2) |
15(2) |
298(2) |
100(2) |
| Wavelength/Å |
0.71073 |
0.71073 |
0.38745 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
Orthorhombic |
| Space group |
P21/c |
P21/c |
P21/c |
P212121 |
P212121 |
|
a/Å |
19.3340(7) |
19.0809(7) |
19.165(3) |
7.5422(3) |
7.5011(6) |
|
b/Å |
11.4560(3) |
11.3904(4) |
11.6749(16) |
13.5725(5) |
13.0676(10) |
|
c/Å |
23.8310(9) |
23.6108(4) |
22.449(3) |
24.6626(9) |
24.704(53) |
|
α/° |
90 |
90 |
90 |
90 |
90 |
|
β/° |
107.131(4) |
107.420(2) |
107.772(3) |
90 |
90 |
|
γ/° |
90 |
90 |
90 |
90 |
90 |
|
V/Å3 |
5044.2(3) |
4896.2(3) |
4783.3(11) |
2524.62(16) |
2421.5(4) |
|
Z, ρcalc/g cm−3 |
8, 1.384 |
8, 1.426 |
8, 1.459 |
4, 1.414 |
4, 1.474 |
| Radiation type |
Mo-Kα |
Mo-Kα |
Synchrotron |
Mo-Kα |
Mo-Kα |
|
μ/mm−1 |
0.718 |
0.739 |
0.122 |
0.713 |
0.744 |
|
F(000) |
2200 |
2200 |
2200 |
1116 |
1116 |
| Crystal size/mm |
0.50 × 0.23 × 0.21 |
0.38 × 0.23 × 0.20 |
0.38 × 0.23 × 0.20 |
0.32 × 0.09 × 0.09 |
0.32 × 0.09 × 0.09 |
|
θ range for the data collection/° |
2.58 to 27.81 |
2.16 to 27.13 |
1.03 to 14.39 |
2.90 to 25.00 |
2.92 to 25.00 |
| Final R indices [I > 2σ(I)]a |
R
1 = 0.0571, wR2 = 0.0933 |
R
1 = 0.0322, wR2 = 0.0699 |
R
1 = 0.0352, wR2 = 0.0698 |
R
1 = 0.0330, wR2 = 0.0578 |
R
1 = 0.0498, wR2 = 0.0857 |
|
R indices (all data)a |
R
1 = 0.1083, wR2 = 0.1073 |
R
1 = 0.0478, wR2 = 0.0764 |
R
1 = 0.0577, wR2 = 0.0781 |
R
1 = 0.0498, wR2 = 0.0606 |
R
1 = 0.0790, wR2 = 0.0919 |
| GoF |
1.046 |
1.029 |
1.002 |
0.897 |
0.935 |
| CCDC number |
995957
|
995955
|
995956
|
995959
|
995958
|
| |
Compound 3 |
Compound 4 |
Compound 5 |
Compound 6 |
| Formula |
C20H19FeN6O6S |
C23H29FeN6O4 |
C36H53FeN4O2S |
C23H27FeN4O4S |
| Formula weight per g mol−1 |
527.32 |
509.37 |
661.73 |
511.39 |
| Crystal colour |
Black |
Black |
Black |
Black |
| Temperature/K |
150(2) |
100(2) |
150(2) |
100(2) |
| Wavelength/Å |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Orthorhombic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
Pbcn
|
P21/n |
P21/c |
P21/n |
|
a/Å |
15.9030(5) |
11.0321(9) |
18.2879(7) |
11.5785(6) |
|
b/Å |
14.2180(5) |
11.3322(9) |
11.8974(4) |
11.6541(5) |
|
c/Å |
19.6342(5) |
18.9441(15) |
17.3763(7) |
18.2132(8) |
|
α
|
90 |
90 |
90 |
90 |
|
β
|
90 |
96.974(3) |
96.903(4) |
106.508(2) |
|
γ
|
90 |
90 |
90 |
90 |
|
V/Å3 |
4439.5(2) |
2350.8(3) |
3753.3(2) |
2356.33(19) |
|
Z, ρcalc/g cm−3 |
8, 1.578 |
4, 1.439 |
4, 1.171 |
4, 1.442 |
| Radiation type |
Mo-Kα |
Mo-Kα |
Mo-Kα |
Mo-Kα |
|
μ (Mo-Kα)/mm−1 |
0.825 |
0.684 |
0.492 |
0.766 |
|
F(000) |
2168 |
1068 |
1420 |
1068 |
| Crystal size/mm |
0.37 × 0.22 × 0.16 |
— |
0.29 × 0.22 × 0.21 |
— |
|
θ range for the data collection/° |
3.05 to 25.00 |
4.13 to 26.37 |
2.92 to 25.00 |
4.13 to 26.37 |
| Final R indices [I > 2σ(I)]a |
R
1 = 0.0383, wR2 = 0.0910 |
R
1 = 0.0249, wR2 = 0.0656 |
R
1 = 0.0393, wR2 = 0.0884 |
R
1 = 0.0318, wR2 = 0.0790 |
|
R indices (all data)a |
R
1 = 0.0533, wR2 = 0.0943 |
R
1 = 0.0281, wR2 = 0.0679 |
R
1 = 0.0671, wR2 = 0.0940 |
R
1 = 0.0421, wR2 = 0.0847 |
| GoF |
1.052 |
1.062 |
0.889 |
1.037 |
| CCDC number |
995960
|
995961
|
995962
|
995963
|
Table 2 Selected bond distances, angles and structural parameters of the reported compounds. Fe–N(im), Fe–N(am), Fe–N(ps) denotation represent the distances for the bonds between the iron(III) central atom and the corresponding imino (im), amino (am) and pseudohalido (ps) nitrogen donor atoms
|
|
1
|
| Fe1 |
Fe2 |
|
T/K |
293 |
100 |
15 |
293 |
100 |
15 |
|
O1–Fe–N3(im) equatorial angle does not exist due to the facial conformation of two N(im) and one N(am) in the crystal structure of 6.
|
| Fe–N(im)/Å |
2.084(3) |
2.083(2) |
1.922(2) |
2.087(3) |
2.091(2) |
1.923(2) |
| Fe–N(im)/Å |
2.077(3) |
2.082(2) |
1.958(2) |
2.082(3) |
2.089(2) |
1.962(2) |
| Fe–N(am)/Å |
2.194(3) |
2.182(2) |
2.013(2) |
2.201(3) |
2.200(2) |
2.015(2) |
| Fe–N(ps)/Å |
2.080(3) |
2.082(1) |
1.947 |
2.069(3) |
2.076(2) |
1.938(2) |
| Fe–O1/Å |
1.916(2) |
1.920(1) |
1.904(2) |
1.916(3) |
1.929(1) |
1.916(2) |
| Fe–O2/Å |
1.940(2) |
1.937(1) |
1.867(1) |
1.940(2) |
1.943(1) |
1.866(2) |
| O1FeN(im)/° |
106.0(1) |
105.35(6) |
90.36(7) |
107.4(1) |
107.42(6) |
91.34(7) |
|
Σ/° |
50.5 |
50.4 |
22.5 |
58.2 |
59.3 |
28.4 |
|
d(Fe⋯Fe)/Å |
7.0268(7) |
7.0127(5) |
7.2597(9) |
— |
— |
— |
|
|
2
|
3
|
4
|
5
|
6
|
|
T/K |
298 |
100 |
150 |
100 |
150 |
100 |
| Fe–N(im)/Å |
2.059(3) |
1.910(5) |
2.085(2) |
2.104(1) |
2.095(2) |
2.118(2) |
| Fe–N(im)/Å |
2.057(3) |
1.950(5) |
2.095(2) |
2.094(1) |
2.090(2) |
2.162(2) |
| Fe–N(am)/Å |
2.167(3) |
1.992(5) |
2.203(2) |
2.213(1) |
2.198(2) |
2.235(2) |
| Fe–N(ps)/Å |
2.128(3) |
1.979(6) |
2.079(2) |
2.055(1) |
2.097(2) |
2.084(2) |
| Fe–O1/Å |
1.902(2) |
1.889(4) |
1.935(2) |
1.971(1) |
1.896(2) |
1.948(1) |
| Fe–O2/Å |
1.956(2) |
1.881(4) |
1.953(2) |
1.931(1) |
1.943(2) |
1.918(1) |
| O1FeN(im)/° |
106.55(8) |
91.89(19) |
111.12(8) |
108.75(4) |
107.47(8) |
—a |
|
Σ/° |
51.7 |
23.0 |
54.2 |
64.3 |
55.4 |
94.0 |
|
d(Fe⋯Fe)/Å |
7.6669(5) |
7.267(2) |
5.6735(5) |
6.4909(5) |
7.3521(5) |
5.0220(4) |
The structural investigation revealed that 2 is a solvent free analogue of [Fe(L2)(NCS)]·CH3CN and it is isostructural with the [Fe(L2)(NCO)] spin crossover complex reported previously.18a
The diffraction study of 2 was performed at two temperatures (100 K, 298 K) in order to determine the crystal structures close to the pure LS or the HS state of the [Fe(L2)(NCS)] molecules (vide infra). Both measurements revealed the orthorhombic P212121 space group with the unit cell parameters listed in Table 1.The most significant prolongation with the temperature change was detected for the b lattice constant (increase about 3.7%). The unit cell volume difference Vuc(298 K)–Vuc(100 K) yields the volume of SCO VSCO = 103 Å3. The asymmetric unit contains one molecule of the complex with the general formula [Fe(L2)(NCS)] (Fig. 1). The metal–ligand bond lengths (Table 2) from the measurement performed at 100 K correspond to the LS state. The presence of the residual HS fraction in 2 at 100 K does not affect the bond lengths noticeably. The room temperature Fe–N(am) and Fe–N(im) bond lengths do not fully adopt the HS values with d(Fe–N2(am)) = 2.167(3) Å (typically 2.18–2.20 Å) and d(Fe–N(im)) = 2.059 Å (pair bond length average, typically 2.07–2.09 Å). In contrast, the iron-isothiocyanato nitrogen bond is significantly longer than usually reported for the HS ferric complexes (d(Fe–N(ps)) = 2.128(3) Å).18 Also, the octahedral distortion parameter Σ is a little bit lower than the usual value reported for the HS compounds when Σ = 51.7° (typically 54–60°), but it must be noted that similar values were observed also in the HS state of 1 (Fe1 coordination centre) and for the almost pure HS structure of the [Fe(L2)(NCSe)]·CH3CN compound.18a In conclusion, the molecular structure of 2 can be considered as the special case of the HS structure in the [Fe(L)X] series and this is in accordance with the magnetic data at the room temperature (Fig. 5, μeff = 6 μB).
In the crystal structure of 2 one dimensional chains of the [Fe(L2)(NCS)] molecules are formed by hydrogen bonding between the amino nitrogen N2 (donor) and sulfur atom S1. The comparison between the LS and HS structure reveals a significant difference: dLS(N2⋯S1) = 3.270(5) Å and dHS(N2⋯S1) = 3.440(3) Å; Δd(N2⋯S1) = 0.17 Å. The crystal structure of 2 lacks the straight stacking interactions of the aromatic rings, only twisted conformation of π–π and CH⋯π interactions can be found, similar to [Fe(L2)(NCO)] (Fig. 1).
The structural investigations of the purely HS complexes 3–6 (Fig. 2 and 3) revealed the orthorhombic Pbcn space group for 3, monoclinic P21/n for 4 and 6 and monoclinic P21/c for 5, with the unit cell parameters shown in Table 1. All kinds of the Fe–N bond distances (Table 2) in the crystal structures of 3–5 vary in the expected range typical for the HS iron(III) coordination centre. The facial conformation of the N-donor atoms in compound 6 is most probably responsible for the slightly higher Fe–N(am) and Fe–N(im) bond lengths as well as for the axial arrangement of the O1–Fe1–N3(im) angle, which in the equatorial case of 1–5 is very dependent on the spin state of the coordination centre (Table 2). For 3–5, the Σ structural parameters vary in the range of 54.2–64.3° and undoubtedly confirm the HS state of the related compounds at the temperature of the measurement. The facial arrangement of the N-donor atoms in the structure of 6 causes significantly stronger deformation of the {FeN3O2N′} chromophore expressed by the elevated value of Σ = 94°.
 |
| | Fig. 2 Asymmetric units of complex 3 (a) and complex 4 (b). | |
 |
| | Fig. 3 Asymmetric units of complex 5 (a) and complex 6 (b). | |
A careful study of the non-covalent interactions in the crystal structure of 3 revealed the presence of interesting contacts between the neighbouring molecules within the crystal lattice (Fig. S2–S4, ESI†). The neighbouring molecules of 3 are interconnected into pseudodimers through short contacts between the oxygen/nitrogen atoms of the nitro functional groups (d(O4⋯O6) = 2.922(3) Å, d(O4⋯N6) = 2.914(4) Å, sum of the van der Waals radii for the O⋯O pair is 3.04 Å and for the O⋯N pair is 3.07 Å). In addition, the pseudodimers are linked with other neighbouring dimers through different π–π interactions of the nitro oxygen atoms (d(O4⋯O4′) = 3.443(3) Å) and through a short contact between the NO2 oxygen and sulfur atom (d(O3⋯S1) = 3.265(2) Å, the sum of van der Waals radii for the O⋯S pair is 3.32 Å) along the bc plane. Moreover, such a two dimensional network (Fig. S3, ESI†) is linked with the third direction through hydrogen bonds between the Schiff base ligand amino nitrogen atoms and oxygen of the nitro functional group (d(O3⋯N2) = 3.071(3) Å; Fig. S4, ESI†).
In the crystal structure of 5, the amino nitrogen atom connects the sulfur atoms of neighbouring molecules through very weak hydrogen bonds (3.529(2) Å) and in such a manner they form a one-dimensional supramolecular chain similar to that already mentioned in the case of the crystal structure of 2 (Fig. S5, ESI†). Non-covalent contacts between the phenolato oxygen and amino nitrogen donor atoms with the distance d(N2⋯O1) = 2.878(2) Å were found also in the crystal structure of 6. Such rather strong hydrogen bonds interconnect two neighbouring complexes (Fig. S7, ESI†) and therefore compound 6 can be considered as a pseudodimer.
Magnetic properties
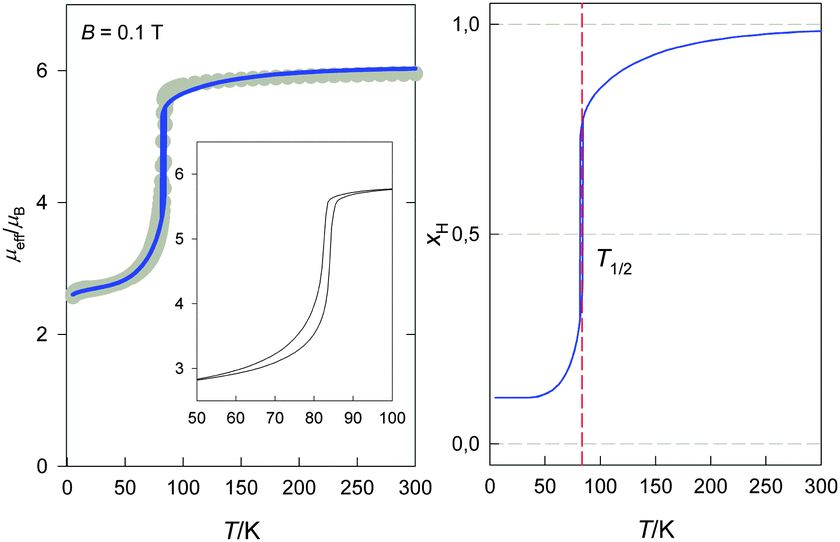
Magnetic investigation of 1 showed an abrupt spin crossover located below 100 K (Fig. 4). The measurement performed in the heating and cooling mode revealed the presence of a thermal hysteresis of the width ΔT = 2 K with transition temperatures Tc↑ = 84 K and Tc↓ = 82 K, respectively. While the high temperature value of the effective magnetic moment of about 5.95 μB is in good agreement with that expected for the S = 5/2 spin system (5.92 μB, gH = 2.00), the low temperature value of about 2.7 μB suggests the presence of a small amount of a remnant HS fraction. The small decrease of the effective magnetic moment below 20 K can be attributed to the intermolecular antiferromagnetic interactions (molecular field effect) and/or to the zero field splitting (abbr. ZFS) of the residual HS molecules of 1.
 |
| | Fig. 4 Magnetic data for 1: effective magnetic moment vs. temperature (left), calculated high-spin fraction vs. temperature (right); grey circles: experimental data, solid line: fitted, dashed line: calculated. | |
The experimental curves were interpreted quantitatively at the level of the spin Hamiltonian combined with the Ising-like model allowing a reliable reconstruction of the system behaviour in the whole temperature interval (see section Theoretical analysis). The final parameters for the SCO system 1 are presented in Table 3. In the frame of the used model it was proved that 13% of the molecules preserved the HS state upon cooling down. The deduced transition enthalpy is ΔH = 0.98 kJ mol−1 and the transition entropy is ΔS = 11.8 J K−1 mol−1. The calculated transition temperature is Tc = 83.1 K which lies within the experimental hysteresis loop. Both the above mentioned hypotheses for the low temperature decrease of the magnetic moment were tested and better fits were obtained with the employment of the molecular field for the LS state. A relatively high value of temperature-independent magnetism (TIM) had to be considered for 1 in order to grasp satisfactorily the slope of the LS region of the curve. The rare shape of the hysteresis loop can be a consequence of the grain distribution of the powder sample.27
Table 3 Summary of magnetic parameters for spin crossover complexes 1 and 2
| Complex |
g
L
|
χ
TIM,L
|
(zj)/cm−1 |
g
H
|
χ
TIM,H
|
x
frz
|
E/K |
γ/K |
ν
H/cm−1 |
ν
L/νH |
R(χ) |
|
In units of 10−9 m3 mol−1.
|
|
1, [Fe(L1)(NCS)] |
1.95 |
19.5 |
−0.48 |
2.05 |
— |
0.13 |
118 |
90.2 |
200 |
1.32 |
0.033 |
|
2, [Fe(L2)(NCS)] |
2.32 |
1.2 |
−1.27 |
2.03 |
15.1 |
0.13 |
370 |
135 |
255 |
1.29 |
0.004 |
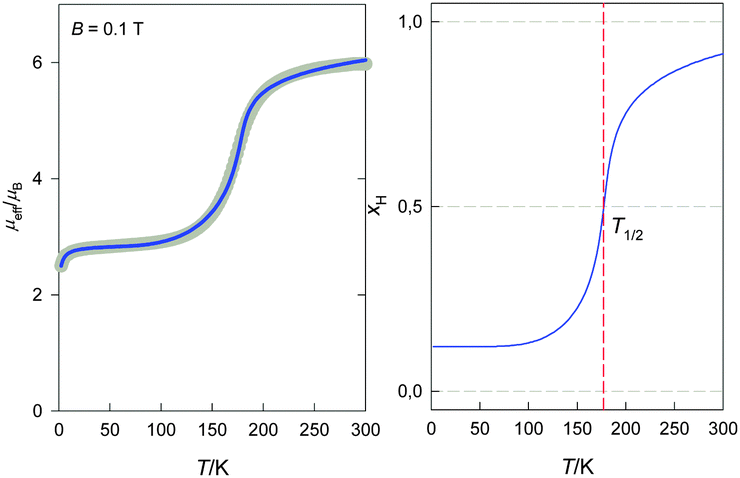
A gradual spin crossover was observed for 2. Magnetic investigation revealed non-hysteretic spin transition with Tc = 174 K (Fig. 5). The effective magnetic moment at the room temperature is slightly higher (ca. 6 μB) than the spin-only value for S = 5/2 and it slowly decreases upon cooling. The main decrease of μeff takes place between 230 K and 150 K and the low temperature value of μeff is almost constant between 20–120 K at ca. 2.9 μB. One can conclude that 2 also exhibits incomplete spin crossover, similar to that was observed for its previously reported solvated analogues [Fe(L2)(NCS)]·CH3CN, (Tc = 151 K) and for [Fe(L2)(NCSe)]·CH3CN (Tc = 170 K). On the other hand, the complete SCO has been observed in the case of next two compounds with L22− ligand dianions – for the isostructural [Fe(L2)(NCO)] (T1/2 = 155 K) and for [Fe(L2)(N3)] where SCO is accompanied by the 5 K hysteresis loop (T1/2↑ = 122 K and T1/2↓ = 117 K).18a Again, the decrease of μeff below 20 K is present and attributed to the molecular field on the basis of the fitting outcomes (Table 3). In this case the remnant HS fraction was again 13%, the thermodynamic characteristics were determined to be ΔH = 3.08 kJ mol−1, ΔS = 17.1 J K−1 mol−1 and the calculated transition temperature Tc = 180 K. At this temperature the HS fraction is 56%, differing from expected 50% due to the frozen HS molecules.
 |
| | Fig. 5 Magnetic data for 2: effective magnetic moment vs. temperature (left), calculated high-spin fraction vs. temperature (right); grey circles: experimental data, solid line: fitted, dashed line: calculated. | |
The magnetic investigation of complexes 3–6 revealed a HS paramagnetic behaviour (Fig. 6–9) and in the temperature range of 300–50 K the compounds obey the Currie law. In order to provide higher reliability of the resulted parameters the temperature and magnetic field dependencies were fitted simultaneously. Similar to that in the previous two systems ZFS and the molecular field were taken into account, however, for 6 also an additional “pseudodimer” antiferromagentic exchange interaction was necessarily included. The optimum parameters are shown in Table 4. The expected weak axial zero-field splitting (reflected in the low value of D) and g-factor values close to 2.0 were proven in all cases. Small magnetic anisotropy justifies also the used approximation of the isotropic g-factor. For these low values of the D parameter its sign cannot be reliably acquired from magnetic susceptibility as a function of temperature, however, the shape of the magnetization as a function of the magnetic field is more sensitive in this sense even for small values of D.22b The alternative calculation with opposite sign provided less accurate fits for magnetization than those presented in Table 4 in cases 3, 5 and 6, in the case 4 it was of similar quality and the correct sign stays questionable (Fig. S9–S12 and Table S1, ESI†). The lower value of D for 4 can be ascribed to a different pseudohalido (azido) ligand in the chromophore, while the negative splitting for 6 is probably a consequence of its different geometry. For 3–5 a molecular field correction of an antiferromagnetic nature improved the quality of the fits, while for 6 it resulted negligibly small.
 |
| | Fig. 6 Magnetic functions for 3: effective magnetic moment vs. temperature (left), magnetization vs. magnetic field (right), magnetic susceptibility vs. temperature (inset); grey circles: experimental data, solid line: fitted. | |
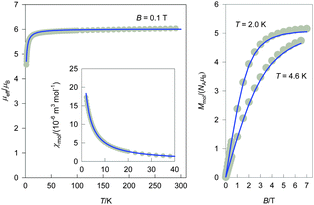 |
| | Fig. 7 Magnetic functions for 4: effective magnetic moment vs. temperature (left), magnetization vs. magnetic field (right), magnetic susceptibility vs. temperature (inset); grey circles: experimental data, solid line: fitted. | |
 |
| | Fig. 8 Magnetic functions for 5: effective magnetic moment vs. temperature (left), magnetization vs. magnetic field (right), magnetic susceptibility vs. temperature (inset); grey circles: experimental data, solid line: fitted. | |
 |
| | Fig. 9 Magnetic functions for 6 per monomeric unit: effective magnetic moment vs. temperature (left), magnetization vs. magnetic field (right), magnetic susceptibility vs. temperature (inset); grey circles: experimental data, solid line: fitted. | |
Table 4 Summary of magnetic parameters for high spin complexes 3–6, alternative fits are provided (Fig. S9–S12 and Table S1, ESI)
| Complex |
J/cm−1 |
g
|
D/cm−1 |
(zj)/cm−1 |
R(χ)/R(M) |
|
3, [Fe(L3)(NCS)] |
— |
1.98 |
+1.07 |
−0.26 |
0.018/0.025 |
|
4, [Fe(L1)(N3)] |
— |
2.03 |
+0.11 |
−0.28 |
0.026/0.020 |
|
5, [Fe(L5)(NCS)] |
— |
2.01 |
+0.60 |
−0.05 |
0.012/0.015 |
|
6, [Fe(L6)(NCS)] |
−0.56 |
2.03 |
−0.50 |
≈0 |
0.018/0.036 |
Conclusions
Six new mononuclear iron(III) complexes of the general formula [Fe(L)X] have been synthesized, structurally and spectrally characterized, and their magnetic behaviour was investigated. The iron(III) central atom is surrounded by the N3O2 donor atoms of the doubly deprotonated Schiff base ligand L and by the additional N-donor atom of the terminal pseudohalido X = NCS− or N3− ligand. In the iron(III) complexes with the N3N′O2 coordination environment, there is a high possibility of spin crossover phenomenon observation and indeed, two compounds exhibit the spin equilibrium located below the room temperature. Compound 1 shows cooperative and abrupt spin crossover with transition temperatures Tc↑ = 84 K and Tc↓ = 82 K and represents the second reported case of 1/2 ↔ 5/2 hysteretic spin crossover for the [Fe(L)X] family of mononuclear compounds. The spin transition in 2 of the formula [Fe(L2)(NCS)] is gradual and without the presence of a thermal hysteresis. The transition temperature of the orthorhombic and solvent-free compound 2 is noticeably elevated in comparison to the previously reported solvated analogue [Fe(L2)(NCS)]·CH3CN (Tc = 151 K) and to the isostructural compound [Fe(L2)(NCO)] (Tc = 155 K).18a As was shown previously, isostructural iron complexes might exhibit similar spin transition properties and parameters,25 therefore 20 K increase in Tc between the herein reported compound 2 and isostructural complex [Fe(L2)(NCO)] can be assigned to the NCO− < NCS− succession in the spectrochemical series.26 The fitting of the magnetic data showed that in both cases about 13% of the system preserves its spin state as remnant HS molecules.
Comparison of HS and LS structures of both spin crossover complexes 1 and 2 allowed identifying the most affected bond distances and angles of the coordination polyhedron, which change upon the spin crossover transformation. The intermolecular hydrogen bonds between amino nitrogen and methoxy oxygen are most probably responsible for the presence of thermal hysteresis in 1. On the other hand, the presence of weak hydrogen bonds in the crystal structure of 2 is apparently not capable of activating the thermal hysteresis effect; thus the lack of vigorous π–π interactions is probably responsible for the gradual and hysteresis free spin crossover in 2.
Magnetic and structural investigation of mononuclear complexes 3–6 revealed and confirmed the HS state of the iron(III) central atom. The quantitative analysis of the magnetic measurements showed that in all compounds a small zero field splitting is present, accompanied in 4 and 5 by an antiferromagnetic molecular field and in the system 6 also by the dimer antiferromagnetic exchange interaction as agreed with its crystal structure.
Experimental
General
All purchased chemicals were used as received. Methanol, acetone, acetonitrile and diethyl ether were used as solvents without any purification. Elemental analysis of carbon, hydrogen, and nitrogen was carried out using an automated analyser (Vario, Micro Cube). IR spectra were measured by the ATR technique or in KBr pellets in the 4000–400 cm−1 region (Magna FTIR 750, Nicolet). Electronic spectra were measured in acetonitrile solutions (Specord 250 plus, Analytical Jena) in the range of 800–200 nm.
Synthesis
The synthesis of mononuclear neutral complexes 1–6 of the general formula [Fe(L)X] consists of three steps (i) the preparation of the pentadentate Schiff base ligands H2L; (ii) the preparation of the mononuclear [Fe(L)Cl] complexes; and (iii) coordination of the pseudohalide monodentate ligands X (X = NCS− for 1, 2, 3, 5, 6 and X = N3− for 4).
Methanol solutions of aromatic 2-hydroxycarbaldehyde (17 mmol, 2 eq., 30 cm3; 3-ethoxysalicylaldehyde for 1, 4 and 6; 2-hydroxy-1-naphthaldehyde for 2, 5-nitrosalicylaldehyde for 3, and 3,5-di-tert-butylsalicylaldehyde for 5) were combined with corresponding triamine dissolved in 10 cm3 of methanol (8.5 mmol, 1 eq.; N,N′-bis(2-aminoethyl)-1,3-propanediamine for 1–5; 2,2′-diaminodiethylamine for 6). The Schiff base condensation was accompanied by a colour change from colourless to yellow and the mixture was stirred at room temperature for 2 hours. The in situ prepared Schiff base ligands (H2L1 = N,N′-bis(3-ethoxy-2-hydroxybenzylidene)-1,6-diamino-3-azahexane; H2L2 = N,N′-bis(-2-hydroxynaphtyllidene)-1,6-diamino-3-azahexane; H2L3 = N,N′-bis(2-hydroxy-5-nitro-benzylidene)-1,6-diamino-3-azahexane; H2L5 = N,N′-bis(3,5-di-tertbutyl 2-hydroxybenzylidene)-1,6-diamino-3-azahexane, H2L6 = N,N′-bis(3-ethoxy-2-hydroxybenzylidene)-1,-diamino-3-azapentane) were used without any further purification or separation in the next step, where a stoichiometric amount of FeCl3·6H2O dissolved in 10 cm3 of methanol was added at the room temperature. Iron(III) complexation is followed by an abrupt colour change to dark-violet, which is the characteristic colour for this family of the HS [Fe(L)Cl] complexes (where L is a deprotonated form of a N3O2 donor Schiff base ligand). After the addition of triethylamine (20 mmol, 10 cm3 CH3OH), the reaction mixture was stirred at 80 °C for next 1 h, cooled down to 0 °C and filtered. The Dark violet product was washed with a small portion of cold methanol, diethyl ether and dried in a vacuum.
[Fe(L1)Cl].
IR (ATR; ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) /cm−1): 3202 (m, N–H); 3059, 3037, 3026, 3012 (w, C–Harom); 2981, 2967, 2953, 2900, 2858 (w, C–Halif); 1632, 1615, 1595, 1547 (s, C
/cm−1): 3202 (m, N–H); 3059, 3037, 3026, 3012 (w, C–Harom); 2981, 2967, 2953, 2900, 2858 (w, C–Halif); 1632, 1615, 1595, 1547 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC). Elemental analysis for C23H29ClFeN3O4 (Mw = 502.79 g mol−1) found % (expected %): C 54.12 (54.94); N 8.02 (8.36); H 5.24 (5.81). Yield 85%.
N and CC). Elemental analysis for C23H29ClFeN3O4 (Mw = 502.79 g mol−1) found % (expected %): C 54.12 (54.94); N 8.02 (8.36); H 5.24 (5.81). Yield 85%.
[Fe(L2)Cl].
IR (ATR; /cm−1): 3276 (m, N–H); 3045, 3033 (w, C–Harom); 2968, 2933, 2907, 2864, 2846 (w, C–Halif); 1614, 1599, 1585, 1539 (s, CN and CC). Elemental analysis for C27H25ClFeN3O2 (Mw = 514.80 g mol−1) found % (expected %): C 62.58 (62.99); N 8.05 (8.16); H 4.77 (4.89). Yield 81%.
[Fe(L3)Cl].
IR (ATR; /cm−1): 3267 (m, N–H); 3054, 3043 (w, C–Harom); 2973, 2941, 2905, 2842 (w, C–Halif); 1612, 1595, 1586, 1541 (s, CN and CC). Elemental analysis for C19H19ClFeN5O6 (Mw = 504.68 g mol−1) found % (expected %): C 45.33 (45.22); N13.56 (13.88); H 3.82 (3.79). Yield 80%.
[Fe(L5)Cl].
IR (ATR; /cm−1): 3262 (m, N–H); 3057, 3041 (w, C–Harom); 2975, 2946, 2911, 2837 (w, C–Halif); 1611, 1595, 1588, 1541 (s, CN and CC). Elemental analysis for C35H53ClFeN3O2 (Mw = 639.11 g mol−1) found % (expected %): C 65.42 (65.77); N 6.39 (6.57); H8.27 (8.36). Yield 90%.
[Fe(L6)Cl].
IR (ATR; /cm−1): 3181 (m, N–H); 3055, 3028 (w, C–Harom); 2970, 2947, 2916, 2896, 2871, 2861, 2812 (w, C–Halif); 1636,1622, 1594, 1548 (s, CN and CC). Elemental analysis for C35H53ClFeN3O2 (Mw = 488.77 g mol−1) found % (expected %): C 53.92 (54.06); N 8.39 (8.60); H 5.20 (5.57). Yield 73%.
General synthesis of 1, 3, 5 and 6: a solution of the corresponding mononuclear complex [Fe(L)Cl] in acetone (0.4 mmol, 30 cm3) was combined with a solution of potassium thiocyanate (0.5 mmol, 1.2 eq. in 8 cm3 of acetone and water 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The reaction mixture was refluxed at 60 °C for 3 h, cooled down to room temperature and filtered. In the next step, the solution of the corresponding [Fe(L)|(NCS)] complex was retained for slow evaporation at room temperature. In a couple of days, small black crystals suitable for single crystal X-ray analysis were collected.
:3). The reaction mixture was refluxed at 60 °C for 3 h, cooled down to room temperature and filtered. In the next step, the solution of the corresponding [Fe(L)|(NCS)] complex was retained for slow evaporation at room temperature. In a couple of days, small black crystals suitable for single crystal X-ray analysis were collected.
Compound 2 can be prepared by a modified procedure for the preparation of the [Fe(L)Cl] precursor complexes. A solution of the in situ prepared ligand H2L2 (0.01 mol) in 50 cm3 of methanol was treated by 0.01 mol of FeCl3·6H2O. An immediate colour change of the solution to dark black occurred. Then, potassium thiocyanate (0.015 mol) in 10 cm3 of methanol was added into the mixture. After 20 min of stirring with heating, the solution of triethylamine (0.02 mol in 10 cm3 methanol) was added. Finally, the resulting slurry was stirred with heating for next 20 min. The dark grey microcrystalline product was filtered off, washed with methanol/water (v/v, 1:1), methanol and diethylether and dried in a vacuum. Single-crystals were obtained by recrystallization of the microcrystalline product from the methanol/water solution (v/v, 9:1).
Compound 1.
IR (KBr; /cm−1): 3213 (m, N–H), 3055 (w, C–Harom); 2976, 2925, 2902, 2870 (w, C–Halif); 2057 (s, NCS); 1633, 1615, 1597, 1547 (s, CN). Elemental analysis calc. For C24H29FeN4O4S (M = 525.42 g mol−1) found % (expected %): C 55.19 (54.86); H 5.58 (5.56); N 10.68 (10.66); S 6.18 (6.10) %. Yield 76%.
Compound 2.
IR (KBr; /cm−1): compound 2: 3198 (m, broad, N–H); 3051, (w, C–Harom); 2921, 2863 (w, C–Halif); 2047 (s, NCS); 1614, 1602, 1538 (s, CN and CC). Elemental analysis calc. for C28H25FeN4O2S (M = 537.43 g mol−1) found % (expected %): C 62.34 (62.58); H 4.58 (4.69); N 10.33 (10.42); S 5.55 (5.97) %. Yield 76%.
Compound 3.
IR (KBr; /cm−1): 3218 (m, N–H); 2918, 2914, 2870 (w, C–Halif); 2044 (s, NCS); 1641, 1627, 1598, 1551 (s, CN and CC). Elemental analysis calc. for C20H19FeN6O6S (M = 527.31 g mol−1) found % (expected %): C 45.62 (45.55); H 3.54 (3.63); N 15.87 (15.94); S 6.21 (6.08) %. Yield 50%.
Compound 5.
IR (KBr; /cm−1): 3201 (m, N–H), 2955, 2904, 2868 (w, C–Halif); 2070 (s, NCS); 1627, 1611, 1603 (s, CN and CC). Elemental analysis calc. for C36H53FeN4O2S (M = 661.74 g mol−1) found % (expected %): C 65.23 (65.34); H 8.15 (8.07); N 8.31 (8.47); S 4.66 (4.85) %. Yield 48%.
Compound 6.
IR (KBr; /cm−1): 3158 (m, N–H); 3072, 3032 (w, C–Harom); 2979, 2967, 2943, 2924, 2910, 2873 (w, C–Halif); 2047 (s, NCS); 1638, 1621, 1601, 1552 (s, CN). Elemental analysis calc. for C23H27FeN4O4S (M = 511.40 g mol−1) found % (expected %): C 52.54 (54.02); H 5.44 (5.32); N 10.67 (10.96); S 5.96 (6.27). Yield 49%.
Complex 4.
A solution of [Fe(L1)Cl] in methanol (0.4 mmol, 30 cm3) was mixed with a solution of sodium azide in methanol (0.5mmol; 15 cm3 of methanol:water = 2:1) and refluxed at 80 °C for about 3.5 hours, cooled down to room temperature and filtered. The solution of [Fe(L1)N3] was retained to evaporate at room temperature and after a few days dark crystals of 6 were separated by filtration, washed with diethyl ether and dried in a vacuum. IR (KBr; /cm−1): 3253 (m, N–H); 3059, 3049, 3020 (w, C–Harom); 2969, 2933, 2922, 2914 (w, C–Halif); 2056 (s, N3−); 1633, 1616, 1598, 1546 (s, CN and CC). Elemental analysis calc. for C23H29FeN6O4 (M = 509.36 g mol−1) found % (expected %): C: 54.29 (54.23); H: 5.80 (5.74); N: 16.65 (16.50). Yield 44%.
Crystal structure determination
Single crystals of 1 were mounted to an Xcalibur 2 diffractometer with a CCD sapphire 2 detector with MoKα radiation at 293 K. The diffraction data at 100 K were collected on a Bruker Kappa APEX-II diffractometer with Triumph monochromator MoKα radiation. Intensity data at 15 K were collected on a kappa goniostat equipped with an APEX-II CCD detector at the ID-15-B beamline at an APS Chicago synchrotron source. Single crystals of 2, 3 and 5 were mounted to an Xcalibur 2 diffractometer with a CCD sapphire 2 detector with graphite monochromator MoKα radiation at 293 K.
Single crystals of 4 and 6 were mounted to a Bruker X8 APEX-II Ultra diffractometer with an APEX-II CCD detector with a Mo micro-focused rotating anode and multilayer monochromator at 100 K.
The structures were solved by direct methods using the programs SIR-2011 (ref. 23) or SHELXS-2013 (ref. 24) and refined by the full-matrix least-squares method on all F2 data using the program SHELXL-2013 (ref. 24). All hydrogen atoms were found in differential Fourier maps and their parameters were refined using the riding model with Uiso(H) = 1.2 (CH, CH2, OH) or 1.5 Ueq (CH3).
Magnetic measurements
Magnetic investigations were performed using a SQUID magnetometer (MPMS-XL7, Quantum Design) in the RSO mode of detection. In all cases, the temperature dependence of the magnetic moment was recorded at 0.1 T as an external magnetic field and the temperature sweeping rate was 1 K min−1 with 120 second stabilisation of every temperature data point. The gelatine-made capsules as sample holders were used and their small diamagnetic contribution is negligible in the overall magnetization, which was dominated by the sample. The diamagnetic corrections to the molar magnetic susceptibilities were applied using Pascal constants.22
Theoretical analysis
The spin Hamiltonian appropriate for the description of the energy spectrum of the HS or the LS state of the presented systems was postulated in the form| | | Ĥ = −(J/2)ℏ−2Ŝ2 + Dℏ−2(Ŝz2 − Ŝ2/3) + μBℏ−1gB·Ŝz + (zj)ℏ−2〈Sz〉TŜz | (1) |
where J is the isotropic exchange coupling constant in the eventual pseudodimer, D is the parameter of the axial zero-field splitting applicable to the high-spin state, and (zj) is the molecular field interaction constant; 〈Sz〉T represents the canonical ensemble mean of the spin eigenvalue in the z-direction. An isotropic g-factor was considered, which is justified by small ZFS (|D| ≤ 5 cm−1).
For the given value of the magnetic field B the eigenvalues of Hamiltonian (1) along with their field derivatives are found for each spatial direction. Subsequently, the molar magnetic susceptibility as a function of temperature χmol,a = f(T) and the molar magnetization as a function of magnetic field Mmol,a = f(B) can be enumerated and presented as spatial averages with regard to the powder state of the samples.22a On this level the temperature independent magnetism χTIM is introduced as an empirical correction which has the same origin as ZFS22b and helps to adjust the slope of the χmol = f(T) function while its effect on the magnetization is negligible. For easier interpretation the magnetic susceptibility is transformed into the effective magnetic moment in units of Bohr magneton as
| |  | (2) |
The above approach is appropriate to complexes
3–6 where
S = 5/2. For
1 and
2, where the spin crossover
S = 1/2 →
S = 5/2 occurs, it must be combined with a model for the conversion curve
xH =
f(
T) where
xH is the HS mole fraction. The Ising-like model with vibrations was employed for this purpose
28 that results in an implicit equation
where
| | | F = revexp[−(E − 2γxH)/kBT] | (4) |
Here
E is an effective energy gap proportional to the enthalpy of the spin transition and
γ stands for the solid-state cooperativeness. The spin state degeneracy ratio is
| |  | (5) |
where
hH and
hL are the averaged vibration energies. Although it may appear that the model is overparametrized, the parameters tune different parts of the curve and the convergence of the iterative procedure is fast. The transition enthalpy and entropy are calculated as
| | | ΔS = R·lnrev|x(HS)=1/2 | (6) |
and the transition temperature
| |  | (7) |
It has to be stressed that
eqn (5) and (6) are valid for systems without a remnant HS phase and the experimental values of Δ
H and Δ
S can be sample-dependent, since the frozen HS state can be present due to the grain distribution of the powder sample.
27 We therefore supposed that the remnant HS molecules are identical to the high temperature HS molecules and also that they are dissolved sufficiently, so that the molecular field effect is relevant only for the LS part of the system. The resulting HS fraction is thus renormalized due to the frozen HS fraction
xfrz as
| | 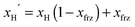 | (8) |
and the susceptibility for
1 and
2 is calculated through
| |  | (9) |
Acknowledgements
Grant Agencies (Slovakia: VEGA 1/0522/14, APVV-0014-11, APVV-0132-11; Czech Republic: CZ.1.05/2.1.00/03.0058, IGA_PrF_2014009) are acknowledged for the financial support. Mrs. Ingrid Svoboda (TU Darmstadt) is greatly appreciated for the single crystal diffraction experiment of compound 1 at room temperature.
Notes and references
-
(a) O. Kahn and C. Jay Martinez, Science, 1998, 279, 44 CrossRef CAS;
(b) O. Kahn, J. Krober and C. Jay, Adv. Mater., 1992, 4, 718 CrossRef CAS.
-
(a) S. Bonhommeau, T. Guillon, L. M. L. Daku, P. Demont, J. S. Costa, J. F. Letard, G. Molnar and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 1625 CrossRef CAS PubMed;
(b) A. Bousseksou, G. Molnar, P. Demont and J. Menegotto, J. Mater. Chem., 2003, 13, 2069 RSC;
(c) F. Varret, K. Boukheddaden, E. Codjovi and A. Goujon, Hyperfine Interact., 2005, 165, 37 CrossRef.
- J. Linares, E. Codjovi and Y. Garcia, Sensors, 2012, 12, 4479 CrossRef CAS PubMed.
- S. Shi, G. Schmerber, J. Arabski, J. B. Beaufrand, D. J. Kim, S. Boukari, M. Bowen, N. T. Kemp, N. Viart, G. Rogez, E. Beaurepaire, H. Aubriet, J. Petersen, C. Becker and D. Ruch, Appl. Phys. Lett., 2009, 95, 043303 CrossRef.
- M. Matsuda, H. Isozaki and H. Tajima, Chem. Lett., 2008, 37, 374 CrossRef CAS.
- I. Šalitroš, N. T. Madhu, R. Boča, J. Pavlik and M. Ruben, Monatsh. Chem., 2009, 140, 695 CrossRef.
- H. J. Shepherd, G. Molniiár, W. Nicolazzi, L. Salmon and A. Bousseksou, Eur. J. Inorg. Chem., 2013, 653 CrossRef CAS.
-
(a) S. Basak, P. Hui and R. Chandrasekar, Chem. Mater., 2013, 25, 3408 CrossRef CAS.
- M. Cavallini, I. Bergenti, S. Milita, G. Ruani, I. Salitros, Z. R. Qu, R. Chandrasekar and M. Ruben, Angew. Chem., Int. Ed., 2008, 47, 8596 CrossRef CAS PubMed.
-
(a) B. Schaefer, C. Rajnak, I. Salitros, O. Fuhr, D. Klar, C. Schmitz-Antoniak, E. Weschke, H. Wende and M. Ruben, Chem. Commun., 2013, 49, 10986 RSC;
(b) T. Palamarciuc, J. C. Oberg, F. E. Hallak, C. F. Hirjibehedin, M. Serri, S. Heutz, J. F. Léthard and P. Rosa, J. Mater. Chem., 2012, 22, 9690 RSC.
-
(a) S. M. Park, Y. Kim and S.-J. Kim, Eur. J. Inorg. Chem., 2003, 4117 CrossRef CAS;
(b) G. Rogez, A. Marvilliers, E. Riviére, J. P. Audiére, F. Lloret, F. Varret, A. Goujon, N. Mendenez, J. J. Girerd and T. Mallah, Angew. Chem., Int. Ed., 2000, 39, 2885 CrossRef CAS;
(c) G. Rogez, S. Parsons, C. Paulsen, V. Villar and T. Mallah, Inorg. Chem., 2001, 40, 3836 CrossRef CAS PubMed;
(d) I. Šalitroš, R. Boča, R. Herchel, J. Moncol, I. Nemec, M. Ruben and F. Renz, Inorg. Chem., 2012, 51, 12755 CrossRef PubMed;
(e) R. Boča and I. Šalitroš, Chem. Pap., 2008, 62, 575 Search PubMed.
-
(a) I. Šalitroš, R. Boča, L. Dlháň, M. Gembický, J. Kožíšek, J. Linares, J. Moncol, I. Nemec, L. Perašínová, F. Renz, I. Svoboda and H. Fuess, Eur. J. Inorg. Chem., 2009, 3141 CrossRef;
(b) R. Boča, I. Šalitroš, J. Kožíšek, J. Linares, J. Moncol and F. Renz, Dalton Trans., 2010, 39, 2198 RSC;
(c) R. Herchel, R. Boča, M. Gembický, J. Kožíšek and F. Renz, Inorg. Chem., 2004, 43, 4103 CrossRef CAS PubMed.
-
(a) I. Nemec, R. Boča, R. Herchel, Z. Trávníček, M. Gembický and W. Linert, Monatsh. Chem., 2009, 140, 815 CrossRef CAS;
(b) S. Imatomi, T. Sato, T. Hamamatsu, R. Kitashima and N. Matsumoto, Bull. Chem. Soc. Jpn., 2007, 80, 2375 CrossRef CAS.
-
(a) R. Kitashima, S. Imatomi, M. Yamada, N. Matsumoto and Y. Maeda, Chem. Lett., 2005, 34, 1388 CrossRef CAS;
(b) T. Fujinami, K. Nishi, R. Kitashima, K. Murakami, N. Matsumoto, S. Iijima and K. Toriumi, Inorg. Chim. Acta, 2011, 376, 136 CrossRef CAS.
-
(a) S. Hayami, Y. Hosokoshi, K. Inoue, Y. Einaga, O. Sato and Y. Maeda, Bull. Chem. Soc. Jpn., 2001, 74, 2361–2368 CrossRef CAS;
(b) R. Herchel and Z. Trávníček, Dalton Trans., 2013, 42, 16279 RSC;
(c) S. Hayami, K. Inoue and Y. Maeda, Mol. Cryst. Liq. Cryst., 1999, 335, 573 CrossRef.
-
(a) A. Sour, M. L. Boillot, E. Riviére and P. Lesot, Eur. J. Inorg. Chem., 1999, 2117 CrossRef CAS;
(b) C. Faulmann, S. Dorbes, B. G. de Bonneval, G. Molnár, A. Bousseksou, C. J. Gomez-Garcia, E. Coronado and L. Valade, Eur. J. Inorg. Chem., 2005, 3261 CrossRef CAS;
(c) A. Bannwarth, S. O. Schmidt, G. Peters, G. D. Sönnichsen, W. Thimm, R. Herges and F. Tuczek, Eur. J. Inorg. Chem., 2012, 2776 CrossRef CAS.
-
(a) C. R. Mayer, G. Cucchiaro, J. Jullien, F. Dumur, J. Marrot, E. Dumas and F. Sécheresse, Eur. J. Inorg. Chem., 2008, 3614 CAS;
(b) H. Jacob, K. Kathirvel, F. Petersen, T. Strunskus, A. Bannwarth, S. Meyer and F. Tuczek, Langmuir, 2013, 29, 8534 CrossRef CAS PubMed.
-
(a) I. Nemec, R. Herchel, R. Boča, Z. Trávníček, I. Svoboda, H. Fuess and W. Linert, Dalton Trans., 2011, 40, 10090 RSC;
(b) C. Krüger, P. Augustín, I. Nemec, Z. Trávníček, H. Oshio, R. Boča and F. Renz, Eur. J. Inorg. Chem., 2013, 902 CrossRef.
- K. Murray, Coord. Chem. Rev., 1974, 12, 1–35 CrossRef CAS.
-
(a) B. P. Gaber, V. Miskowski and T. G. Spiroc, J. Am. Chem.
Soc., 1974, 96, 6868 CAS;
(b) R. M. Wallace and E. K. Dukes, J. Phys. Chem., 1961, 65, 2094 CrossRef CAS;
(c) H. S. Frank and R. L Oswalt, J. Am. Chem. Soc., 1947, 69, 1321 CrossRef CAS.
-
 ; where φ is value of N–Fe–N octahedron angle P. Guionneau, M. Marchivie, G. Bravic, J. F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97 CrossRef CAS.
; where φ is value of N–Fe–N octahedron angle P. Guionneau, M. Marchivie, G. Bravic, J. F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97 CrossRef CAS.
-
(a)
R. Boča, Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed;
(b) R. Boča, Coord. Chem. Rev., 2004, 248, 757 CrossRef.
- M. C. Burla, R. Caliandro, M. Camalli, B. Varrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
-
(a) C. Rajadurai, Z. Qu, O. Fuhr, B. Gopalan, R. Kruk, M. Ghafari and M. Ruben, Dalton Trans., 2007, 3531 RSC;
(b) J. Elhaik, D. J. Evans, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2005, 1693 RSC;
(c) I. Salitros, O. Fuhr, A. Eichhöfer, R. Kruk, J. Pavlik, L. Dlháň, R. Boča and M. Ruben, Dalton Trans., 2012, 41, 5163 RSC;
(d) I. Salitros, J. Pavlik, R. Boča, O. Fuhr, C. Rajadurai and M. Ruben, CrystEngComm, 2010, 12, 2361 RSC.
-
A. M. Golub, H. Koehler and V. V. Skopenko, Chemistry of Pseudohalides, Elsevier, Amsterdam, 1986 Search PubMed.
-
M. A. Halcrow, Spin-Crossover Materials, Properties and Applications, John Wiley & Sons, Hoboken, 2013 Search PubMed.
- J. Pavlik and R. Boča, Eur. J. Inorg. Chem., 2013, 697 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 995955–995963. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01363h |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Ján
Pavlik
a,
Milan
Gembický
c,
Zdeněk
Trávníček
b,
Roman
Boča
d and
Ivan
Šalitroš
*a
b,
Ján
Pavlik
a,
Milan
Gembický
c,
Zdeněk
Trávníček
b,
Roman
Boča
d and
Ivan
Šalitroš
*a
 .
.